
Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways

Abstract
Simple Summary
Abstract
1. Introduction
2. Diagnosis of Glaucoma and Screening for Potential Biomarkers
2.1. Proteins Involved in Cytoskeleton Organization
2.2. SERPIN Gene Family
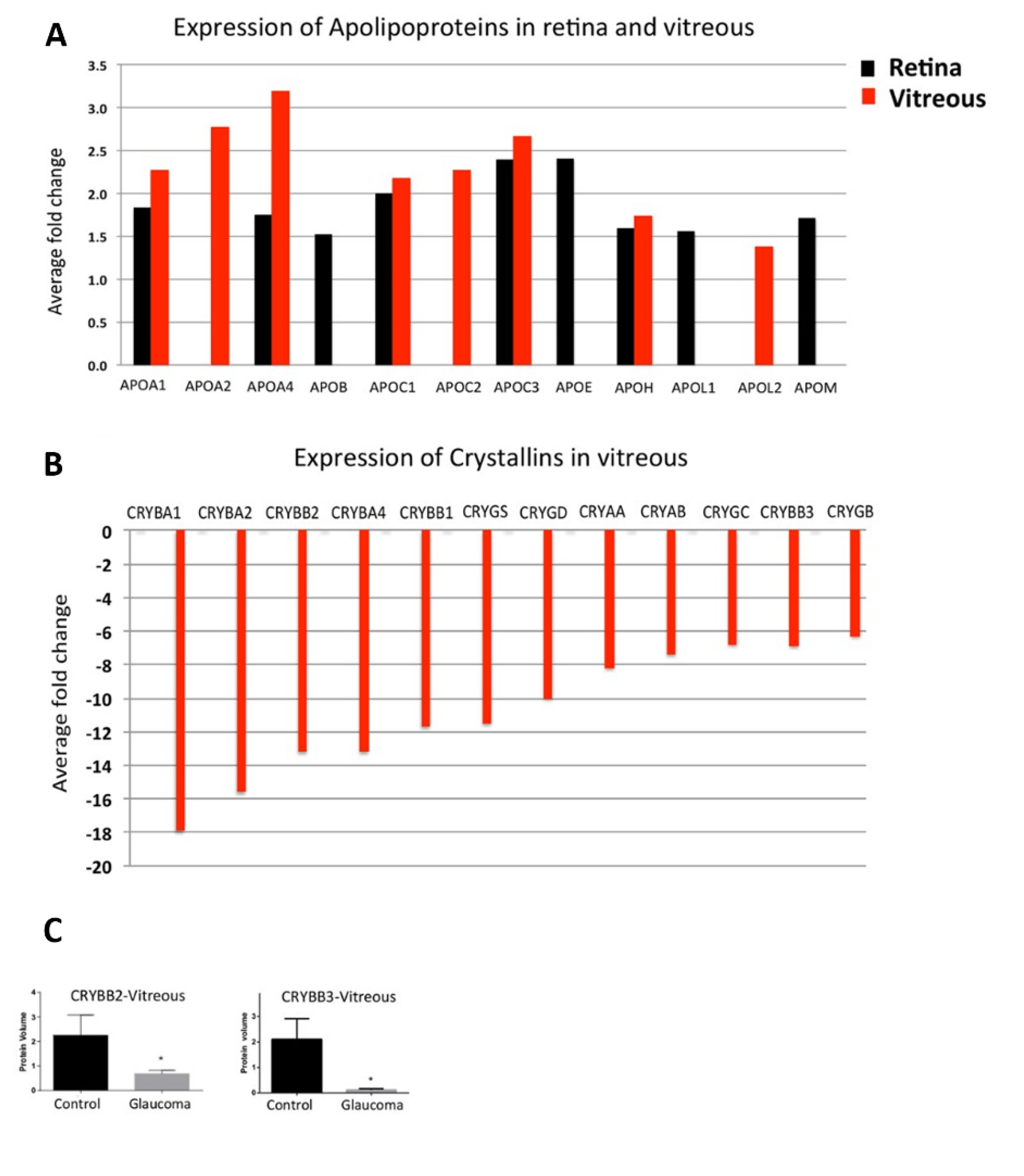
2.3. Apolipoproteins
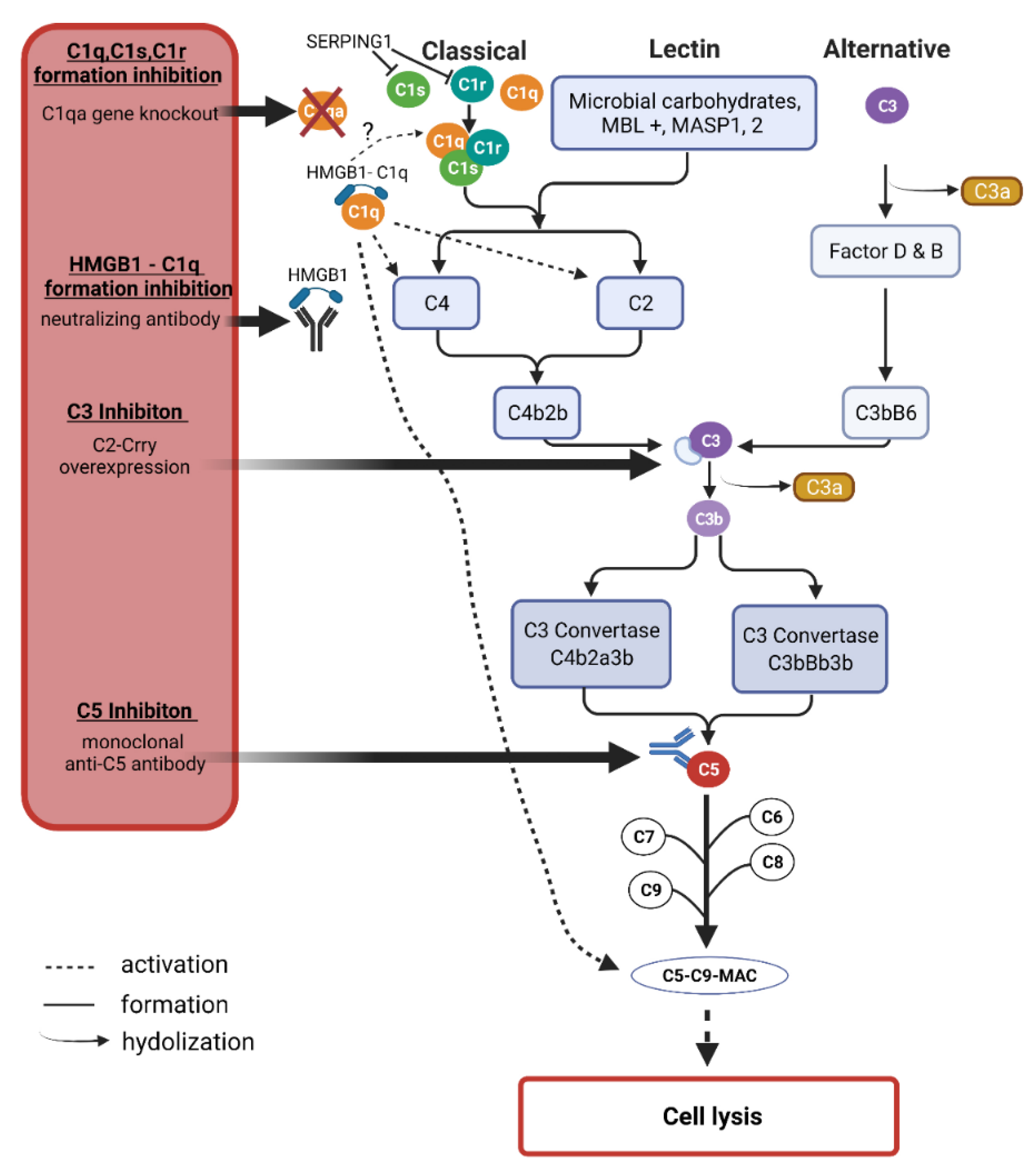
2.4. Complement System
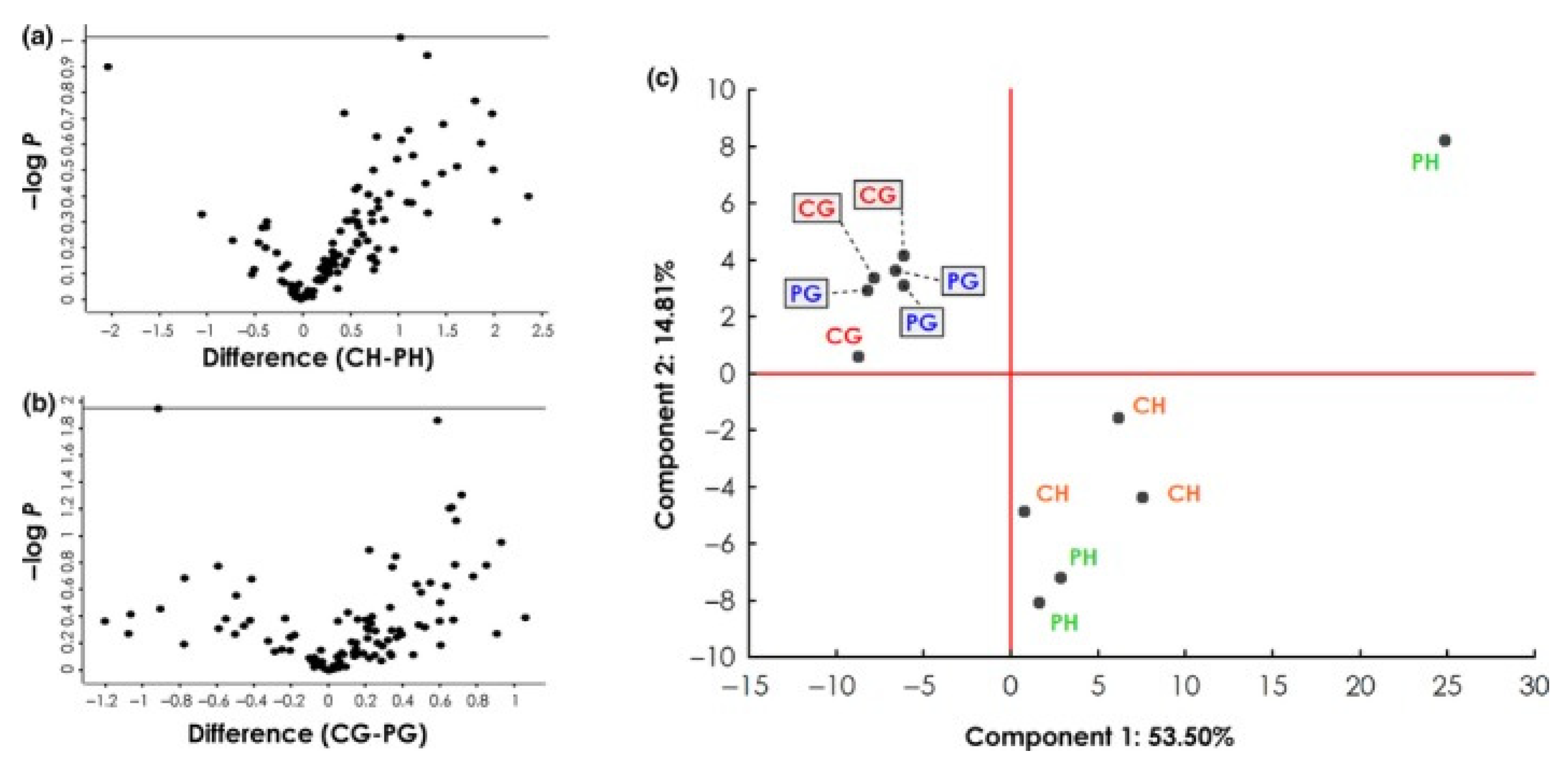
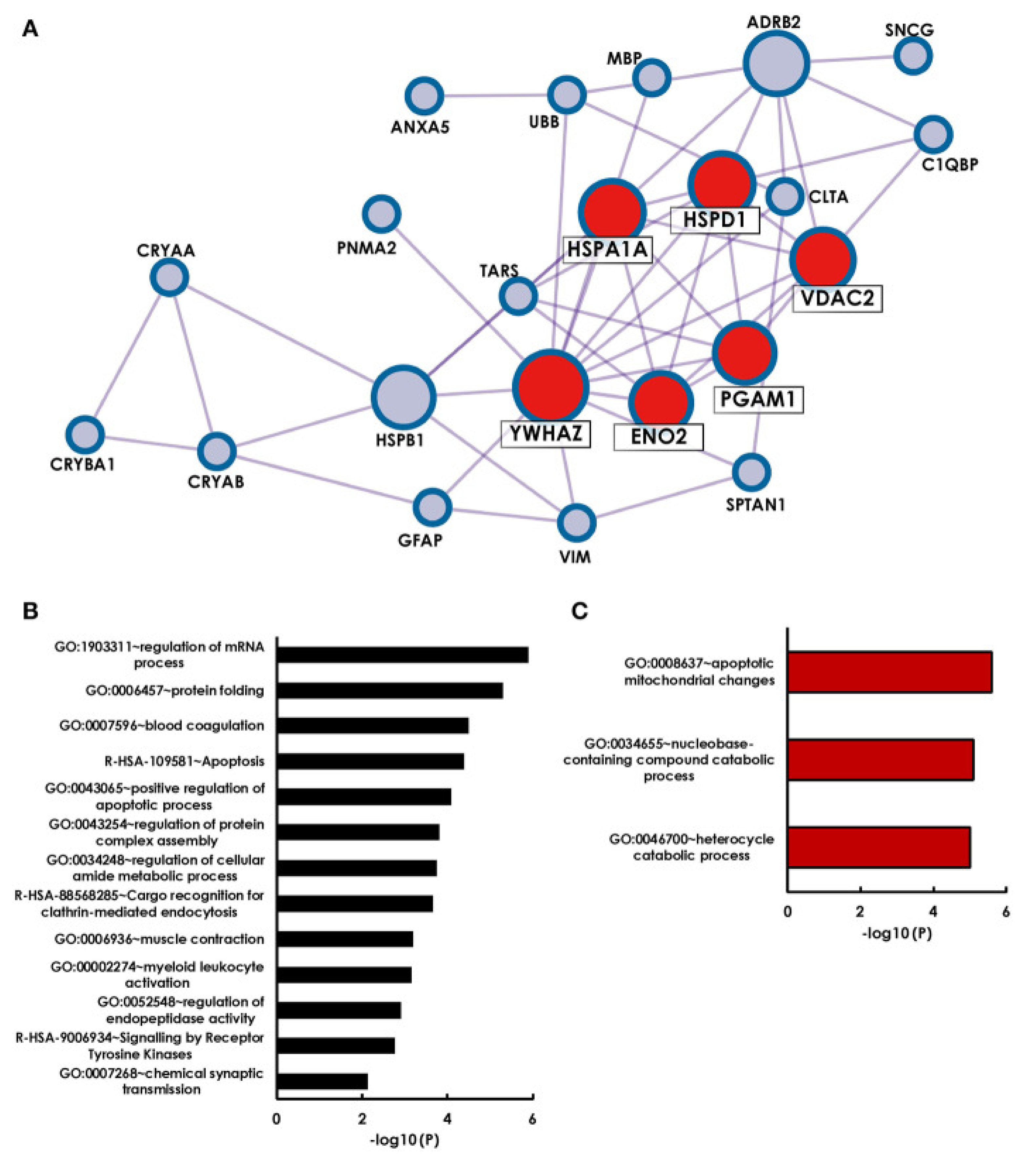
2.5. Autoantibodies
3. Therapeutical Approaches for Future Glaucoma Therapy
3.1. Crystallins
3.2. GFAP
3.3. The Complement System
3.4. High-Mobility Group Protein 1 (HMGB1)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blanco, A.A.; Bagnasco, L.; Bagnis, A.; Barton, K.; Baudouin, C.; Bengtsson, B.; Bron, A.; Cordeiro, F. European Glaucoma Society Terminology and Guidelines for Glaucoma, 4th Edition—Chapter 2: Classification and terminology Supported by the EGS Foundation: Part 1: Foreword; Introduction; Glossary; Chapter 2 Classification and Terminology. Br. J. Ophthalmol. 2017, 101, 73–127. [Google Scholar] [CrossRef]
- Allison, K.; Patel, D.G.; Greene, L. Racial and Ethnic Disparities in Primary Open-Angle Glaucoma Clinical Trials: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4, e218348. [Google Scholar] [CrossRef]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bosl, M.; Bosserhoff, A.; Kostler, J.; Wagner, R.; Tamm, E.R.; et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef]
- Yang, Y.F.; Sun, Y.Y.; Acott, T.S.; Keller, K.E. Effects of induction and inhibition of matrix cross-linking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells. Sci. Rep. 2016, 6, 30505. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- De Gaetano, A.; Gibellini, L.; Zanini, G.; Nasi, M.; Cossarizza, A.; Pinti, M. Mitophagy and Oxidative Stress: The Role of Aging. Antioxidants 2021, 10, 794. [Google Scholar] [CrossRef]
- Gorbatyuk, M.S.; Starr, C.R.; Gorbatyuk, O.S. Endoplasmic reticulum stress: New insights into the pathogenesis and treatment of retinal degenerative diseases. Prog. Retin. Eye Res. 2020, 79, 100860. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Barsoum, M.J.; Godzik, A.; Schwarzenbacher, R.; Lipton, S.A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003, 15, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramirez, A.I.; Salinas-Navarro, M.; Ortin-Martinez, A.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9, 92. [Google Scholar] [CrossRef]
- Soto, I.; Oglesby, E.; Buckingham, B.P.; Son, J.L.; Roberson, E.D.; Steele, M.R.; Inman, D.M.; Vetter, M.L.; Horner, P.J.; Marsh-Armstrong, N. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 548–561. [Google Scholar] [CrossRef][Green Version]
- Grus, F.H.; Joachim, S.C.; Hoffmann, E.M.; Pfeiffer, N. Complex autoantibody repertoires in patients with glaucoma. Mol. Vis. 2004, 10, 132–137. [Google Scholar]
- Tezel, G.; Edward, D.P.; Wax, M.B. Serum autoantibodies to optic nerve head glycosaminoglycans in patients with glaucoma. Arch. Ophthalmol. 1999, 117, 917–924. [Google Scholar] [CrossRef]
- Wax, M.B. Is there a role for the immune system in glaucomatous optic neuropathy? Curr. Opin. Ophthalmol. 2000, 11, 145–150. [Google Scholar] [CrossRef]
- Romano, C.; Li, Z.; Arendt, A.; Hargrave, P.A.; Wax, M.B. Epitope mapping of anti-rhodopsin antibodies from patients with normal pressure glaucoma. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1275–1280. [Google Scholar]
- Crabb, D.P.; Smith, N.D.; Glen, F.C.; Burton, R.; Garway-Heath, D.F. How does glaucoma look? Patient perception of visual field loss. Ophthalmology 2013, 120, 1120–1126. [Google Scholar] [CrossRef]
- Chan, M.P.Y.; Khawaja, A.P.; Broadway, D.C.; Yip, J.; Luben, R.; Hayat, S.; Peto, T.; Khaw, K.T.; Foster, P.J. Risk factors for previously undiagnosed primary open-angle glaucoma: The EPIC-Norfolk Eye Study. Br. J. Ophthalmol. 2021, 104, 220–224. [Google Scholar] [CrossRef]
- Mitchell, S.L.; Kiely, D.K.; Kiel, D.P.; Lipsitz, L.A. The epidemiology, clinical characteristics, and natural history of older nursing home residents with a diagnosis of Parkinson’s disease. J. Am. Geriatr. Soc. 1996, 44, 394–399. [Google Scholar] [CrossRef]
- Frank, T.S.; Deffenbaugh, A.M.; Reid, J.E.; Hulick, M.; Ward, B.E.; Lingenfelter, B.; Gumpper, K.L.; Scholl, T.; Tavtigian, S.V.; Pruss, D.R.; et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. J. Clin. Oncol. 2002, 20, 1480–1490. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Jiang, F.; You, C.; Mao, C.; Yu, J.; Han, J.; Zhang, Z.; Yan, H. Vitreous and plasma VEGF levels as predictive factors in the progression of proliferative diabetic retinopathy after vitrectomy. PLoS ONE 2014, 9, e110531. [Google Scholar] [CrossRef]
- Yan, H.; Cui, J.; Yu, J.G.; Han, J.D.; Chen, S.; Zhang, J.K.; Xu, Y.H. The expression of vascular endothelial growth factor of vitreous in patients with proliferative diabetic retinopathy. Zhonghua Yan Ke Za Zhi 2009, 45, 206–209. [Google Scholar] [PubMed]
- Fingert, J.H.; Heon, E.; Liebmann, J.M.; Yamamoto, T.; Craig, J.E.; Rait, J.; Kawase, K.; Hoh, S.T.; Buys, Y.M.; Dickinson, J.; et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum. Mol. Genet. 1999, 8, 899–905. [Google Scholar] [CrossRef]
- Pasutto, F.; Matsumoto, T.; Mardin, C.Y.; Sticht, H.; Brandstatter, J.H.; Michels-Rautenstrauss, K.; Weisschuh, N.; Gramer, E.; Ramdas, W.D.; van Koolwijk, L.M.; et al. Heterozygous NTF4 mutations impairing neurotrophin-4 signaling in patients with primary open-angle glaucoma. Am. J. Hum. Genet. 2009, 85, 447–456. [Google Scholar] [CrossRef]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Heon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Monemi, S.; Spaeth, G.; DaSilva, A.; Popinchalk, S.; Ilitchev, E.; Liebmann, J.; Ritch, R.; Heon, E.; Crick, R.P.; Child, A.; et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum. Mol. Genet. 2005, 14, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.K.; Lee, R.K.; Grus, F.H. Molecular biomarkers in glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Kass, M.A.; Kolker, A.E.; Becker, B.; Wax, M.B. Plasma and aqueous humor endothelin levels in primary open-angle glaucoma. J. Glaucoma 1997, 6, 83–89. [Google Scholar] [CrossRef]
- Kee, C.; Son, S.; Ahn, B.H. The relationship between gelatinase A activity in aqueous humor and glaucoma. J. Glaucoma 1999, 8, 51–55. [Google Scholar] [CrossRef]
- Ahmad, Y.; Arya, A.; Gangwar, A.; Paul, S.; Bhargava, K. Proteomics in diagnosis: Past, present and future. J. Proteom. Genom. 2014, 1, 103. [Google Scholar]
