
Soil Rehabilitation Promotes Resilient Microbiome with Enriched Keystone Taxa than Agricultural Infestation in Barren Soils on the Loess Plateau

 ,
,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description
2.2. DNA Extraction and PCR Amplification
2.3. Sequencing on Illumina MiSeq Platform
2.4. Network and Keystone Taxa Analyses
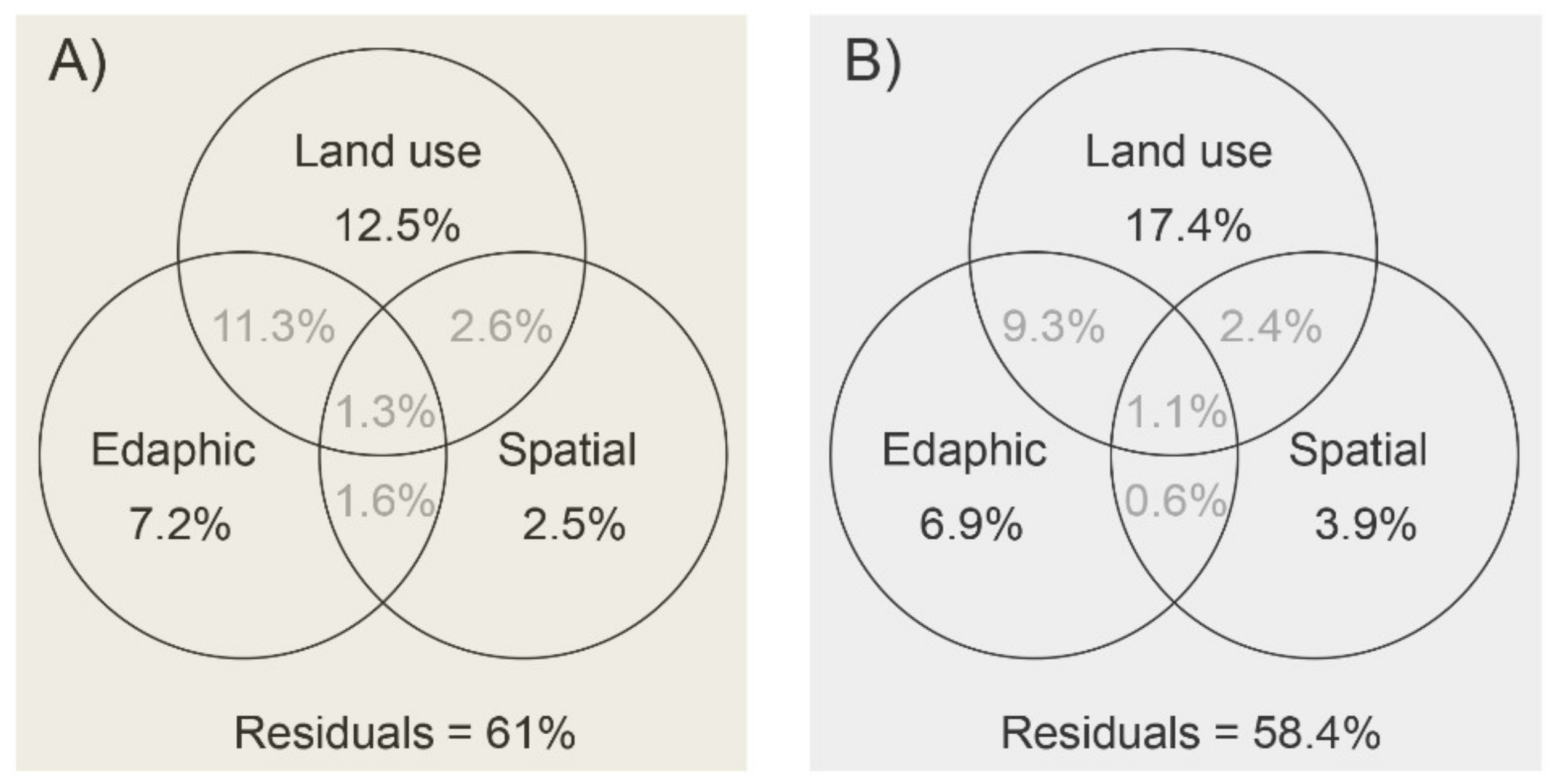
2.5. The Relationship between Network Topology and Environmental Variables
