
Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Microglia Cultures
2.4. BV2 Cell Culture
2.5. Ex Vivo Microglia Isolation
2.6. Isolation of Peritoneal Macrophages (PMs) and Bone Marrow-Derived Monocytes (BMMCs)
2.7. Flow Cytometry
2.8. RNA Isolation, Reverse Transcription and Quantitative RT-PCR
2.9. Statistics
3. Results
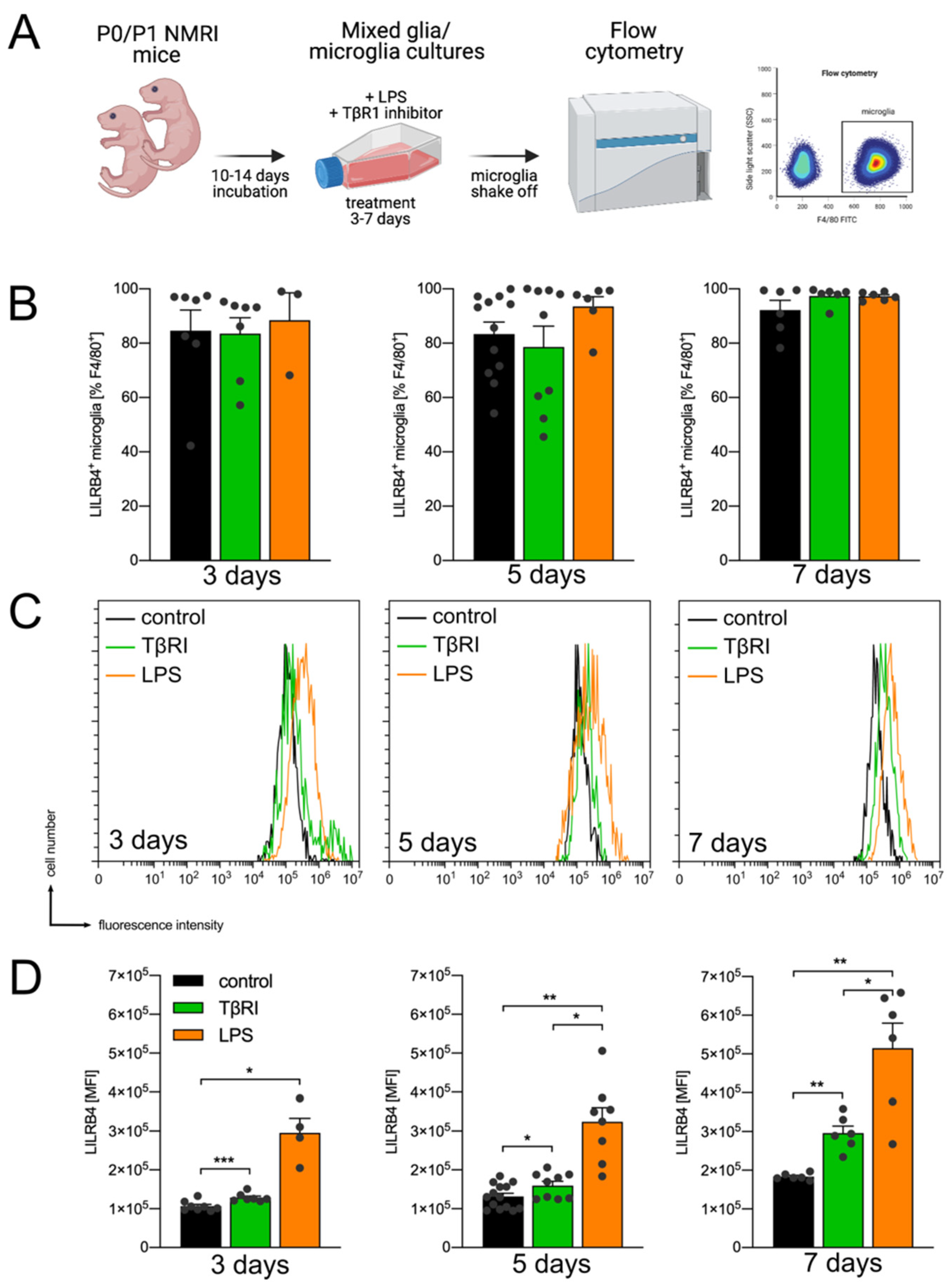
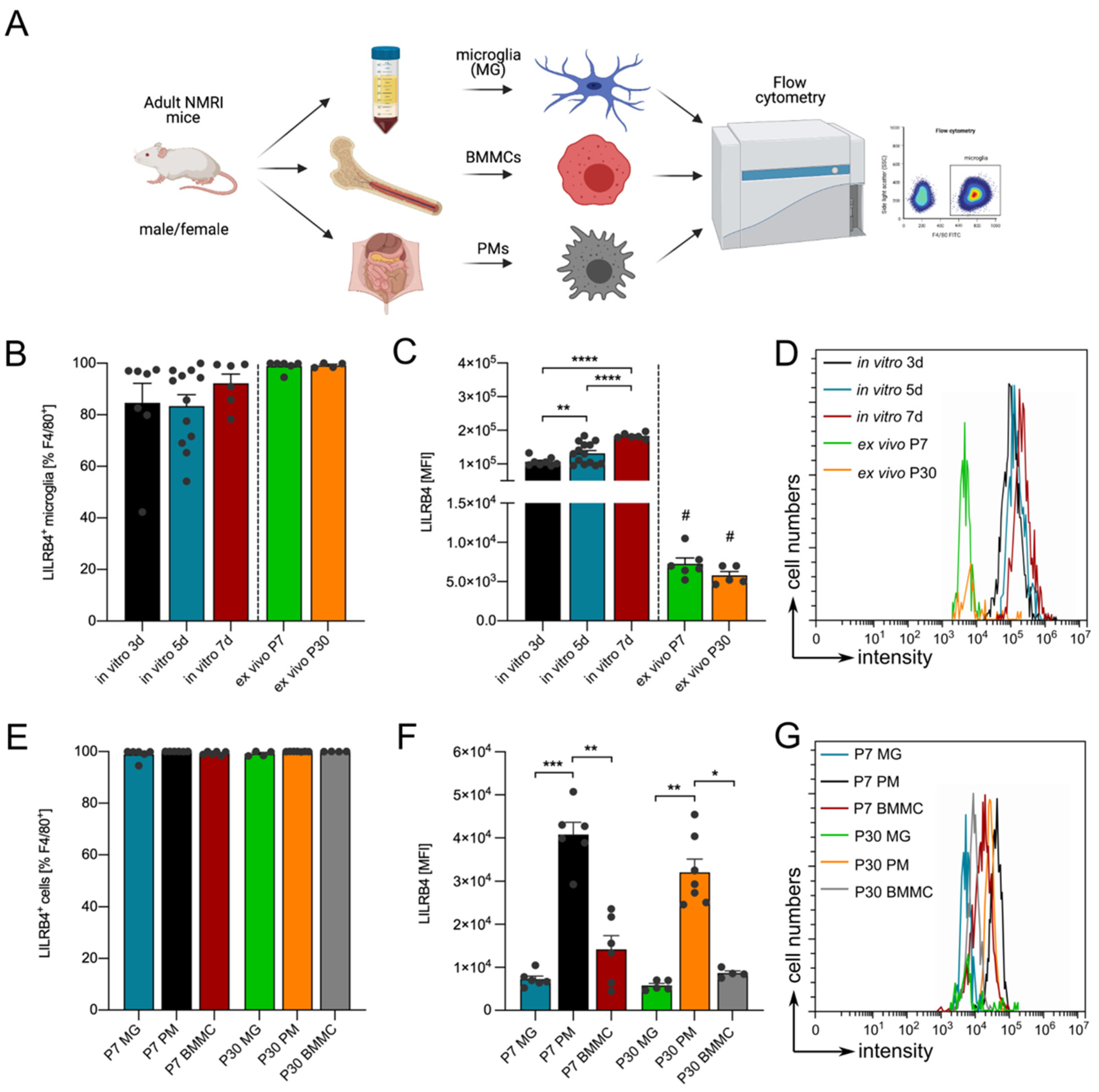
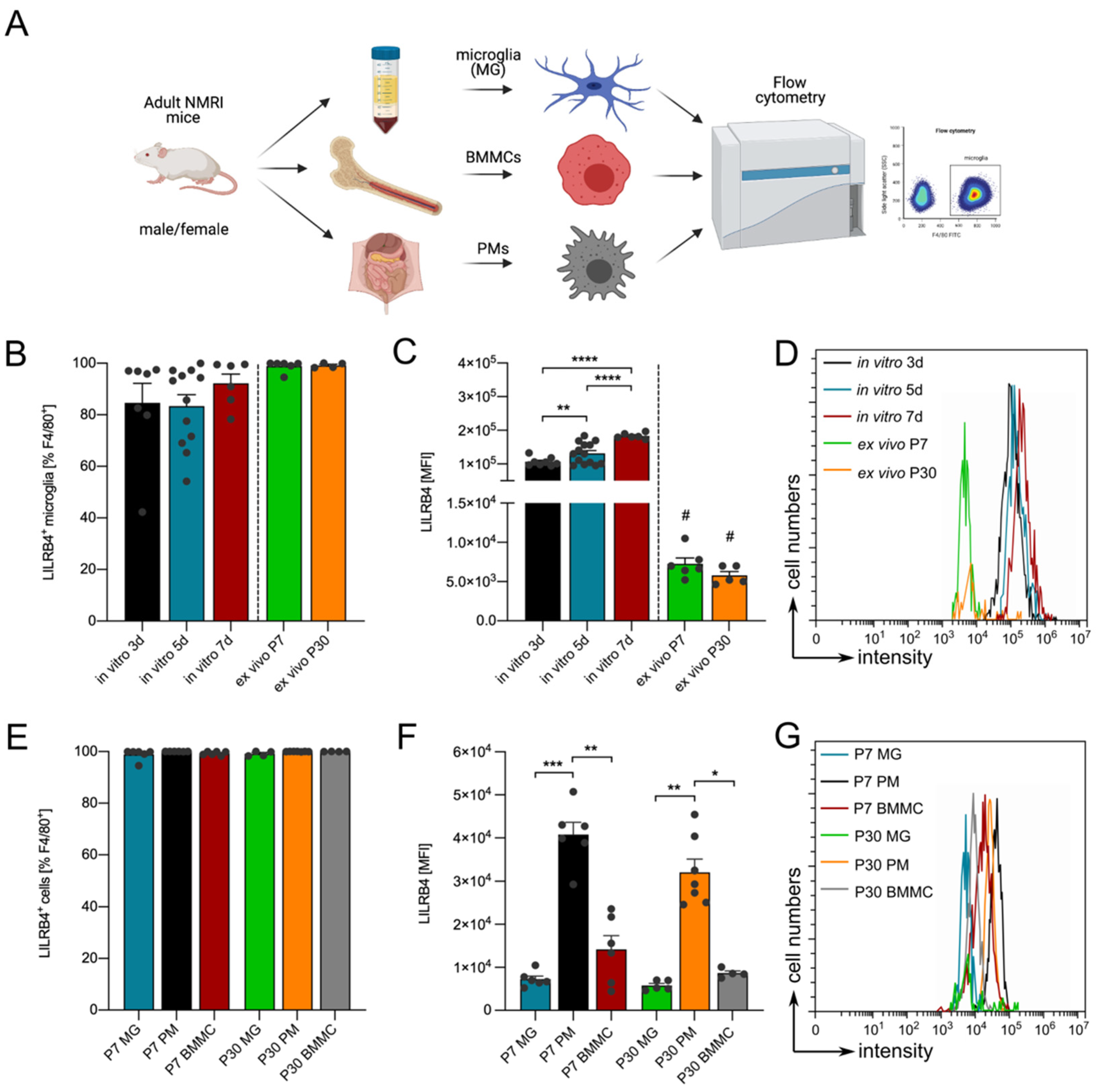
3.1. Expression of Lilrb4 in Primary Microglia In Vitro
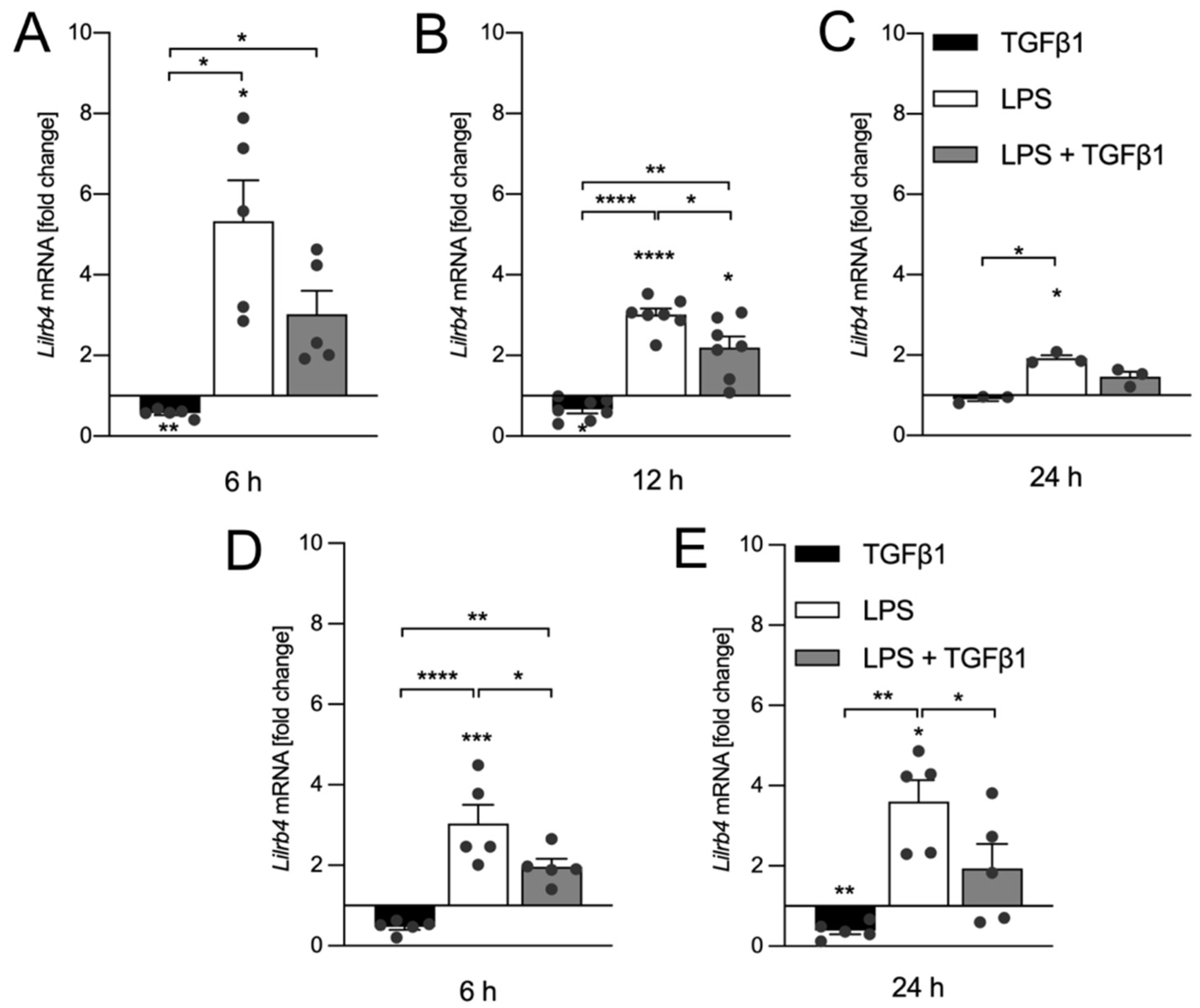
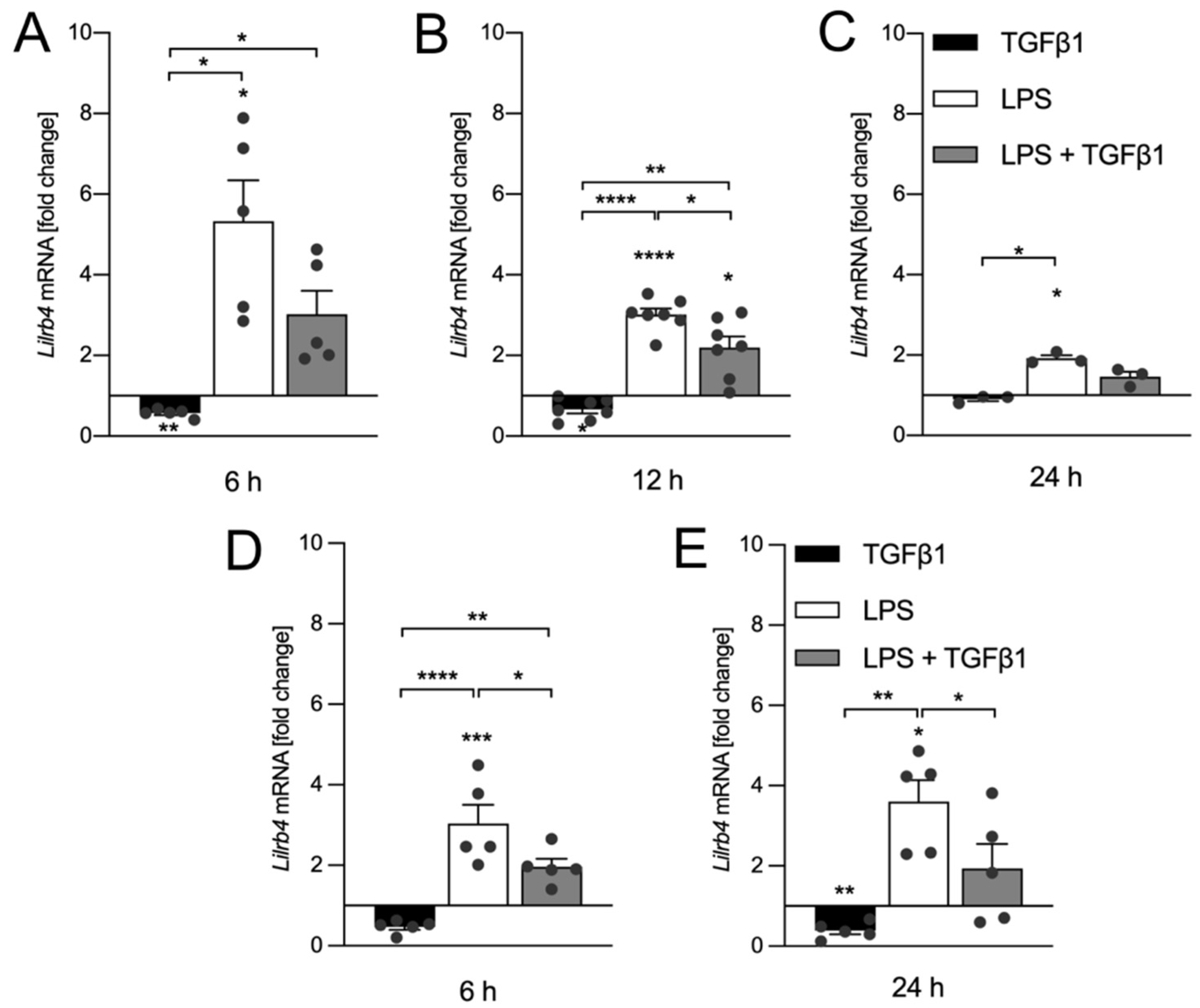
3.2. TGFβ1 Inhibits LPS-Mediated Transcriptional Upregulation of Lilrb4
3.3. Lilrb4 Is Highly Expressed in Peripheral Macrophages
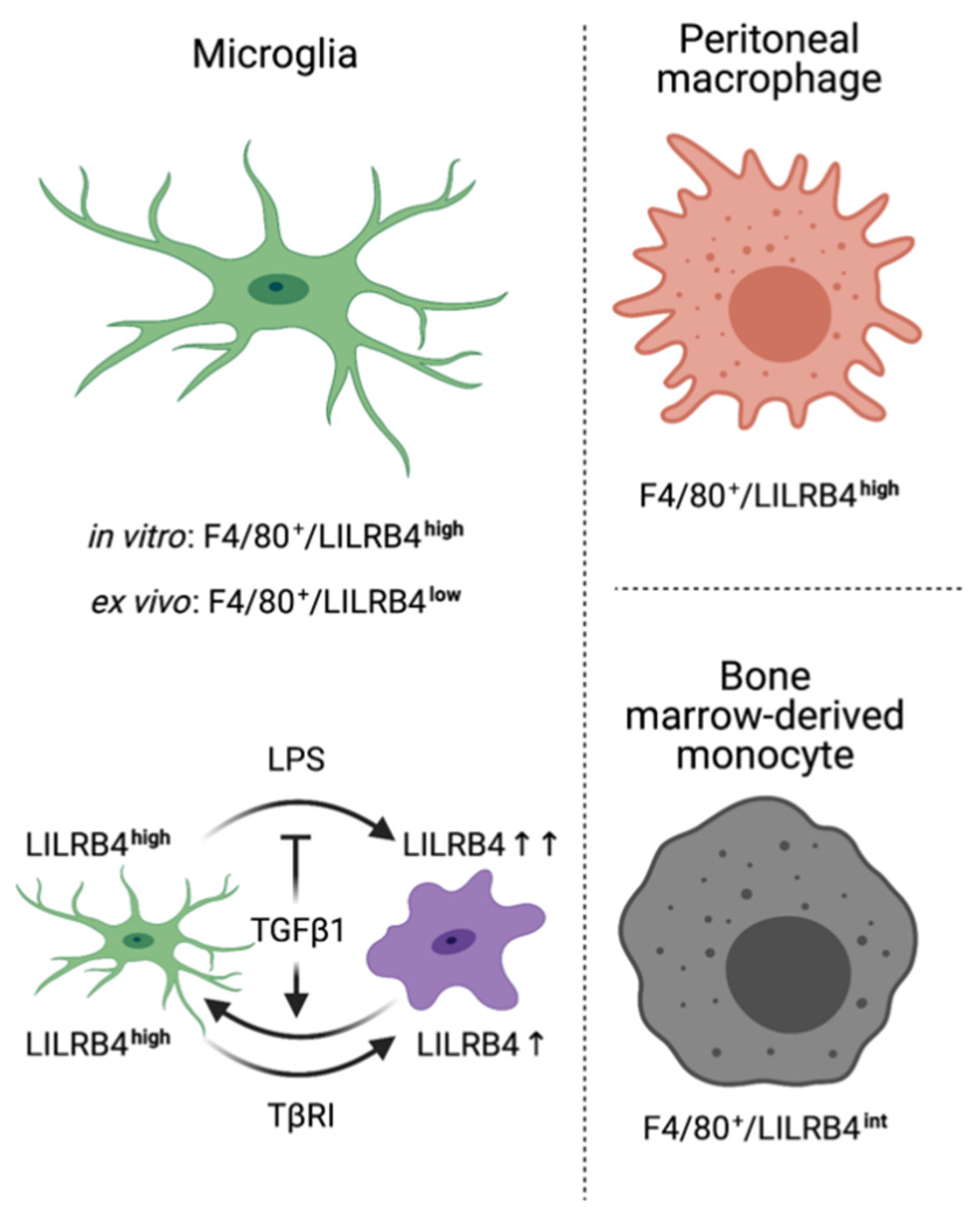
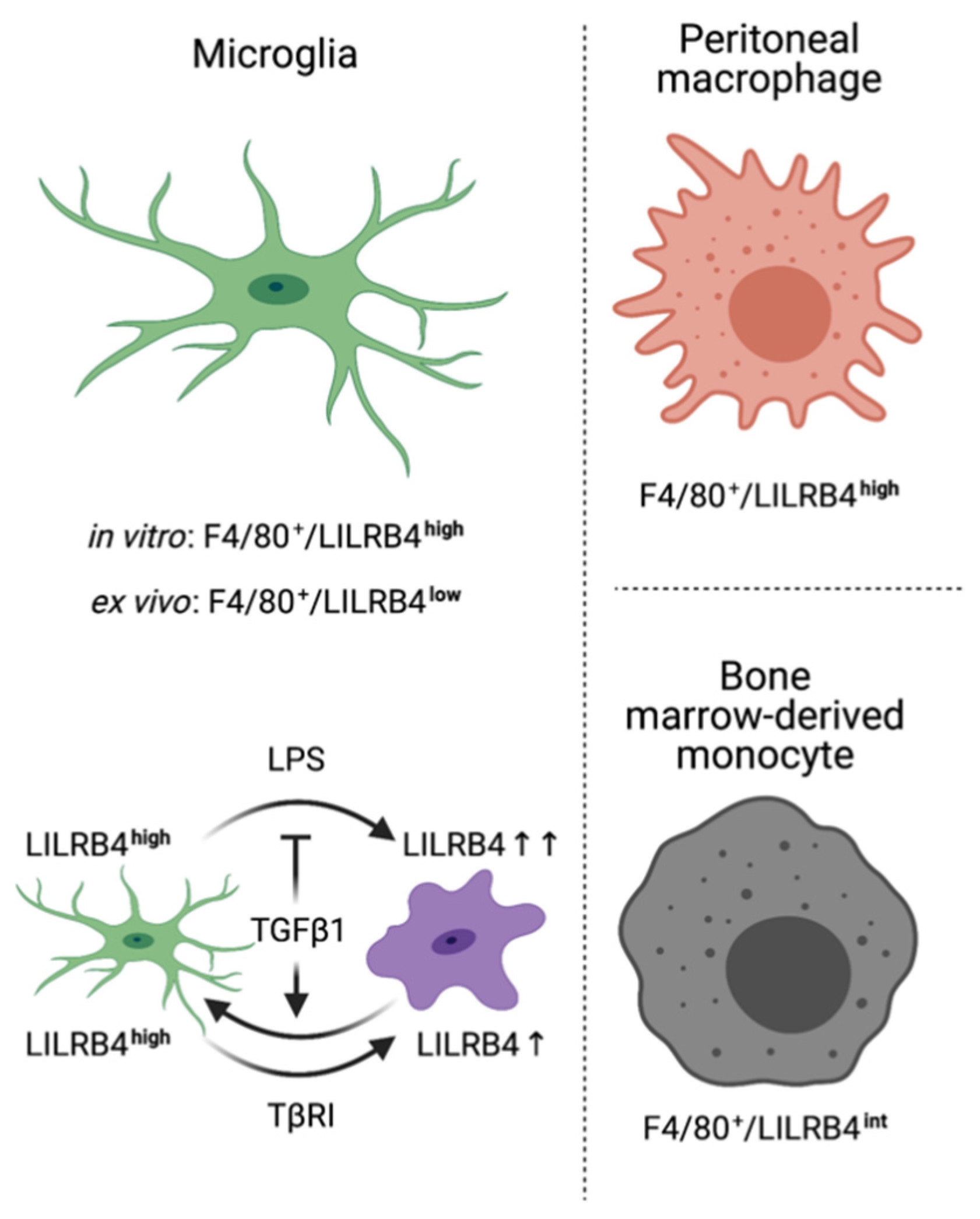
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M.; et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Attaai, A.; Neidert, N.; von