
A Conservative Replacement in the Transmembrane Domain of SARS-CoV-2 ORF7a as a Putative Risk Factor in COVID-19

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Preparation and Sequencing
2.3. Sequencing Data Processing, Data Availability, and Mutation Analysis
2.4. ORF7a Protein Modeling
2.5. Molecular Dynamics (MD) Simulation Protocols and Analysis
3. Results
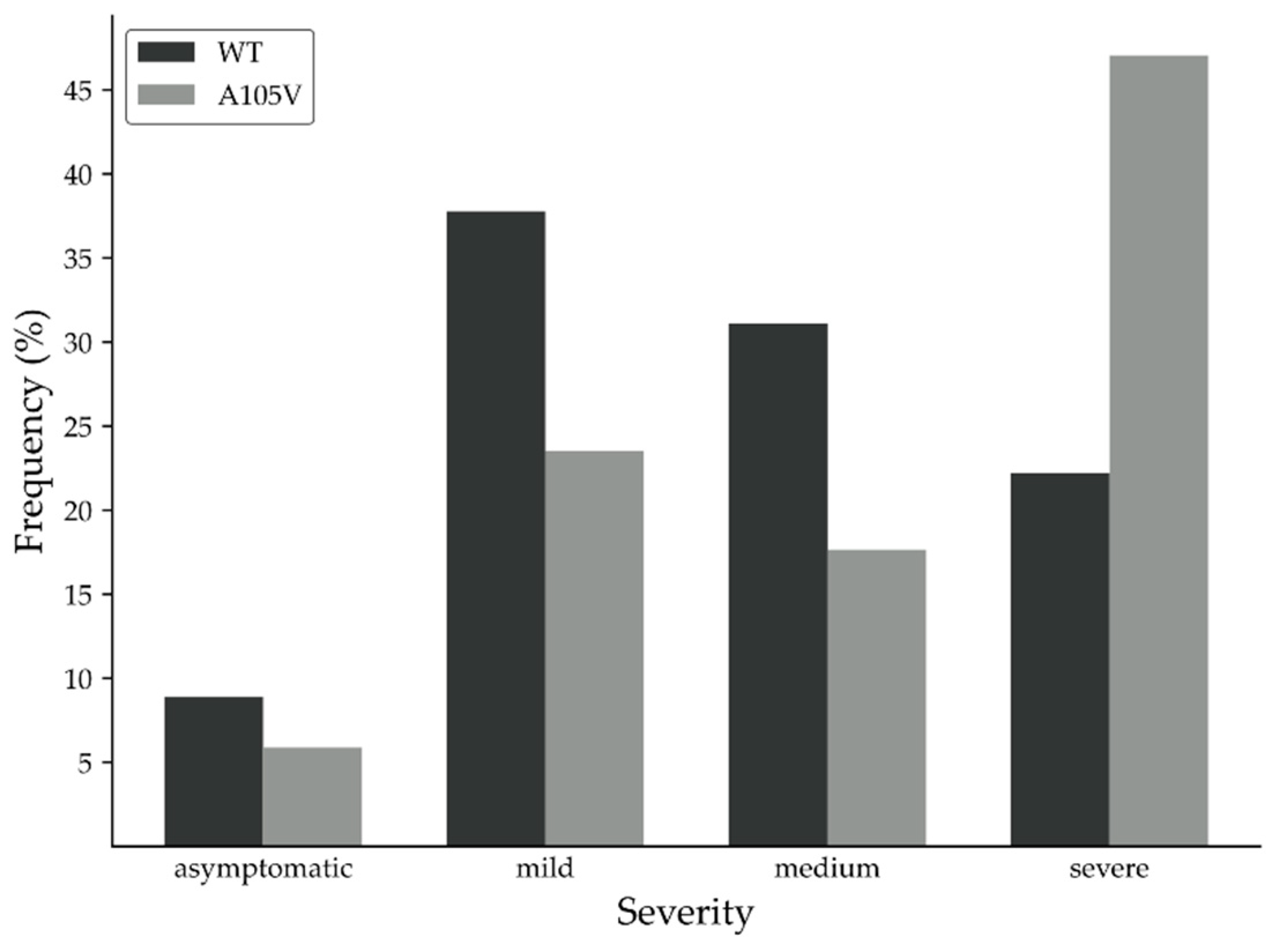
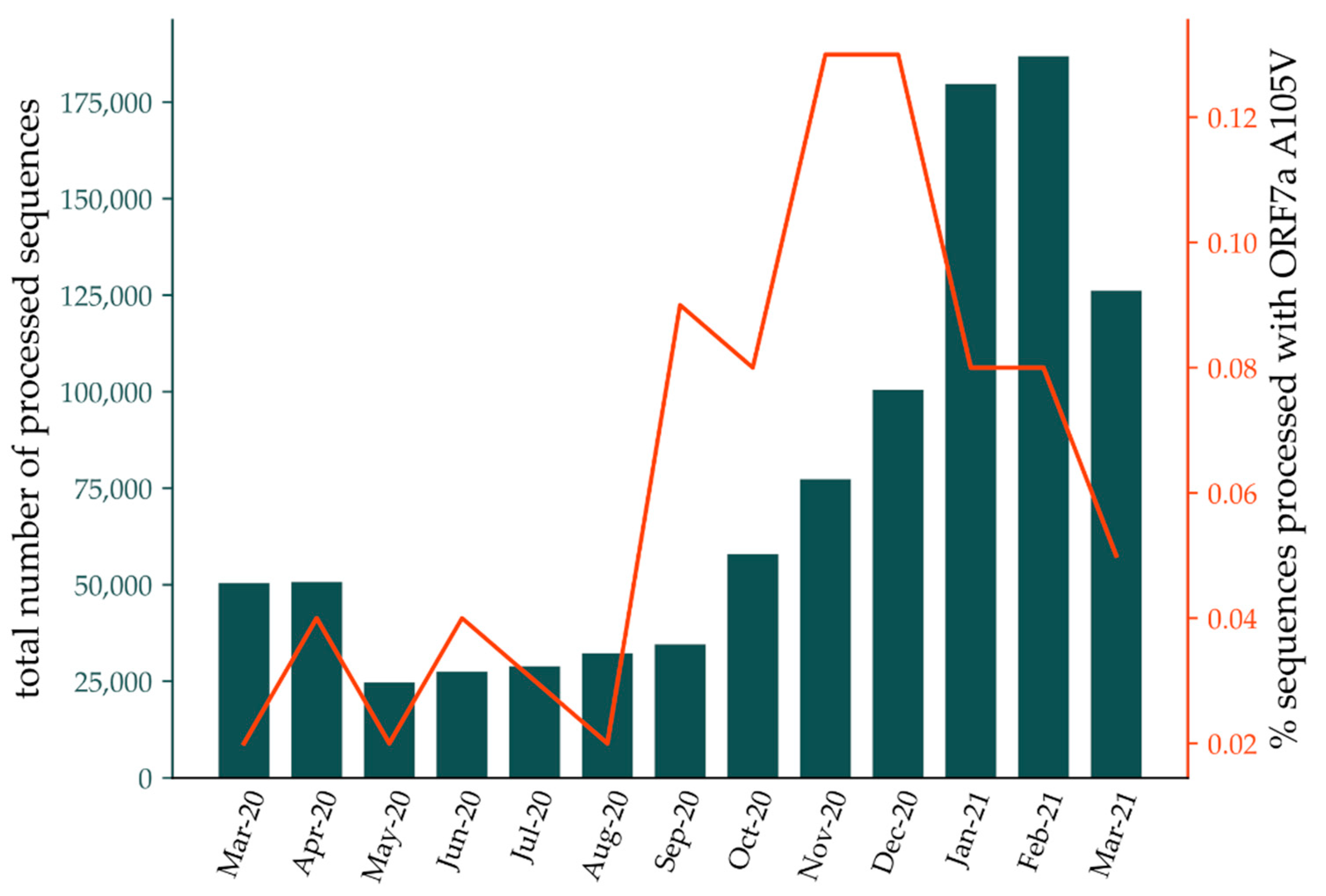
3.1. Clinical Data and Putative Disease-Enhancing Role of ORF7a A105V