2.6. Statistical Analysis
3. Results
3.1. Variations in Edaphic Properties and Microbial Communities across Different Land-Use Types
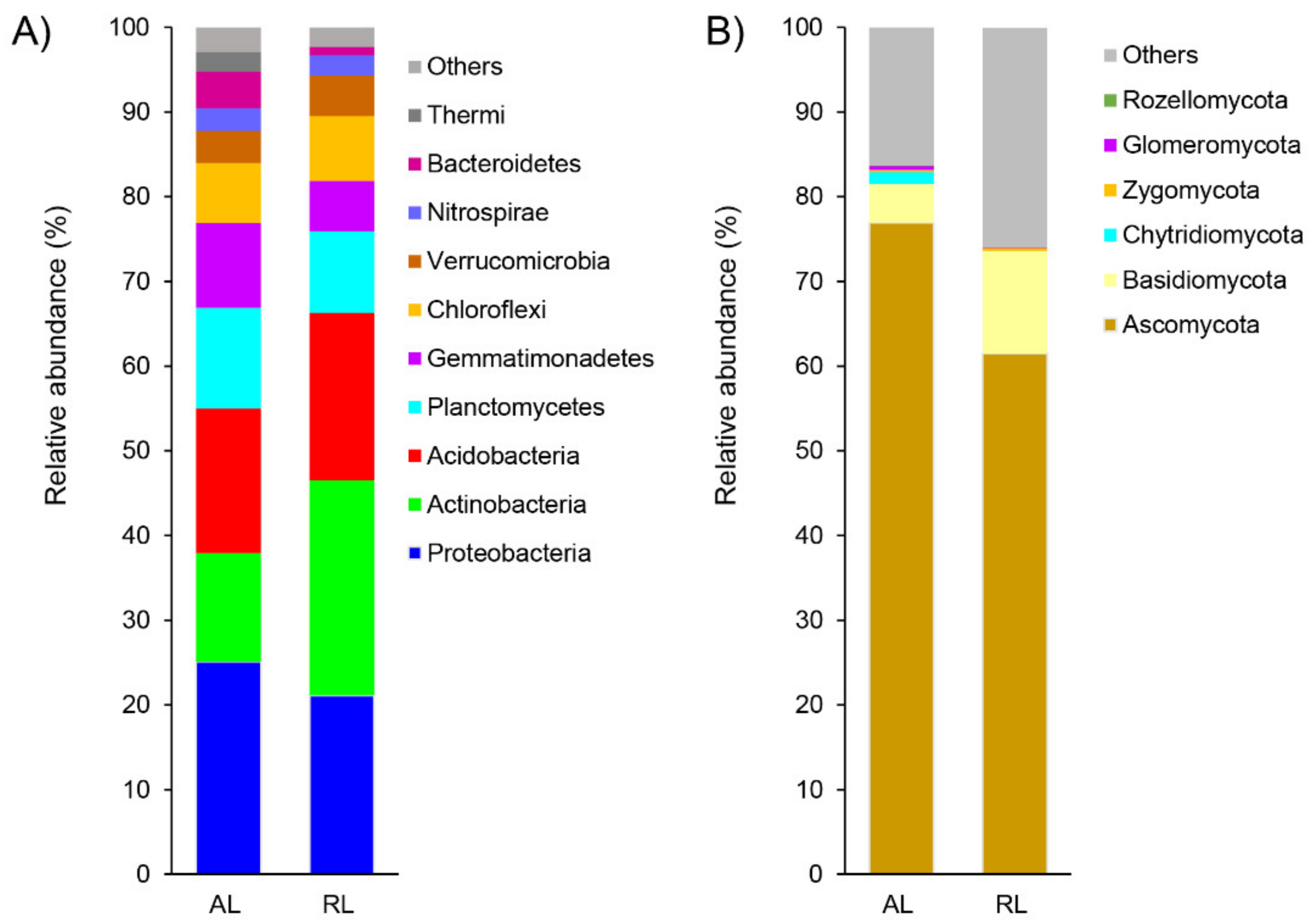