- Lauwen, S.; de Jong, E.K.; Lefeber, D.J.; den Hollander, A. Omics Biomarkers in Ophthalmology. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO88–BIO98. [Google Scholar] [CrossRef]
- Rao, P.V.; Horwitz, J.; Zigler, J.S., Jr. Alpha-crystallin, a molecular chaperone, forms a stable complex with carbonic anhydrase upon heat denaturation. Biochem. Biophys. Res. Commun. 1993, 190, 786–793. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Singh, B.N.; Rao, K.S.; Rao, C.M. AlphaB-crystallin, a small heat shock protein, modulates NF-kappaB activity in a phosphorylation-dependent manner and protects muscle myoblasts from TNF-alpha induced cytotoxicity. Biochim. Biophys. Acta 2011, 1813, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Gupta, V.B.; Chick, J.M.; Greco, T.M.; Wu, Y.; Chitranshi, N.; Wall, R.V.; Hone, E.; Deng, L.; Dheer, Y.; et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci. Rep. 2017, 7, 12685. [Google Scholar] [CrossRef] [PubMed]
- Kaeslin, M.A.; Killer, H.E.; Fuhrer, C.A.; Zeleny, N.; Huber, A.R.; Neutzner, A. Changes to the Aqueous Humor Proteome during Glaucoma. PLoS ONE 2016, 11, e0165314. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.; Perumal, N.; Beck, S.; Gabel-Scheurich, S.; Schmelter, C.; Teister, J.; Gerbig, C.; Gramlich, O.W.; Pfeiffer, N.; Grus, F.H. Glaucoma related Proteomic Alterations in Human Retina Samples. Sci. Rep. 2016, 6, 29759. [Google Scholar] [CrossRef]
- Mirzaei, M.; Gupta, V.K.; Chitranshi, N.; Deng, L.; Pushpitha, K.; Abbasi, M.; Chick, J.M.; Rajput, R.; Wu, Y.; McKay, M.J.; et al. Retinal proteomics of experimental glaucoma model reveal intraocular pressure-induced mediators of neurodegenerative changes. J. Cell. Biochem. 2020, 121, 4931–4944. [Google Scholar] [CrossRef]
- Anders, F.; Teister, J.; Funke, S.; Pfeiffer, N.; Grus, F.; Solon, T.; Prokosch, V. Proteomic profiling reveals crucial retinal protein alterations in the early phase of an experimental glaucoma model. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol. 2017, 255, 1395–1407. [Google Scholar] [CrossRef]
- Piri, N.; Song, M.; Kwong, J.M.K.; Caprioli, J. Modulation of alpha and beta crystallin expression in rat retinas with ocular hypertension-induced ganglion cell degeneration. Brain Res. 2007, 1141, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Rosignol, I.; Sierra-Filardi, E.; Rodriguez-Muela, N.; Schmelter, C.; Cecconi, F.; Grus, F.; Boya, P. Age related retinal Ganglion cell susceptibility in context of autophagy deficiency. Cell Death Discov. 2020, 6, 21. [Google Scholar] [CrossRef]
- Graw, J. Genetics of crystallins: Cataract and beyond. Exp. Eye Res. 2009, 88, 173–189. [Google Scholar] [CrossRef]
- Mackay, D.S.; Andley, U.P.; Shiels, A. Cell death triggered by a novel mutation in the alphaA-crystallin gene underlies autosomal dominant cataract linked to chromosome 21q. Eur. J. Hum. Genet. 2003, 11, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [PubMed]
- Kase, S.; Ishida, S.; Rao, N.A. Increased expression of alphaA-crystallin in human diabetic eye. Int. J. Mol. Med. 2011, 28, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Renkawek, K.; Stege, G.J.; Bosman, G.J. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 1999, 10, 2273–2276. [Google Scholar] [CrossRef] [PubMed]
- Head, M.W.; Corbin, E.; Goldman, J.E. Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am. J. Pathol. 1993, 143, 1743–1753. [Google Scholar] [PubMed]
- Rao, N.A.; Saraswathy, S.; Wu, G.S.; Katselis, G.S.; Wawrousek, E.F.; Bhat, S. Elevated retina-specific expression of the small heat shock protein, alphaA-crystallin, is associated with photoreceptor protection in experimental uveitis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Tomooka, B.H.; van Noort, J.M.; Wawrousek, E.F.; O’Connor, K.C.; Hafler, D.A.; Sobel, R.A.; Robinson, W.H.; Steinman, L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature 2007, 448, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Rong, X.; Ye, H.; Zhang, K.; Lu, Y. Proteomic analysis of aqueous humor proteins associated with cataract development. Clin. Biochem. 2015, 48, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Lu, P.; Zhang, W.F.; Lu, J.H. High myopia as a risk factor in primary open angle glaucoma. Int. J. Ophthalmol. 2012, 5, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Anders, F.; Funke, S.; Mercieca, K.; Grus, F.; Prokosch, V. Proteome alterations in aqueous humour of primary open angle glaucoma patients. Int. J. Ophthalmol. 2020, 13, 176–179. [Google Scholar] [CrossRef]
- Gonzalez-Iglesias, H.; Alvarez, L.; Garcia, M.; Escribano, J.; Rodriguez-Calvo, P.P.; Fernandez-Vega, L.; Coca-Prados, M. Comparative proteomic study in serum of patients with primary open-angle glaucoma and pseudoexfoliation glaucoma. J. Proteom. 2014, 98, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Mirzaei, M.; Gupta, V.B.; Chitranshi, N.; Dheer, Y.; Vander Wall, R.; Abbasi, M.; You, Y.; Chung, R.; Graham, S. Glaucoma is associated with plasmin proteolytic activation mediated through oxidative inactivation of neuroserpin. Sci. Rep. 2017, 7, 8412. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Kawaji, T.; Tanihara, H. Elevated levels of multiple biomarkers of Alzheimer’s disease in the aqueous humor of eyes with open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5353–5358. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Kaplan, H.J. Oxidative stress and the regulation of complement activation in human glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, O.W.; Beck, S.; Hohenstein-Blaul, N.V.T.U.; Boehm, N.; Ziegler, A.; Vetter, J.M.; Pfeiffer, N.; Grus, F.H. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PLoS ONE 2013, 8, e57557. [Google Scholar] [CrossRef]
- Reinehr, S.; Reinhard, J.; Gandej, M.; Gottschalk, I.; Stute, G.; Faissner, A.; Dick, H.B.; Joachim, S.C. S100B immunization triggers NFkappaB and complement activation in an autoimmune glaucoma model. Sci. Rep. 2018, 8, 9821. [Google Scholar] [CrossRef] [PubMed]