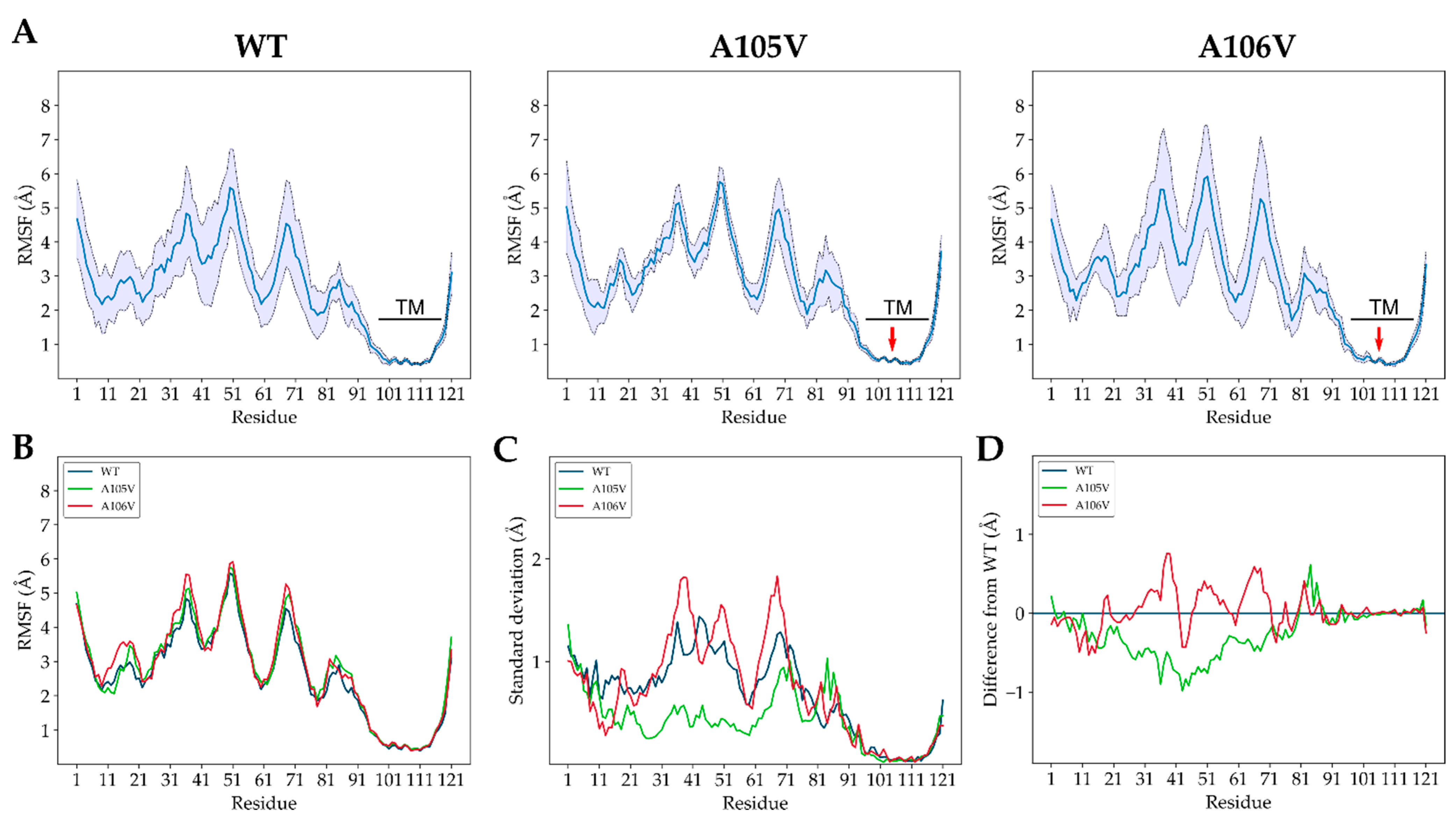
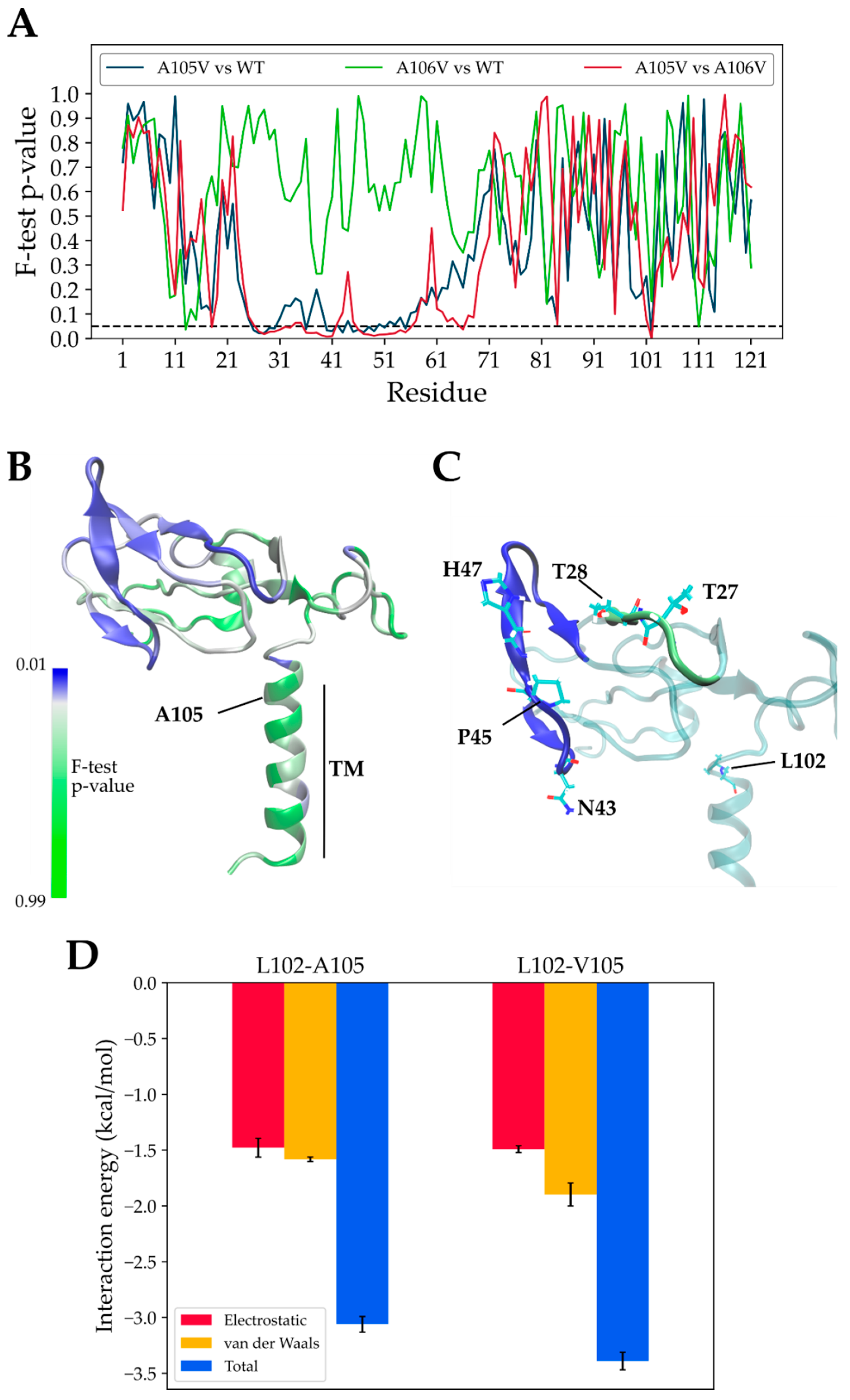
3.2. Structural Dynamics of Mutant and Native ORF7a Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drosten, C.; Preiser, W.; Günther, S.; Schmitz, H.; Doerr, H.W. Severe Acute Respiratory Syndrome: Identification of the Etiological Agent. Trends Mol. Med. 2003, 9, 325–327. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- WHO. Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 11 March 2021).