Ehr, A.; Potru, P.S.; Zöller, T.; Spittau, B. Postnatal maturation of microglia is associated with alternative activation and activated TGFβ signaling. Glia 2018, 66, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, T.D.; Lizama, C.O.; Cautivo, K.M.; Santander, N.; Lin, L.; Qiu, H.; Huang, E.J.; Liu, C.; Mukouyama, Y.-S.; Reichardt, L.F.; et al. Impaired αVβ8 and TGFβ signaling lead to microglial dysmaturation and neuromotor dysfunction. J. Exp. Med. 2019, 216, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Garrison, B.S.; Ma, W.; Wang, R.; Jiang, A.; Li, J.; Mistry, M.; Bronson, R.T.; Santoro, D.; Franco, C.; et al. A Milieu Molecule for TGF-β Required for Microglia Function in the Nervous System. Cell 2018, 174, 156–171.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spittau, B.; Dokalis, N.; Prinz, M. The Role of TGFβ Signaling in Microglia Maturation and Activation. Trends Immunol. 2020, 41, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Spittau, B.; Wullkopf, L.; Zhou, X.; Rilka, J.; Pfeifer, D.; Krieglstein, K. Endogenous transforming growth factor-beta promotes quiescence of primary microglia in vitro. Glia 2013, 61, 287–300. [Google Scholar] [CrossRef]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Spittau, B.; Krieglstein, K. TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. J. Neuroinflamm. 2012, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zöller, T.; Krieglstein, K.; Spittau, B. TGFβ1 inhibits IFNγ-mediated microglia activation and protects mDA neurons from IFNγ-driven neurotoxicity. J. Neurochem. 2015, 134, 125–134. [Google Scholar] [CrossRef]
- Zlotnik, A.; Spittau, B. GDNF fails to inhibit LPS-mediated activation of mouse microglia. J. Neuroimmunol. 2014, 270, 22–28. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Zöller, T.; Attaai, A.; Potru, P.S.; Ruß, T.; Spittau, B. Aged Mouse Cortical Microglia Display an Activation Profile Suggesting Immunotolerogenic Functions. Int. J. Mol. Sci. 2018, 19, 706. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 1847–1860. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wu, Q.; Shi, J.; Guo, W.; Jiang, X.; Zhou, B.; Ren, C. LILRB4, from the immune system to the disease target. Am. J. Transl. Res. 2020, 12, 3149–3166. [Google Scholar] [PubMed]
- Bąbolewska, E.; Brzezińska-Błaszczyk, E. Mast cell inhibitory receptors. Postepy Hig. Med. Dosw. 2012, 66, 739–751. [Google Scholar] [CrossRef]
- Park, M.; Liu, R.W.; An, H.; Geczy, C.L.; Thomas, P.S.; Tedla, N. A dual positive and negative regulation of monocyte activation by leukocyte Ig-like receptor B4 depends on the position of the tyrosine residues in its ITIMs. Innate Immun. 2017, 23, 381–391. [Google Scholar] [CrossRef]
- Zhou, J.S.; Friend, D.S.; Lee, D.M.; Li, L.; Austen, K.F.; Katz, H.R. gp49B1 deficiency is associated with increases in cytokine and chemokine production and severity of proliferative synovitis induced by anti-type II collagen mAb. Eur. J. Immunol. 2005, 35, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Qiu, T.; Zhou, J.; Wang, T.; Chen, Z.; Ma, X.; Zhang, L.; Zou, J. Leukocyte immunoglobulin-like receptor B4 deficiency exacerbates acute lung injury via NF-κB signaling in bone marrow-derived macrophages. Biosci. Rep. 2019, 39, BSR20181888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.A.; Cardona, S.M.; Cardona, A.E. Isolation and analysis of mouse microglial cells. Curr. Protoc. Immunol. 2014, 104, 14.35.1–14.35.15. [Google Scholar] [CrossRef] [Green Version]
- Koerten, H.K.; Ploem, J.S.; Daems, W.T. Ingestion of latex beads by filopodia of adherent mouse peritoneal macrophages. A scanning electron microscopical and reflection contrast microscopical study. Exp. Cell Res. 1980, 128, 470–475. [Google Scholar] [CrossRef]