- Hastings, G.A.; Coleman, T.A.; Haudenschild, C.C.; Stefansson, S.; Smith, E.P.; Barthlow, R.; Cherry, S.; Sandkvist, M.; Lawrence, D.A. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J. Biol. Chem. 1997, 272, 33062–33067. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, Z.; Zhang, B.; Jiang, S.; Wang, Q.; Du, L.; Xue, H.; Zhang, Y.; Jin, M.; Zhu, X.; et al. Circular RNA sequencing indicates circ-IQGAP2 and circ-ZC3H6 as noninvasive biomarkers of primary Sjogren’s syndrome. Rheumatology 2020, 59, 2603–2615. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, L.; Hou, H.; Zhou, J.; Li, X. Epigenetic regulation of IQGAP2 promotes ovarian cancer progression via activating Wnt/beta-catenin signaling. Int. J. Oncol. 2016, 48, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Webber, H.C.; Bermudez, J.Y.; Millar, J.C.; Mao, W.; Clark, A.F. The Role of Wnt/beta-Catenin Signaling and K-Cadherin in the Regulation of Intraocular Pressure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1454–1466. [Google Scholar] [CrossRef]
- Garcia, A.L.; Udeh, A.; Kalahasty, K.; Hackam, A.S. A growing field: The regulation of axonal regeneration by Wnt signaling. Neural Regen. Res. 2018, 13, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Arnes, M.; Casas Tinto, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and Wnt/beta-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; McLeod, D.S.; Hasegawa, T.; Kim, S.Y.; Merges, C.; Tong, P.; Lutty, G.A. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp. Eye Res. 2006, 82, 99–110. [Google Scholar] [CrossRef]
- Ogata, N.; Nishikawa, M.; Nishimura, T.; Mitsuma, Y.; Matsumura, M. Unbalanced vitreous levels of pigment epithelium-derived factor and vascular endothelial growth factor in diabetic retinopathy. Am. J. Ophthalmol. 2002, 134, 348–353. [Google Scholar] [CrossRef]
- Ma, J.Y.W.; Sze, Y.H.; Bian, J.F.; Lam, T.C. Critical role of mass spectrometry proteomics in tear biomarker discovery for multifactorial ocular diseases (Review). Int. J. Mol. Med. 2021, 47, 83. [Google Scholar] [CrossRef]
- Burger, S.; Meng, J.; Zwanzig, A.; Beck, M.; Pankonin, M.; Wiedemann, P.; Eichler, W.; Unterlauft, J.D. Pigment Epithelium-Derived Factor (PEDF) Receptors Are Involved in Survival of Retinal Neurons. Int. J. Mol. Sci. 2020, 22, 369. [Google Scholar] [CrossRef]
- Unterlauft, J.D.; Eichler, W.; Kuhne, K.; Yang, X.M.; Yafai, Y.; Wiedemann, P.; Reichenbach, A.; Claudepierre, T. Pigment epithelium-derived factor released by Muller glial cells exerts neuroprotective effects on retinal ganglion cells. Neurochem. Res. 2012, 37, 1524–1533. [Google Scholar] [CrossRef]
- Vigneswara, V.; Ahmed, Z. Pigment epithelium-derived factor mediates retinal ganglion cell neuroprotection by suppression of caspase-2. Cell Death Dis. 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Boersma, M.C.; Dresselhaus, E.C.; De Biase, L.M.; Mihalas, A.B.; Bergles, D.E.; Meffert, M.K. A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef] [PubMed]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4220–4233. [Google Scholar] [CrossRef]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8442–8454. [Google Scholar] [CrossRef]
- Leung, L.L.; Morser, J. Plasmin as a complement C5 convertase. EBioMedicine 2016, 5, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Kroksveen, A.C.; Jaffe, J.D.; Aasebo, E.; Barsnes, H.; Bjorlykke, Y.; Franciotta, D.; Keshishian, H.; Myhr, K.M.; Opsahl, J.A.; van Pesch, V.; et al. Quantitative proteomics suggests decrease in the secretogranin-1 cerebrospinal fluid levels during the disease course of multiple sclerosis. Proteomics 2015, 15, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; Lefterova, M.I.; Lazo, J.S. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity. Biochemistry 2001, 40, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N. Role of Apolipoproteins and alpha-Synuclein in Parkinson’s Disease. J. Mol. Neurosci. 2017, 62, 344–355. [Google Scholar] [CrossRef]
- Vitali, C.; Wellington, C.L.; Calabresi, L. HDL and cholesterol handling in the brain. Cardiovasc. Res. 2014, 103, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; Do Carmo, S.; Lora, J.M.; Torres-Schumann, S.; Vogel, M.; Allhorn, M.; Gonzalez, C.; Bastiani, M.J.; Rassart, E.; Sanchez, D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell 2008, 7, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; Do Carmo, S.; Martinez, E.; Tolivia, J.; Navarro, A.; Rassart, E.; Sanchez, D. ApoD, a glia-derived apolipoprotein, is required for peripheral nerve functional integrity and a timely response to injury. Glia 2010, 58, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Kliuchnikova, A.A.; Samokhina, N.I.; Ilina, I.Y.; Karpov, D.S.; Pyatnitskiy, M.A.; Kuznetsova, K.G.; Toropygin, I.Y.; Kochergin, S.A.; Alekseev, I.B.; Zgoda, V.G.; et al. Human aqueous humor proteome in cataract, glaucoma, and pseudoexfoliation syndrome. Proteomics 2016, 16, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.X.; Bu, G.J. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Malek, G.; Johnson, L.V.; Mace, B.E.; Saloupis, P.; Schmechel, D.E.; Rickman, D.W.; Toth, C.A.; Sullivan, P.M.; Rickman, C.B. Apolipoprotein E allele-dependent pathogenesis: A model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 11900–11905. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Cerebrovascular Effects of Apolipoprotein E Implications for Alzheimer Disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef]
- Ling, M.; Murali, M. Analysis of the Complement System in the Clinical Immunology Laboratory. Clin. Lab. Med. 2019, 39, 579–590. [Google Scholar] [CrossRef]
- Hubens, W.H.G.; Beckers, H.J.M.; Gorgels, T.; Webers, C.A.B. Increased ratios of complement factors C3a to C3 in aqueous humor and serum mark glaucoma progression. Exp. Eye Res. 2021, 204, 108460. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.C.; Xu, D.; Li, T.T.; Zhang, J.F.; Liu, F. iTRAQ-based proteomics analysis of aqueous humor in patients with dry age-related macular degeneration. Int. J. Ophthalmol. 2019, 12, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Mou, S.; Ge, J.; To, C.H.; Hui, Y.; Liu, A.; Wang, Z.; Long, C.; Tan, J. A new strategy to replace the natural vitreous by a novel capsular artificial vitreous body with pressure-control valve. Eye 2008, 22, 461–468. [Google Scholar] [CrossRef]
- Bell, K.; Gramlich, O.W.; Hohenstein-Blaul, N.V.T.U.; Beck, S.; Funke, S.; Wilding, C.; Pfeiffer, N.; Grus, F.H. Does autoimmunity play a part in the pathogenesis of glaucoma? Prog. Retin. Eye Res. 2013, 36, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Kamhieh-Milz, J.; Sterzer, V.; Celik, H.; Khorramshahi, O.; Fadl Hassan Moftah, R.; Salama, A. Identification of novel autoantigens via mass spectroscopy-based antibody-mediated identification of autoantigens (MS-AMIDA) using immune thrombocytopenic purpura (ITP) as a model disease. J. Proteom. 2017, 157, 59–70. [Google Scholar] [CrossRef]