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and Influenza Pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Nelson, C.A.; Pekosz, A.; Lee, C.A.; Diamond, M.S.; Fremont, D.H. Structure and Intracellular Targeting of the SARS-Coronavirus ORF7a Accessory Protein. Structure 2005, 13, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaecher, S.R.; Touchette, E.; Schriewer, J.; Buller, R.M.; Pekosz, A. Severe Acute Respiratory Syndrome Coronavirus Gene 7 Products Contribute to Virus-Induced Apoptosis. J. Virol. 2007, 81, 11054–11068. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wu, J.; Shan, Y.; Yao, Z.; Dong, B.; Chen, B.; Zhao, Z.; Wang, S.; Chen, J.; Cong, Y. SARS Coronavirus 7a Protein Blocks Cell Cycle Progression at G0/G1 Phase via the Cyclin D3/pRb Pathway. Virology 2006, 346, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaecher, S.R.; Pekosz, A. SARS Coronavirus Accessory Gene Expression and Function. In Molecular Biology of the SARS-Coronavirus; Springer: Berlin/Heidelberg, Germany, 2010; pp. 153–166. ISBN 978-3-64203-682-8. [Google Scholar]
- Zhou, Z.; Huang, C.; Zhou, Z.; Huang, Z.; Su, L.; Kang, S.; Chen, X.; Chen, Q.; He, S.; Rong, X.; et al. Structural Insight Reveals SARS-CoV-2 ORF7a as an Immunomodulating Factor for Human CD14+ Monocytes. iScience 2021, 24, 102187. [Google Scholar] [CrossRef] [PubMed]
- Nizamudeen, Z.A.; Xu, E.R.; Karthik, V.; Halawa, M.; Arkill, K.P.; Jackson, A.M.; Bates, D.O.; Emsley, J. Structural Assessment of SARS-CoV2 Accessory Protein ORF7a Predicts LFA-1 and Mac-1 Binding Potential. Biosci. Rep. 2021, 41, BSR20203837. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a Promotes Antagonism of Interferon Response. Cell. Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Addetia, A.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.; Huang, M.L.; Jerome, K.R.; Greninger, A.L. Identification of Multiple Large Deletions in ORF7a Resulting in In-frame Gene Fusions in Clinical SARS-CoV-2 Isolates. J. Clin. Virol. 2020, 129, 104523. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Kaelin, E.A.; Maqsood, R.; Estifanos, B.; Wu, L.I.; Varsani, A.; Halden, R.U.; Hogue, B.G.; Scotch, M.; Lim, E.S. An 81 Nucleotide Deletion in SARS-CoV-2 ORF7a Identified from Sentinel Surveillance in Arizona (January–March 2020). J. Virol. 2020, 94, e00711-20. [Google Scholar] [CrossRef]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Nichols, J.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; Lee, H.; Vanderwood, K.K.; Bimczok, D.; et al. SARS-CoV-2 Genomic Surveillance Identifies Naturally Occurring Truncation of ORF7a that Limits Immune Suppression. Cell Rep. 2021, 35, 109197. [Google Scholar] [CrossRef]
- Panzera, Y.; Ramos, N.; Frabasile, S.; Calleros, L.; Marandino, A.; Tomás, G.; Techera, C.; Grecco, S.; Fuques, E.; Goñi, N.; et al. A Deletion in SARS-CoV-2 ORF7 Identified in COVID-19 Outbreak in Uruguay. Transbound. Emerg. Dis. 2021, 68, 3075–3082. [Google Scholar] [CrossRef]