- Wagner, M.; Koester, H.; Deffge, C.; Weinert, S.; Lauf, J.; Francke, A.; Lee, J.; Braun-Dullaeus, R.C.; Herold, J. Isolation and intravenous injection of murine bone marrow derived monocytes. J. Vis. Exp. 2014, 94, 52347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Liu, Z.; Cao, B.-B.; Qiu, Y.-H.; Peng, Y.-P. TGF-β1 Neuroprotection via Inhibition of Microglial Activation in a Rat Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 433–446. [Google Scholar] [CrossRef]
- Taylor, R.A.; Chang, C.-F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Qin, J.-J.; Zhang, Y.; Cheng, W.-L.; Ji, Y.-X.; Gong, F.-H.; Zhu, X.-Y.; Zhang, Y.; She, Z.-G.; Huang, Z.; et al. LILRB4 deficiency aggravates the development of atherosclerosis and plaque instability by increasing the macrophage inflammatory response via NF-κB signaling. Clin. Sci. 2017, 131, 2275–2288. [Google Scholar] [CrossRef]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10, 1043. [Google Scholar] [CrossRef]
- Horvat, A.; Schwaiger, F.; Hager, G.; Brocker, F.; Streif, R.; Knyazev, P.; Ullrich, A.; Kreutzberg, G.W. A novel role for protein tyrosine phosphatase shp1 in controlling glial activation in the normal and injured nervous system. J. Neurosci. 2001, 21, 865–874. [Google Scholar] [CrossRef]
- Zhao, J.; Brooks, D.M.; Lurie, D.I. Lipopolysaccharide-activated SHP-1-deficient motheaten microglia release increased nitric oxide, TNF-alpha, and IL-1beta. Glia 2006, 53, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Raftery, M.J.; Thomas, P.S.; Geczy, C.L.; Bryant, K.; Tedla, N. Leukocyte immunoglobulin-like receptor B4 regulates key signalling molecules involved in FcγRI-mediated clathrin-dependent endocytosis and phagocytosis. Sci. Rep. 2016, 6, 35085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salih, D.A.; Bayram, S.; Guelfi, S.; Reynolds, R.H.; Shoai, M.; Ryten, M.; Brenton, J.W.; Zhang, D.; Matarin, M.; Botia, J.A.; et al. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 2019, 1, fcz022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; d´Errico, P.; Snaidero, N.; Costa Jordão, M.J.; Böttcher, C.; Kierdorf, K.; et al. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzschmar, F.; Piecha, R.; Jahn, J.; Potru, P.S.; Spittau, B. Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia. Biology 2021, 10, 1300. https://doi.org/10.3390/biology10121300
Kretzschmar F, Piecha R, Jahn J, Potru PS, Spittau B. Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia. Biology. 2021; 10(12):1300. https://doi.org/10.3390/biology10121300
Chicago/Turabian StyleKretzschmar, Felix, Robin Piecha, Jannik Jahn, Phani Sankar Potru, and Björn Spittau. 2021. "Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia" Biology 10, no. 12: 1300. https://doi.org/10.3390/biology10121300
APA StyleKretzschmar, F., Piecha, R., Jahn, J., Potru, P. S., & Spittau, B. (2021). Characterization of the Leucocyte Immunoglobulin-like Receptor B4 (Lilrb4) Expression in Microglia. Biology, 10(12), 1300. https://doi.org/10.3390/biology10121300