- Sadam, H.; Pihlak, A.; Jaago, M.; Pupina, N.; Rahni, A.; Toots, M.; Vaheri, A.; Nieminen, J.K.; Siuko, M.; Tienari, P.J.; et al. Identification of two highly antigenic epitope markers predicting multiple sclerosis in optic neuritis patients. EBioMedicine 2021, 64, 103211. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Seigel, G.M.; Wax, M.B. Autoantibodies to small heat shock proteins in glaucoma. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2277–2287. [Google Scholar]
- Maruyama, I.; Ohguro, H.; Ikeda, Y. Retinal ganglion cells recognized by serum autoantibody against gamma-enolase found in glaucoma patients. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1657–1665. [Google Scholar]
- Grus, F.H.; Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Wax, M.B. Serum autoantibodies to alpha-fodrin are present in glaucoma patients from Germany and the United States. Investig. Ophthalmol. Vis. Sci. 2006, 47, 968–976. [Google Scholar] [CrossRef]
- Joachim, S.C.; Reichelt, J.; Berneiser, S.; Pfeiffer, N.; Grus, F.H. Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2008, 246, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Grotegut, P.; Reinehr, S.; Joachim, S.C. Role of Heat Shock Proteins in Glaucoma. Int. J. Mol. Sci. 2019, 20, 5160. [Google Scholar] [CrossRef] [PubMed]
- Boehm, N.; Wolters, D.; Thiel, U.; Lossbrand, U.; Wiegel, N.; Pfeiffer, N.; Grus, F.H. New insights into autoantibody profiles from immune privileged sites in the eye: A glaucoma study. Brain Behav. Immun. 2012, 26, 96–102. [Google Scholar] [CrossRef]
- Joachim, S.C.; Grus, F.H.; Pfeiffer, N. Analysis of autoantibody repertoires in sera of patients with glaucoma. Eur. J. Ophthalmol. 2003, 13, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to alpha B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Curr. Eye Res. 2007, 32, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Castineiras, S.; Andino Moreno, M.E.; Perez, R.; Vazquez, L.A. Antibodies to lens crystallins after endocapsular cataract surgery. Puerto Rico Health Sci. J. 1993, 12, 123–128. [Google Scholar]
- Frostegard, J.; Hellstrom, C.; Nilsson, P.; Frostegard, A.G.; Ajeganova, S. Autoantibody profiling reveals four protein candidate autoantigens associated with systemic lupus erythematosus. Lupus 2018, 27, 1670–1678. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2007, 245, 619–626. [Google Scholar] [CrossRef]
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in multiple sclerosis. Clin. Immunol. 2015, 161, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Sanna, G.; Piga, M.; Terryberry, J.W.; Peltz, M.T.; Giagheddu, S.; Satta, L.; Ahmed, A.; Cauli, A.; Montaldo, C.; Passiu, G.; et al. Central nervous system involvement in systemic lupus erythematosus: Cerebral imaging and serological profile in patients with and without overt neuropsychiatric manifestations. Lupus 2000, 9, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Poletaev, A.B.; Morozov, S.G.; Gnedenko, B.B.; Zlunikin, V.M.; Korzhenevskey, D.A. Serum anti-S100b, anti-GFAP and anti-NGF autoantibodies of IgG class in healthy persons and patients with mental and neurological disorders. Autoimmunity 2000, 32, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Suliman, H.B.; Siniscalco, D.; Antonucci, N.; ElKafrawy, P. De novo Blood Biomarkers in Autism: Autoantibodies against Neuronal and Glial Proteins. Behav. Sci. 2019, 9, 47. [Google Scholar] [CrossRef]
- Tomczak, A.; Su, E.; Tugizova, M.; Carlson, A.M.; Kipp, L.B.; Feng, H.; Han, M.H. A case of GFAP-astroglial autoimmunity presenting with reversible parkinsonism. Mult. Scler. Relat. Disord. 2019, 39, 101900. [Google Scholar] [CrossRef]
- Beutgen, V.M.; Perumal, N.; Pfeiffer, N.; Grus, F.H. Autoantibody Biomarker Discovery in Primary Open Angle Glaucoma Using Serological Proteome Analysis (SERPA). Front. Immunol. 2019, 10, 381. [Google Scholar] [CrossRef]
- Wax, M.B.; Tezel, G.; Saito, I.; Gupta, R.S.; Harley, J.B.; Li, Z.; Romano, C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am. J. Ophthalmol. 1998, 125, 145–157. [Google Scholar] [CrossRef]
- Reichelt, J.; Joachim, S.C.; Pfeiffer, N.; Grus, F.H. Analysis of autoantibodies against human retinal antigens in sera of patients with glaucoma and ocular hypertension. Curr. Eye Res. 2008, 33, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Beutgen, V.M.; Schmelter, C.; Pfeiffer, N.; Grus, F.H. Autoantigens in the trabecular meshwork and glaucoma-specific alterations in the natural autoantibody repertoire. Clin. Transl. Immunol. 2020, 9, e01101. [Google Scholar] [CrossRef]
- Junemann, A.; Hohberger, B.; Rech, J.; Sheriff, A.; Fu, Q.; Schlotzer-Schrehardt, U.; Voll, R.E.; Bartel, S.; Kalbacher, H.; Hoebeke, J.; et al. Agonistic Autoantibodies to the beta2-Adrenergic Receptor Involved in the Pathogenesis of Open-Angle Glaucoma. Front. Immunol. 2018, 9, 145. [Google Scholar] [CrossRef]
- Coakes, R.L.; Brubaker, R.F. The mechanism of timolol in lowering intraocular pressure. In the normal eye. Arch. Ophthalmol. 1978, 96, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Hohberger, B.; Kunze, R.; Wallukat, G.; Kara, K.; Mardin, C.Y.; Lammer, R.; Schlotzer-Schrehardt, U.; Hosari, S.; Horn, F.; Munoz, L.; et al. Autoantibodies Activating the beta2-Adrenergic Receptor Characterize Patients with Primary and Secondary Glaucoma. Front. Immunol. 2019, 10, 2112. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Wollenberger, A. Effects of the serum gamma globulin fraction of patients with allergic asthma and dilated cardiomyopathy on chronotropic beta adrenoceptor function in cultured neonatal rat heart myocytes. Biomed. Biochim. Acta 1987, 46, S634–S639. [Google Scholar] [PubMed]
- Karczewski, P.; Hempel, P.; Kunze, R.; Bimmler, M. Agonistic autoantibodies to the alpha(1)-adrenergic receptor and the beta(2)-adrenergic receptor in Alzheimer’s and vascular dementia. Scand. J. Immunol. 2012, 75, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Beutgen, V.M.; Pfeiffer, N.; Grus, F.H. Serological Levels of Anti-clathrin Antibodies Are Decreased in Patients with Pseudoexfoliation Glaucoma. Front. Immunol. 2021, 12, 616421. [Google Scholar] [CrossRef]