- Rosenthal, S.H.; Kagan, R.M.; Gerasimova, A.; Anderson, B.; Grover, D.; Hua, M.; Liu, Y.; Owen, R.; Lacbawan, F. Identification of Eight SARS-CoV-2 ORF7a Deletion Variants in 2,726 Clinical Specimens. bioRxiv 2020. [Google Scholar] [CrossRef]
- Thao, T.T.N.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid Reconstruction of SARS-CoV-2 Using a Synthetic Genomics Platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef]
- Lobiuc, A.; Dimian, M.; Gheorghita, R.; Sturdza, O.A.C.; Covasa, M. Introduction and Characteristics of SARS-CoV-2 in North-East of Romania During the First COVID-19 Outbreak. Front. Microbiol. 2021, 12, 654417. [Google Scholar] [CrossRef]
- Nelson, C.A.; Minasov, G.; Shuvalova, L.; Fremont, D.H. RCSB PDB—6W37: Structure of the SARS-CoV-2 ORF7A Encoded Accesory Protein. Available online: https://www.rcsb.org/structure/6W37 (accessed on 8 October 2020).
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Zhang Lab Modeling of the SARS-COV-2 Genome Using I-TASSER. Available online: https://zhanglab.ccmb.med.umich.edu/COVID-19/ (accessed on 8 October 2020).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef] [PubMed]
- Amamuddy, O.S.; Verkhivker, G.M.; Bishop, Ö.T. Impact of Early Pandemic Stage Mutations on Molecular Dynamics of SARS-CoV-2 MPro. J. Chem. Inf. Model. 2020, 60, 5080–5102. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV Spike Protein Interface Reveal Unique Mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Singer, J.B.; Gifford, R.J.; Cotten, M.; Robertson, D.L. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints 2020, 2020060225. [Google Scholar] [CrossRef]
- Tan, Y.-X.; Tan, T.H.P.; Lee, M.J.-R.; Tham, P.-Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of Apoptosis by the Severe Acute Respiratory Syndrome Coronavirus 7a Protein Is Dependent on Its Interaction with the Bcl-XL Protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.K.; Coleman, C.M.; Postel, S.; Sisk, J.M.; Bernbaum, J.G.; Venkataraman, T.; Sundberg, E.J.; Frieman, M.B. Severe Acute Respiratory Syndrome Coronavirus ORF7a Inhibits Bone Marrow Stromal Antigen 2 Virion Tethering through a Novel Mechanism of Glycosylation Interference. J. Virol. 2015, 89, 11820–11833. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional Landscape of SARS-CoV-2 Cellular Restriction. Mol. Cell 2021, 81, 2656–2668.e8. [Google Scholar] [CrossRef]
- Ongaro, A.; Oselladore, E.; Memo, M.; Ribaudo, G.; Gianoncelli, A. Insight into the LFA-1/SARS-CoV-2 Orf7a Complex by Protein–Protein Docking, Molecular Dynamics, and MM-GBSA Calculations. J. Chem. Inf. Model. 2021, 61, 2780–2787. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Fuglebakk, E.; Echave, J.; Reuter, N. Measuring and Comparing Structural Fluctuation Patterns in Large Protein Datasets. Bioinformatics 2012, 28, 2431–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, L.; Chen, Z.; Li, Q.; Liu, R.; Song, W.; Kong, R.; Chang, S. Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. Int. J. Mol. Sci. 2019, 20, 224. [Google Scholar] [CrossRef] [Green Version]