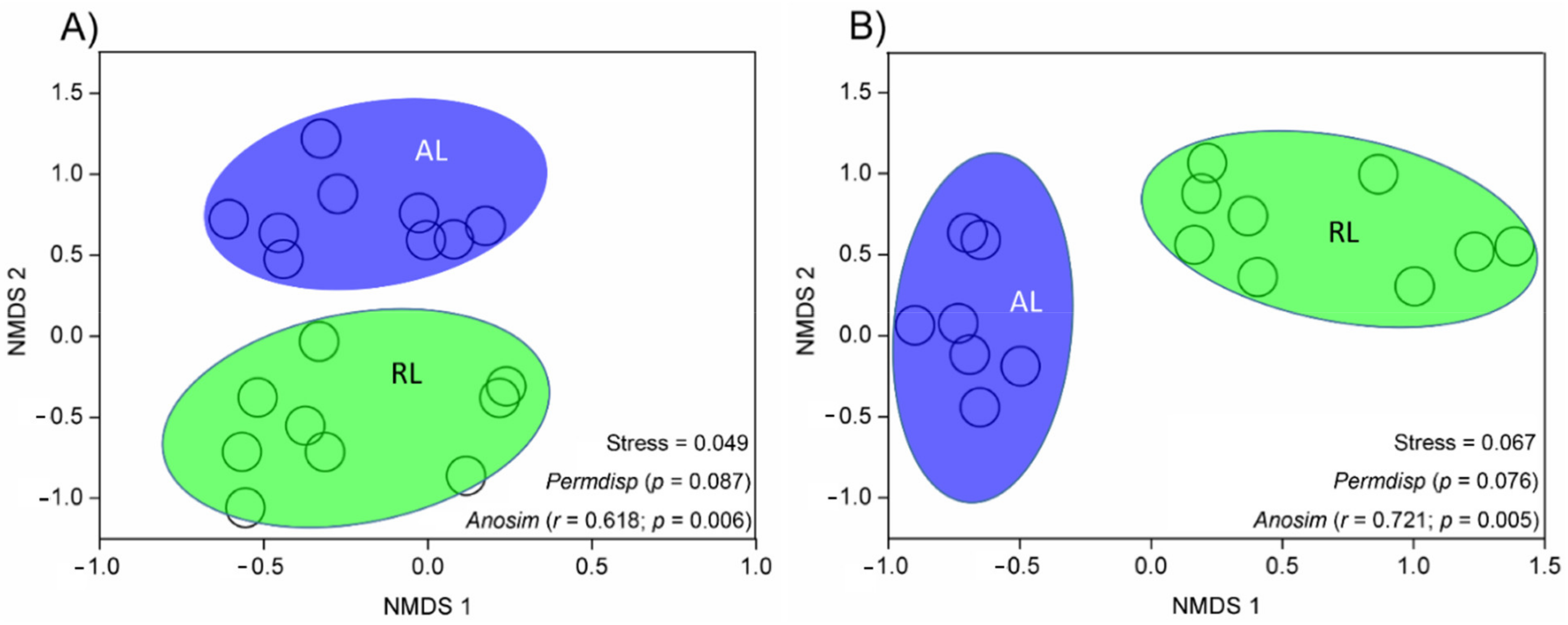
3.2. Variations in Microbial Community Composition, Diversity, and Structure
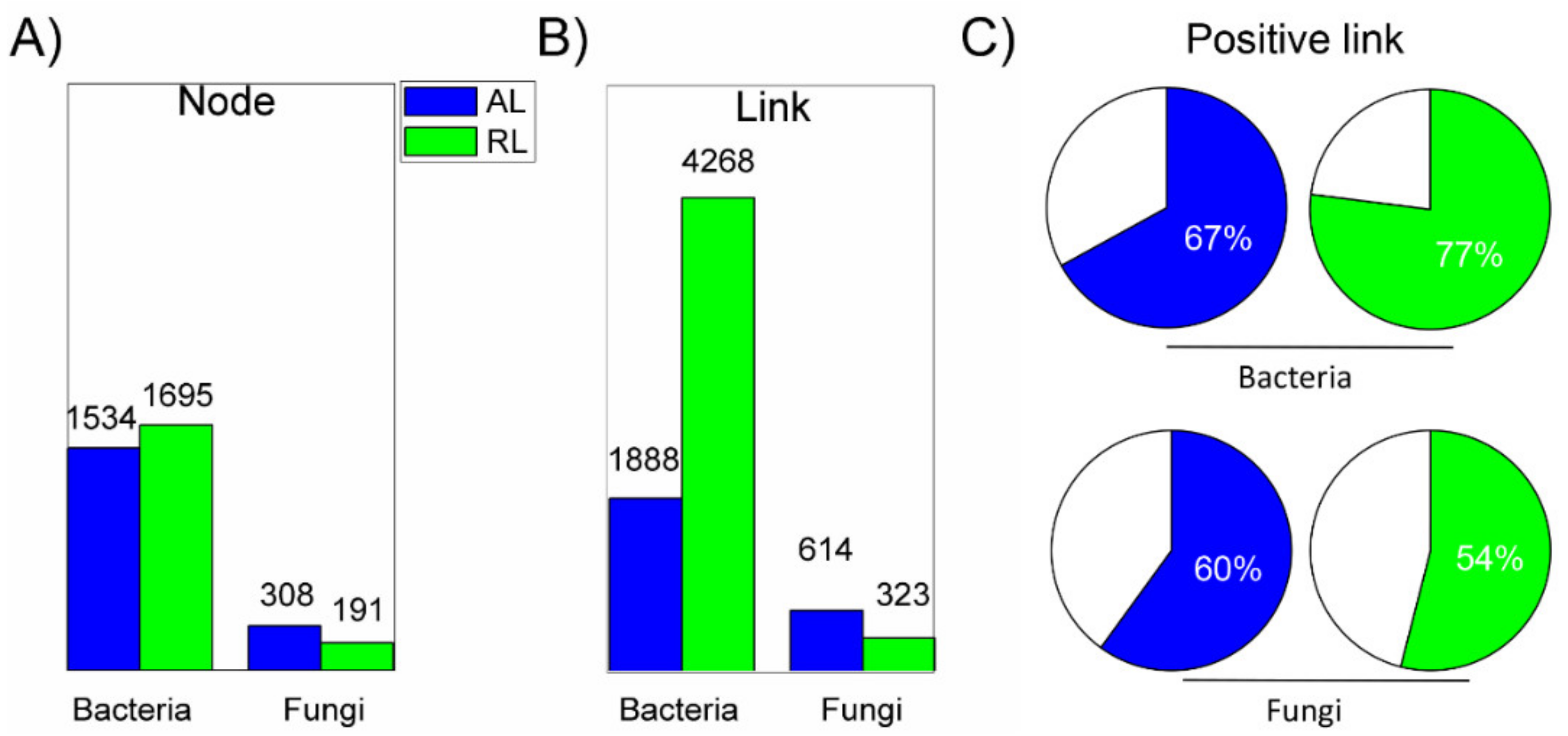
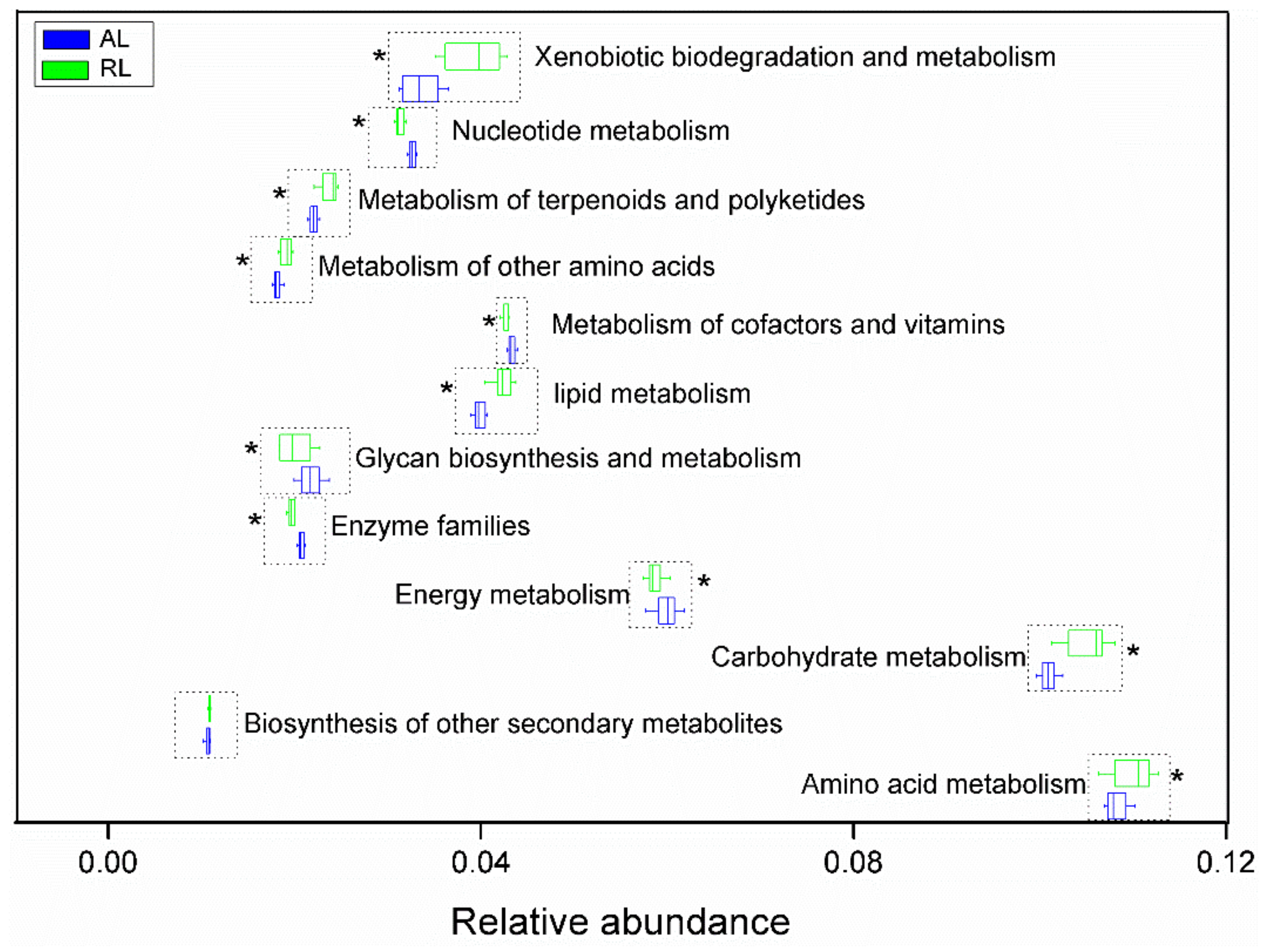