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1999, 79, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Maksimiuk, M.; Sobiborowicz, A.; Tuzimek, A.; Deptala, A.; Czerw, A.; Badowska-Kozakiewicz, A.M. AlphaB-crystallin as a promising target in pathological conditions—A review. Ann. Agric. Environ. Med. AAEM 2020, 27, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Andley, U.P. Crystallins in the eye: Function and pathology. Prog. Retin. Eye Res. 2007, 26, 78–98. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Brown, K.M.; Stephan, D.A.; Morrison, J.C.; Johnson, E.C.; Tomarev, S.I. Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1247–1258. [Google Scholar] [CrossRef][Green Version]
- Zhu, Z.; Yang, F.; Zhang, K.; Cao, W.; Jin, Y.; Wang, G.; Mao, R.; Li, D.; Guo, J.; Liu, X.; et al. Comparative Proteomic Analysis of Wild-Type and SAP Domain Mutant Foot-and-Mouth Disease Virus-Infected Porcine Cells Identifies the Ubiquitin-Activating Enzyme UBE1 Required for Virus Replication. J. Proteome Res. 2015, 14, 4194–4206. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Peng, Y.; Zhang, J.; Wang, X.; Wu, N.; Zeng, Y.; Wang, Y. Endogenous alpha-crystallin inhibits expression of caspase-3 induced by hypoxia in retinal neurons. Life Sci. 2014, 111, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Masilamoni, J.G.; Jesudason, E.P.; Baben, B.; Jebaraj, C.E.; Dhandayuthapani, S.; Jayakumar, R. Molecular chaperone alpha-crystallin prevents detrimental effects of neuroinflammation. Biochim. Biophys. Acta 2006, 1762, 284–293. [Google Scholar] [CrossRef]
- Shao, W.Y.; Liu, X.; Gu, X.L.; Ying, X.; Wu, N.; Xu, H.W.; Wang, Y. Promotion of axon regeneration and inhibition of astrocyte activation by alpha A-crystallin on crushed optic nerve. Int. J. Ophthalmol. 2016, 9, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Ruebsam, A.; Dulle, J.E.; Myers, A.M.; Sakrikar, D.; Green, K.M.; Khan, N.W.; Schey, K.; Fort, P.E. A specific phosphorylation regulates the protective role of alphaA-crystallin in diabetes. JCI Insight 2018, 3, e97919. [Google Scholar] [CrossRef]
- Nath, M.; Shan, Y.; Myers, A.M.; Fort, P.E. HspB4/alphaA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways. J. Clin. Med. 2021, 10, 2384. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Oshitari, T.; Yamamoto, S. Level of vitreous alpha-B crystallin in eyes with rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 2015, 253, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.O.; Osmand, A.; Outeiro, T.F.; Muchowski, P.J.; Finkbeiner, S. alphaB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1677–1689. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, Z.; Lou, B.; Duan, F.; Qiu, S.; Cheng, Z.; Ma, X.; Yang, Y.; Lin, X. alphaB-Crystallin Alleviates Endotoxin-Induced Retinal Inflammation and Inhibits Microglial Activation and Autophagy. Front. Immunol. 2021, 12, 641999. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Reiser, G. Phosphorylation of Ser45 and Ser59 of alphaB-crystallin and p38/extracellular regulated kinase activity determine alphaB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J. Neurochem. 2011, 118, 354–364. [Google Scholar] [CrossRef]
- Bartelt-Kirbach, B.; Wiegreffe, C.; Birk, S.; Baur, T.; Moron, M.; Britsch, S.; Golenhofen, N. HspB5/alphaB-crystallin phosphorylation at S45 and S59 is essential for protection of the dendritic tree of rat hippocampal neurons. J. Neurochem. 2021, 157, 2055–2069. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Zhang, S.Z.; Tang, M.; Zhang, X.H.; Zhou, Z.; Yin, Y.Q.; Zhou, Q.B.; Huang, Y.Y.; Liu, Y.J.; Wawrousek, E.; et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 2013, 494, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Anders, F.; Liu, A.; Mann, C.; Teister, J.; Lauzi, J.; Thanos, S.; Grus, F.H.; Pfeiffer, N.; Prokosch, V. The Small Heat Shock Protein alpha-Crystallin B Shows Neuroprotective Properties in a Glaucoma Animal Model. Int. J. Mol. Sci. 2017, 18, 2418. [Google Scholar] [CrossRef]
- Van Noort, J.M.; Bsibsi, M.; Nacken, P.J.; Verbeek, R.; Venneker, E.H. Therapeutic Intervention in Multiple Sclerosis with Alpha B-Crystallin: A Randomized Controlled Phase IIa Trial. PLoS ONE 2015, 10, e0143366. [Google Scholar] [CrossRef]
- Dulle, J.E.; Rubsam, A.; Garnai, S.J.; Pawar, H.S.; Fort, P.E. BetaB2-crystallin mutations associated with cataract and glaucoma leads to mitochondrial alterations in lens epithelial cells and retinal neurons. Exp. Eye Res. 2017, 155, 85–90. [Google Scholar] [CrossRef]
- Lynch, J.M.; Li, B.; Katoli, P.; Xiang, C.; Leehy, B.; Rangaswamy, N.; Saenz-Vash, V.; Wang, Y.K.; Lei, H.; Nicholson, T.B.; et al. Binding of a glaucoma-associated myocilin variant to the alphaB-crystallin chaperone impedes protein clearance in trabecular meshwork cells. J. Biol. Chem. 2018, 293, 20137–20156. [Google Scholar] [CrossRef]
- Anders, F.; Teister, J.; Liu, A.; Funke, S.; Grus, F.H.; Thanos, S.; von Pein, H.D.; Pfeiffer, N.; Prokosch, V. Intravitreal injection of beta-crystallin B2 improves retinal ganglion cell survival in an experimental animal model of glaucoma. PLoS ONE 2017, 12, e0175451. [Google Scholar] [CrossRef]
- De Leeuw, R.; Gruenbaum, Y.; Medalia, O. Nuclear Lamins: Thin Filaments with Major Functions. Trends Cell Biol. 2018, 28, 34–45. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Rao, M.V.; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef]
- De Hoz, R.; Rojas, B.; Ramirez, A.I.; Salazar, J.J.; Gallego, B.I.; Trivino, A.; Ramirez, J.M. Retinal Macroglial Responses in Health and Disease. BioMed Res. Int. 2016, 2016, 2954721. [Google Scholar] [CrossRef] [PubMed]
- Eng, L.F. Glial fibrillary acidic protein (GFAP): The major protein of glial intermediate filaments in differentiated astrocytes. J. Neuroimmunol. 1985, 8, 203–214. [Google Scholar] [CrossRef]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Robel, S.; Mori, T.; Zoubaa, S.; Schlegel, J.; Sirko, S.; Faissner, A.; Goebbels, S.; Dimou, L.; Gotz, M. Conditional deletion of beta1-integrin in astroglia causes partial reactive gliosis. Glia 2009, 57, 1630–1647. [Google Scholar] [CrossRef]
- Chong, R.S.; Busoy, J.M.F.; Tan, B.; Yeo, S.W.; Lee, Y.S.; Barathi, A.V.; Crowston, J.G.; Schmetterer, L. A Minimally Invasive Experimental Model of Acute Ocular Hypertension with Acute Angle Closure Characteristics. Transl. Vis. Sci. Technol. 2020, 9, 24. [Google Scholar] [CrossRef]