- Fukuyoshi, S.; Kometani, M.; Watanabe, Y.; Hiratsuka, M.; Yamaotsu, N.; Hirono, S.; Manabe, N.; Takahashi, O.; Oda, A. Molecular Dynamics Simulations to Investigate the Influences of Amino Acid Mutations on Protein Three-Dimensional Structures of Cytochrome P450 2D6.1, 2, 10, 14A, 51, and 62. PLoS ONE 2016, 11, e0152946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Liao, M.; Meng, X.; Somero, G.N. Structural Flexibility and Protein Adaptation to Temperature: Molecular Dynamics Analysis of Malate Dehydrogenases of Marine Molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF7a WT | ORF7a A105V | ||||
|---|---|---|---|---|---|
| Values 1 | Patients | Values | Patients | ||
| General data | Age (years) | 53.24 ± 2.88 | (n = 45) | 50.06 ± 4.21 | (n = 17) |
| Sex (%) | 37.78 F 62.22 M | (n = 45) | 35.29 F 64.71 M | (n = 17) | |
| PCR data | CT (N) | 23.75 ± 0.71 | (n = 40) | 25.36 ± 0.97 | (n = 12) |
| CT (S) | 24.9 ± 0.62 | (n = 41) | 26.91 ± 0.9 | (n = 13) | |
| CT (R) | 26.27 ± 0.65 | (n = 34) | 26.7 ± 0.92 | (n = 16) | |
| Blood tests | Leucocytes (cells × 109/L) | 8.76 ± 1.02 | (n = 33) | 10.03 ± 2 | (n = 8) |
| CRP (mg/dL) | 5.59 ± 1.17 | (n = 35) | 9.66 ± 2.7 * | (n = 15) | |
| Thrombocytes (cells × 103/µL) | 272.22 ± 31.61 | (n = 23) | 197.5 ± 29.9 | (n = 6) | |
| Hemoglobin (g/dL) | 12.56 ± 0.44 | (n = 30) | 12.2 ± 0.74 | (n = 10) | |
| ALAT/GPT (U/L) | 55.11 ± 7.29 | (n = 27) | 47.24 ± 9.32 | (n = 10) | |
| Comorbidities (%) | Hypertension | 42.50 | (n = 40) | 21.43 | (n = 14) |
| Obesity | 37.50 | 21.43 | |||
| Diabetes | 17.50 | 14.29 | |||
| Min. 1 comorbidity | 50.00 | 35.71 | |||
| Other clinical data | Days in hospital | 20.27 ± 1.57 | (n = 33) | 20.33 ± 2.45 | (n = 12) |
| Deaths due to COVID-19 (%) | 9.52 | (n = 45) | 18.75 | (n = 17) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobiuc, A.; Șterbuleac, D.; Sturdza, O.; Dimian, M.; Covasa, M. A Conservative Replacement in the Transmembrane Domain of SARS-CoV-2 ORF7a as a Putative Risk Factor in COVID-19. Biology 2021, 10, 1276. https://doi.org/10.3390/biology10121276
Lobiuc A, Șterbuleac D, Sturdza O, Dimian M, Covasa M. A Conservative Replacement in the Transmembrane Domain of SARS-CoV-2 ORF7a as a Putative Risk Factor in COVID-19. Biology. 2021; 10(12):1276. https://doi.org/10.3390/biology10121276
Chicago/Turabian StyleLobiuc, Andrei, Daniel Șterbuleac, Olga Sturdza, Mihai Dimian, and Mihai Covasa. 2021. "A Conservative Replacement in the Transmembrane Domain of SARS-CoV-2 ORF7a as a Putative Risk Factor in COVID-19" Biology 10, no. 12: 1276. https://doi.org/10.3390/biology10121276
APA StyleLobiuc, A., Șterbuleac, D., Sturdza, O., Dimian, M., & Covasa, M. (2021). A Conservative Replacement in the Transmembrane Domain of SARS-CoV-2 ORF7a as a Putative Risk Factor in COVID-19. Biology, 10(12), 1276. https://doi.org/10.3390/biology10121276