- Ling, Y.T.T.; Pease, M.E.; Jefferys, J.L.; Kimball, E.C.; Quigley, H.A.; Nguyen, T.D. Pressure-Induced Changes in Astrocyte GFAP, Actin, and Nuclear Morphology in Mouse Optic Nerve. Investig. Ophthalmol. Vis. Sci. 2020, 61, 14. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, T.H.; Chen, W.C.; Peng, S.M.; Huang, X.S.; Cho, K.S.; Chen, D.F.; Liu, G.S. Optic neuropathy and increased retinal glial fibrillary acidic protein due to microbead-induced ocular hypertension in the rabbit. Int. J. Ophthalmol. 2016, 9, 1732–1739. [Google Scholar] [CrossRef]
- Kanamori, A.; Nakamura, M.; Nakanishi, Y.; Yamada, Y.; Negi, A. Long-term glial reactivity in rat retinas ipsilateral and contralateral to experimental glaucoma. Exp. Eye Res. 2005, 81, 48–56. [Google Scholar] [CrossRef]
- Reinehr, S.; Koch, D.; Weiss, M.; Froemel, F.; Voss, C.; Dick, H.B.; Fuchshofer, R.; Joachim, S.C. Loss of retinal ganglion cells in a new genetic mouse model for primary open-angle glaucoma. J. Cell. Mol. Med. 2019, 23, 5497–5507. [Google Scholar] [CrossRef]
- Quillen, S.; Schaub, J.; Quigley, H.; Pease, M.; Korneva, A.; Kimball, E. Astrocyte responses to experimental glaucoma in mouse optic nerve head. PLoS ONE 2020, 15, e0238104. [Google Scholar] [CrossRef] [PubMed]
- Lozano, D.C.; Choe, T.E.; Cepurna, W.O.; Morrison, J.C.; Johnson, E.C. Early Optic Nerve Head Glial Proliferation and Jak-Stat Pathway Activation in Chronic Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 921–932. [Google Scholar] [CrossRef]
- Cheng, P.Y.; Lin, Y.P.; Chen, Y.L.; Lee, Y.C.; Tai, C.C.; Wang, Y.T.; Chen, Y.J.; Kao, C.F.; Yu, J. Interplay between SIN3A and STAT3 mediates chromatin conformational changes and GFAP expression during cellular differentiation. PLoS ONE 2011, 6, e22018. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, H.; Namihira, M.; Kohyama, J.; Asano, H.; Sanosaka, T.; Nakashima, K. Methyl-CpG binding proteins are involved in restricting differentiation plasticity in neurons. J. Neurosci. Res. 2006, 84, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Calder, C.J.; Albon, J.; Erichsen, J.T.; Boulton, M.E.; Morgan, J.E. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp. Eye Res. 2011, 92, 338–343. [Google Scholar] [CrossRef]
- Hu, X.; Xu, M.X.; Zhou, H.; Cheng, S.; Li, F.; Miao, Y.; Wang, Z. Tumor necrosis factor-alpha aggravates gliosis and inflammation of activated retinal Muller cells. Biochem. Biophys. Res. Commun. 2020, 531, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Choi, J.S.; Yoon, Y.J.; Kim, K.A.; Joo, C.K. KR-31378, a potassium-channel opener, induces the protection of retinal ganglion cells in rat retinal ischemic models. J. Pharmacol. Sci. 2009, 109, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef]
- Unlu, M.; Aktas, Z.; Gocun, P.U.; Ilhan, S.O.; Hasanreisoglu, M.; Hasanreisoglu, B. Neuroprotective effect of systemic and/or intravitreal rosuvastatin administration in rat glaucoma model. Int. J. Ophthalmol. 2016, 9, 340–347. [Google Scholar] [CrossRef]
- Igarashi, T.; Miyake, K.; Kobayashi, M.; Kameya, S.; Fujimoto, C.; Nakamoto, K.; Takahashi, H.; Igarashi, T.; Miyake, N.; Iijima, O.; et al. Tyrosine triple mutated AAV2-BDNF gene therapy in a rat model of transient IOP elevation. Mol. Vis. 2016, 22, 816–826. [Google Scholar]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Geng, X.; Tian, L.; Wang, D.; Wang, Q. Grape seed proanthocyanidins protect retinal ganglion cells by inhibiting oxidative stress and mitochondrial alteration. Arch. Pharm. Res. 2020, 43, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, B.; Hu, Y.; Lu, L.; Lu, X.; Wang, J.; Xu, F.; Yu, S.; Huang, J.; Liang, X. Wogonin prevents TLR4-NF-kappaB-medicated neuro-inflammation and improves retinal ganglion cells survival in retina after optic nerve crush. Oncotarget 2016, 7, 72503–72517. [Google Scholar] [CrossRef] [PubMed]
- Wilding, C.; Bell, K.; Funke, S.; Beck, S.; Pfeiffer, N.; Grus, F.H. GFAP antibodies show protective effect on oxidatively stressed neuroretinal cells via interaction with ERP57. J. Pharmacol. Sci. 2015, 127, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.; Wilding, C.; Funke, S.; Perumal, N.; Beck, S.; Wolters, D.; Holz-Muller, J.; Pfeiffer, N.; Grus, F.H. Neuroprotective effects of antibodies on retinal ganglion cells in an adolescent retina organ culture. J. Neurochem. 2016, 139, 256–269. [Google Scholar] [CrossRef]
- Tatomir, A.; Talpos-Caia, A.; Anselmo, F.; Kruszewski, A.M.; Boodhoo, D.; Rus, V.; Rus, H. The complement system as a biomarker of disease activity and response to treatment in multiple sclerosis. Immunol. Res. 2017, 65, 1103–1109. [Google Scholar] [CrossRef]
- Schroder-Braunstein, J.; Kirschfink, M. Complement deficiencies and dysregulation: Pathophysiological consequences, modern analysis, and clinical management. Mol. Immunol. 2019, 114, 299–311. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science: New York, NY, USA, 2017; pp. 1–904. [Google Scholar]
- Xu, H.; Chen, M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur. J. Pharmacol. 2016, 787, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Muckersie, E.; Forrester, J.V.; Xu, H. Immune activation in retinal aging: A gene expression study. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5888–5896. [Google Scholar] [CrossRef] [PubMed]
- Chovatiya, R.; Medzhitov, R. Stress, inflammation, and defense of homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Anderson, S.R.; Breen, K.T.; Romero, C.O.; Steele, M.R.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Tomlinson, S.; Vetter, M.L. Complement C3-Targeted Gene Therapy Restricts Onset and Progression of Neurodegeneration in Chronic Mouse Glaucoma. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 2379–2396. [Google Scholar] [CrossRef]
- Kuehn, S.; Rodust, C.; Stute, G.; Grotegut, P.; Meissner, W.; Reinehr, S.; Dick, H.B.; Joachim, S.C. Concentration-Dependent Inner Retina Layer Damage and Optic Nerve Degeneration in a NMDA Model. J. Mol. Neurosci. 2017, 63, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Sobrado-Calvo, P.; Vidal-Sanz, M.; Villegas-Perez, M.P. Rat retinal microglial cells under normal conditions, after optic nerve section, and after optic nerve section and intravitreal injection of trophic factors or macrophage inhibitory factor. J. Comp. Neurol. 2007, 501, 866–878. [Google Scholar] [CrossRef]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Stasi, K.; Nagel, D.; Yang, X.; Wang, R.F.; Ren, L.; Podos, S.M.; Mittag, T.; Danias, J. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Naito, A.T.; Sumida, T.; Nomura, S.; Liu, M.L.; Higo, T.; Nakagawa, A.; Okada, K.; Sakai, T.; Hashimoto, A.; Hara, Y.; et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell 2012, 149, 1298–1313. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Barratt-Due, A.; Mollnes, T.E. Complement in clinical medicine: Clinical trials, case reports and therapy monitoring. Mol. Immunol. 2017, 89, 10–21. [Google Scholar] [CrossRef]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef]
- Einck, L.; Bustin, M. The intracellular distribution and function of the high mobility group chromosomal proteins. Exp. Cell Res. 1985, 156, 295–310. [Google Scholar] [CrossRef]
- Landsman, D.; Bustin, M. A signature for the HMG-1 box DNA-binding proteins. BioEssays News Rev. Mol. Cell. Dev. Biol. 1993, 15, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Livesey, K.M.; Zeh, H.J.; Loze, M.T.; Tang, D. HMGB1: A novel Beclin 1-binding protein active in autophagy. Autophagy 2010, 6, 1209–1211. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 110–116. [Google Scholar] [CrossRef]
- Zimmermann, K.; Volkel, D.; Pable, S.; Lindner, T.; Kramberger, F.; Bahrami, S.; Scheiflinger, F. Native versus recombinant high-mobility group B1 proteins: Functional activity in vitro. Inflammation 2004, 28, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [PubMed]
- Haggadone, M.D.; Grailer, J.J.; Fattahi, F.; Zetoune, F.S.; Ward, P.A. Bidirectional Crosstalk between C5a Receptors and the NLRP3 Inflammasome in Macrophages and Monocytes. Mediat. Inflamm. 2016, 2016, 1340156. [Google Scholar] [CrossRef]
- Yu, S.; Wang, D.; Huang, L.; Zhang, Y.; Luo, R.; Adah, D.; Tang, Y.; Zhao, K.; Lu, B. The complement receptor C5aR2 promotes protein kinase R expression and contributes to NLRP3 inflammasome activation and HMGB1 release from macrophages. J. Biol. Chem. 2019, 294, 8384–8394. [Google Scholar] [CrossRef]
- Kim, S.Y.; Son, M.; Lee, S.E.; Park, I.H.; Kwak, M.S.; Han, M.; Lee, H.S.; Kim, E.S.; Kim, J.Y.; Lee, J.E.; et al. High-Mobility Group Box 1-Induced Complement Activation Causes Sterile Inflammation. Front. Immunol. 2018, 9, 705. [Google Scholar] [CrossRef]
- Mohammad, G.; Abdelaziz, G.M.; Siddiquei, M.M.; Ahmad, A.; De Hertogh, G.; Abu El-Asrar, A.M. Cross-Talk between Sirtuin 1 and the Proinflammatory Mediator High-Mobility Group Box-1 in the Regulation of Blood-Retinal Barrier Breakdown in Diabetic Retinopathy. Curr. Eye Res. 2019, 44, 1133–1143. [Google Scholar] [CrossRef]
- Joachim, S.C.; Gramlich, O.W.; Laspas, P.; Schmid, H.; Beck, S.; von Pein, H.D.; Dick, H.B.; Pfeiffer, N.; Grus, F.H. Retinal ganglion cell loss is accompanied by antibody depositions and increased levels of microglia after immunization with retinal antigens. PLoS ONE 2012, 7, e40616. [Google Scholar] [CrossRef]
- Joachim, S.C.; Mondon, C.; Gramlich, O.W.; Grus, F.H.; Dick, H.B. Apoptotic retinal ganglion cell death in an autoimmune glaucoma model is accompanied by antibody depositions. J. Mol. Neurosci. 2014, 52, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Noristani, R.; Kuehn, S.; Stute, G.; Reinehr, S.; Stellbogen, M.; Dick, H.B.; Joachim, S.C. Retinal and Optic Nerve Damage is Associated with Early Glial Responses in an Experimental Autoimmune Glaucoma Model. J. Mol. Neurosci. 2016, 58, 470–482. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Nawaz, M.I.; Siddiquei, M.M.; Al-Kharashi, A.S.; Kangave, D.; Mohammad, G. High-mobility group box-1 induces decreased brain-derived neurotrophic factor-mediated neuroprotection in the diabetic retina. Mediat. Inflamm. 2013, 2013, 863036. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Liu, Z.; Sun, N.; Liu, S.; Li, L.; Shen, Y.; Xiu, J.; Xu, Q. BDNF Alleviates Neuroinflammation in the Hippocampus of Type 1 Diabetic Mice via Blocking the Aberrant HMGB1/RAGE/NF-kappaB Pathway. Aging Dis. 2019, 10, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Wojcik-Gryciuk, A.; Gajewska-Wozniak, O.; Kordecka, K.; Boguszewski, P.M.; Waleszczyk, W.; Skup, M. Neuroprotection of Retinal Ganglion Cells with AAV2-BDNF Pretreatment Restoring Normal TrkB Receptor Protein Levels in Glaucoma. Int. J. Mol. Sci. 2020, 21, 6262. [Google Scholar] [CrossRef]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef]
- Das, S.; Mishra, K.P.; Chanda, S.; Ganju, L.; Singh, S.B. CXCR7: A key neuroprotective molecule against alarmin HMGB1 mediated CNS pathophysiology and subsequent memory impairment. Brain Behav. Immun. 2019, 82, 319–337. [Google Scholar] [CrossRef]
- Werner, L.; Guzner-Gur, H.; Dotan, I. Involvement of CXCR4/CXCR7/CXCL12 Interactions in Inflammatory bowel disease. Theranostics 2013, 3, 40–46. [Google Scholar] [CrossRef]
- Jiang, G.; Sun, D.; Yang, H.; Lu, Q.; Kaplan, H.J.; Shao, H. HMGB1 is an early and critical mediator in an animal model of uveitis induced by IRBP-specific T cells. J. Leukoc. Biol. 2014, 95, 599–607. [Google Scholar] [CrossRef]
- Watanabe, T.; Keino, H.; Sato, Y.; Kudo, A.; Kawakami, H.; Okada, A.A. High mobility group box protein-1 in experimental autoimmune uveoretinitis. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.L.; Poirier-Beaudouin, B.; Durant, J.; Lebrun-Frenay, C.; Saidi, H.; Seffer, V.; Ticchioni, M.; Chanalet, S.; Carsenti, H.; Harvey-Langton, A.; et al. HMGB1/anti-HMGB1 antibodies define a molecular signature of early stages of HIV-Associated Neurocognitive Isorders (HAND). Heliyon 2017, 3, e00245. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; de Leeuw, K.; Horst, G.; Maas, F.; Bootsma, H.; Heeringa, P.; Limburg, P.C.; Westra, J. Autoantibodies to box A of high mobility group box 1 in systemic lupus erythematosus. Clin. Exp. Immunol. 2017, 188, 412–419. [Google Scholar] [CrossRef]
- Wen, Z.; Xu, L.; Chen, X.; Xu, W.; Yin, Z.; Gao, X.; Xiong, S. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J. Immunol. 2013, 190, 5411–5422. [Google Scholar] [CrossRef]
- Grus, F.; Sun, D. Immunological mechanisms in glaucoma. Semin. Immunopathol. 2008, 30, 121–126. [Google Scholar] [CrossRef]
- Tezel, G.; Fourth ARVO/Pfizer Ophthalmics Research Institute Conference Working Group. The role of glia, mitochondria, and the immune system in glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1001–1012. [Google Scholar] [CrossRef]
- Wax, M.B.; Tezel, G. Immunoregulation of retinal ganglion cell fate in glaucoma. Exp. Eye Res. 2009, 88, 825–830. [Google Scholar] [CrossRef]
- Bohm, M.R.; Schallenberg, M.; Brockhaus, K.; Melkonyan, H.; Thanos, S. The pro-inflammatory role of high-mobility group box 1 protein (HMGB-1) in photoreceptors and retinal explants exposed to elevated pressure. Lab. Investig. J. Tech. Methods Pathol. 2016, 96, 409–427. [Google Scholar] [CrossRef][Green Version]
- Sakamoto, K.; Okuwaki, T.; Ushikubo, H.; Mori, A.; Nakahara, T.; Ishii, K. Activation inhibitors of nuclear factor kappa B protect neurons against the NMDA-induced damage in the rat retina. J. Pharmacol. Sci. 2017, 135, 72–80. [Google Scholar] [CrossRef]
- Chi, W.; Chen, H.; Li, F.; Zhu, Y.; Yin, W.; Zhuo, Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J. Neuroinflamm. 2015, 12, 137. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Protein Name | Retina | Aqueous Humor | Vitreous | Serum | Animal Model |
|---|---|---|---|---|---|---|
| CRYAA | α-Crystallin A chain | ↓ [34] | [33] | ↓ TLP [38] ONC [39] | ||
| CRYAB | α-Crystallin B chain | ↑ [35] | ↓ [33] | ↓ MB [36] EVO [37] TLP [38] ONC [39] | ||
| CRYBA1 (CRYBA3) | β-Crystallin A3 | ↓ [35] | ↓ [33] | |||
| CRYBA2 | β-Crystallin A2 | ↓ [33] | ↓ MB [36] EVO [33] | |||
| CRYBA4 | β-Crystallin A4 | ↓ [33] | ↓ MB EVO [33] | |||
| CRYBB1 | β-Crystallin B1 | ↓ [35] | ↓ [33] | ↓ MB [36] EVO [37] | ||
| CRYBB2 | β-Crystallin B2 | ↓ [33] | ↓ MB [36] EVO [33] TLP [38] ONC [39] | |||
| CRYBB3 | β-Crystallin B3 | ↓ [33] | ↓ MB [36] EVO [37] | |||
| CRYGB | γ-Crystallin B | ↓ [33] | ||||
| CRYGD | γ-Crystallin D | ↓ [33] | ||||
| CRYGS | γ-Crystallin S | ↓ [33] | ↓ MB [36] EVO [37] | |||
| TPM1/TPM3 TPM4 | Tropomyosin alpha-1 chain/alpha-3 chain/alpha-4 chain | ↓ [36] | ||||
| IQGAP2 | Ras GTPase-activating-like protein IQGAP2 | ↓ [36] | ↓ MB [36] ↑ EVO [37] | |||
| AHNAK | Neuroblast differentiation-associated protein AHNAK | ↓ [36] | ↓ MB [36] | |||
| DKK3 | Dickkopf-related protein 3 | ↑ [50] | ||||
| WIF1 | Wnt inhibitory factor 1 | ↑ [50] | ||||
| SERPINF1 (PEDF) | Pigment epithelium-derived factor | ↑ [50] | ||||
| SERPINA1 (AAT) | α-1-antitrypsin | ↑ [35] | ↑ [51] | |||
| SERPINA3 (AACT) | α-1-antichymo- trypsin | ↑ [50] | ||||
| SERPINA6 (CBG) | Corticosteroid-binding globulin | ↑ [50] | ||||
| SERPINA7 (TBG) | Thyroxine-binding globulin | ↑ [50] | ||||
| SERPINA8 (AGT) | Angiotensinogen | ↑ [50] | ||||
| SERPINF2 (A2AP) | α-2-antiplasmin | ↑ [50] | ||||
| SERPINI9 (NEUS) | Neuroserpin | ↔ [52] | ↓ [34] | ↔ [52] | ||
| APOA1 | Apolipoprotein A-1 | ↑ [33] | ↑ [53] | ↑ [33] | ↑ [51] | |
| APOA4 | Apolipoprotein A-4 | ↑ [34] | ↑ [51] | |||
| APOD | Apolipoprotein D | ↑ [34] | ||||
| APOE4 | Apolipoprotein E4 | ↑ [33] | ↑ [50] | ↑ [33] | ↓ MB [36] | |
| C1Q | Complement C1q subcomponent | ↑ [54] | ↑ [50] | |||
| C3 | Complement C3 | ↑ [55] | ↑ [50] | ↑ [51] | ↑ EAG [56] | |
| C5 | Complement C5 | ↔ [55] | ↔ EAG [56] | |||
| C8 | Complement component C8 | ↑ [54] | ↑ [52] | |||
| C9 | Complement component C9 | ↑ [57] | ↑ [52] | |||
| VSIG4 | V-set immunoglobulin domain-containing protein 4 | ↑ [52] | ||||
| MASP1/MASP2 | Mannan-binding lectin serine protease 1/2 | ↑ [57] |
| Gene Name | POAG vs. PEXG vs. Control | POAG vs. Control | PEXG vs. Control | POAG vs. PEXG | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CA a | Sens. b | Spec. c | AUC d | CA | Sens. | Spec. | AUC | CA | Sens. | Spec. | AUC | CA | Sens. | Spec. | AUC | |
| APOA4 | 0.8113 | 1.0000 | 0.9118 | 0.9233 | 0.9744 | 1.0000 | 0.9500 | 1.0000 | 0.8788 | 0.9474 | 0.7857 | 0.8609 | 0.7941 | 0.8000 | 0.7857 | 0.8143 |
| C3 | 0.6415 | 0.8421 | 0.8824 | 0.8607 | 0.8718 | 0.8421 | 0.9000 | 0.9763 | 0.7879 | 0.7895 | 0.7857 | 0.8910 | 0.6765 | 0.6500 | 0.7143 | 0.6786 |
| TF | 0.6792 | 0.8947 | 0.8235 | 0.8283 | 0.8718 | 0.9474 | 0.8000 | 0.9526 | 0.8485 | 0.9474 | 0.7143 | 0.8120 | 0.6765 | 0.6500 | 0.7143 | 0.6571 |
| VTN | 0.6038 | 1.0000 | 0.8529 | 0.7786 | 0.9231 | 0.9474 | 0.9000 | 0.9474 | 0.8182 | 0.8421 | 0.7857 | 0.9060 | 0.4706 | 0.6000 | 0.2857 | 0.4464 |
| TTR | 0.5849 | 0.8421 | 0.7941 | 0.7635 | 0.8718 | 0.8421 | 0.9000 | 0.9342 | 0.7576 | 0.8947 | 0.5714 | 0.6617 | 0.5588 | 0.7000 | 0.3571 | 0.6750 |
| SERPINA1 | 0.6038 | 0.8421 | 0.7059 | 0.7203 | 0.8462 | 0.8947 | 0.8000 | 0.8684 | 0.6667 | 0.7368 | 0.5714 | 0.7556 | 0.6176 | 0.8000 | 0.3571 | 0.4821 |
| FBLN1 | 0.6981 | 0.8421 | 0.9118 | 0.8380 | 0.8293 | 0.8421 | 0.9091 | 0.8886 | 0.9091 | 0.8947 | 0.9286 | 0.8797 | 0.6471 | 0.8000 | 0.4286 | 0.5821 |
| APOA1 | 0.4717 | 0.7895 | 0.7647 | 0.6641 | 0.7692 | 0.7895 | 0.7500 | 0.8158 | 0.7576 | 0.7895 | 0.7143 | 0.7857 | 0.4412 | 0.6500 | 0.1429 | 0.3643 |
| FCN3 | 0.5849 | 0.6842 | 0.7059 | 0.7559 | 0.7692 | 0.8421 | 0.7000 | 0.8553 | 0.6970 | 0.7368 | 0.6429 | 0.7293 | 0.6176 | 0.6000 | 0.6429 | 0.6786 |
| CFH | 0.6226 | 0.7895 | 0.8824 | 0.6933 | 0.7949 | 0.6842 | 0.9000 | 0.8079 | 0.7576 | 0.7895 | 0.7143 | 0.7368 | 0.5588 | 0.7000 | 0.3571 | 0.4893 |
| ITIH4 | 0.3396 | 0.4737 | 0.4412 | 0.4590 | 0.4872 | 0.4737 | 0.5000 | 0.6158 | 0.5758 | 0.7368 | 0.3571 | 0.3647 | 0.5294 | 0.8000 | 0.1429 | 0.3679 |
| APOL1 | 0.4906 | 0.6842 | 0.7059 | 0.5972 | 0.6923 | 0.6842 | 0.7000 | 0.7553 | 0.5455 | 0.6842 | 0.3571 | 0.6955 | 0.3824 | 0.6500 | – | 0.2607 |
| ALB | 0.2453 | 0.3158 | 0.5882 | 0.5778 | 0.3846 | 0.3158 | 0.4500 | 0.5263 | 0.7576 | 0.8421 | 0.6429 | 0.7895 | 0.4706 | 0.6000 | 0.2857 | 0.5357 |
| SERPINC1 | 0.3019 | 0.3158 | 0.6765 | 0.4816 | 0.4359 | 0.3158 | 0.5500 | 0.3842 | 0.6667 | 0.7368 | 0.5714 | 0.6466 | 0.4706 | 0.6500 | 0.2143 | 0.4643 |
| IGHG2 | 0.2453 | 0.2632 | 0.5882 | 0.2894 | 0.3333 | 0.2632 | 0.4000 | 0.4000 | 0.2727 | 0.4737 | – | 0.2068 | 0.3235 | 0.5500 | – | 0.2214 |
| C4A | 0.35 85 | 0.5789 | 0.6471 | 0.5346 | 0.4103 | 0.4737 | 0.3500 | 0.3947 | 0.7273 | 0.7895 | 0.6429 | 0.7444 | 0.4706 | 0.6000 | 0.2857 | 0.5500 |
| APCS | 0.3208 | 0.2632 | 0.5294 | 0.3164 | 0.4359 | 0.3158 | 0.5500 | 0.3368 | 0.5152 | 0.7368 | 0.2143 | 0.3722 | 0.6471 | 0.9000 | 0.2857 | 0.2786 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auler, N.; Tonner, H.; Pfeiffer, N.; Grus, F.H. Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways. Biology 2021, 10, 1296. https://doi.org/10.3390/biology10121296
Auler N, Tonner H, Pfeiffer N, Grus FH. Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways. Biology. 2021; 10(12):1296. https://doi.org/10.3390/biology10121296
Chicago/Turabian StyleAuler, Nadine, Henrik Tonner, Norbert Pfeiffer, and Franz H. Grus. 2021. "Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways" Biology 10, no. 12: 1296. https://doi.org/10.3390/biology10121296
APA StyleAuler, N., Tonner, H., Pfeiffer, N., & Grus, F. H. (2021). Antibody and Protein Profiles in Glaucoma: Screening of Biomarkers and Identification of Signaling Pathways. Biology, 10(12), 1296. https://doi.org/10.3390/biology10121296