3.3. Distinct Microbial Networks and Putative Metabolic Profiles
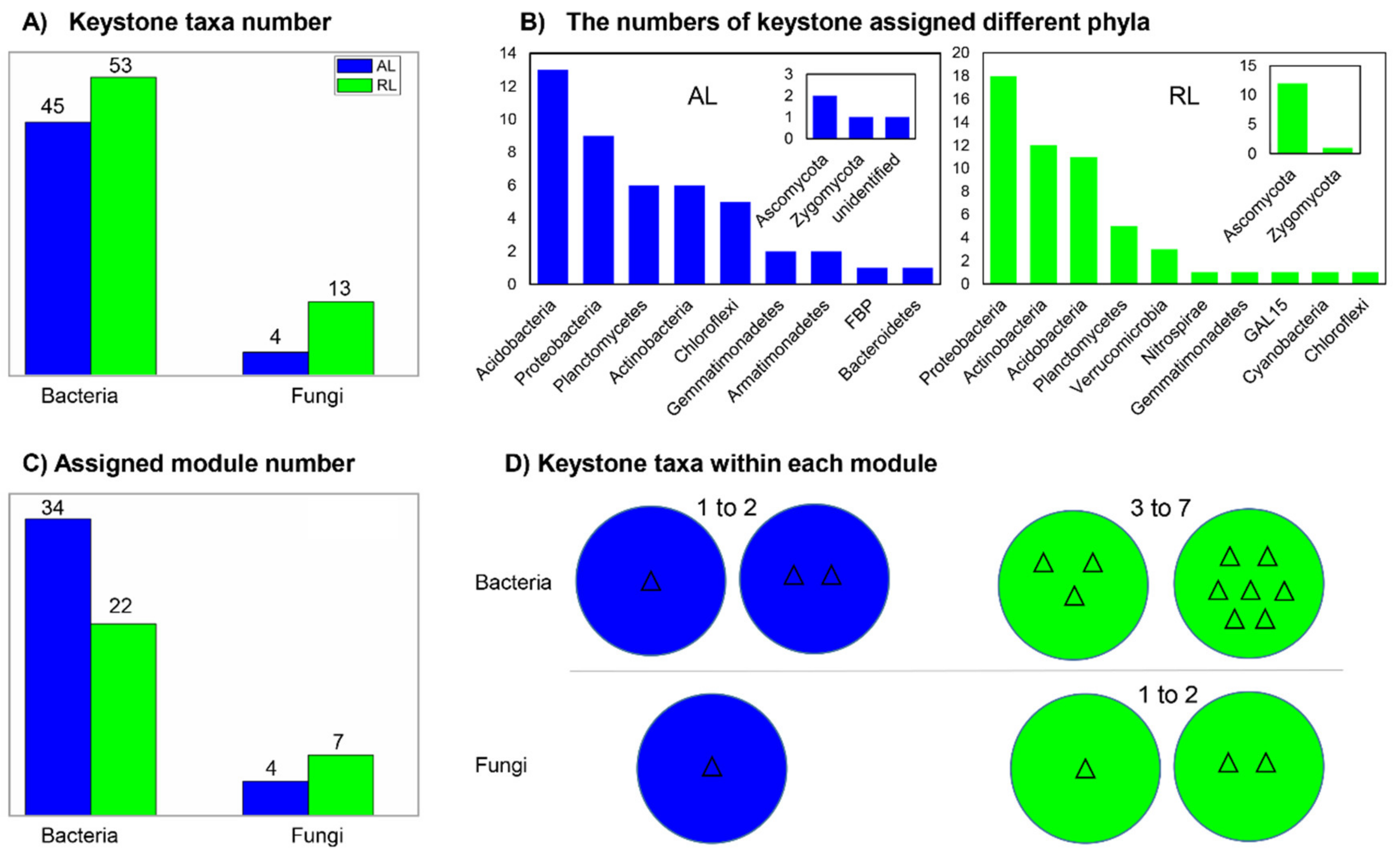
3.4. Keystone Taxa and Their Distributing Feature
4. Discussion
4.1. Microbial Diversity and Community Structure Variation
4.2. Distinct Microbial Networks, Keystone Taxa and Putative Metabolism Profiles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Primmer, E.; Jokinen, P.; Blicharska, M.; Barton, D.N.; Bugter, R.; Potschin, M. Governance of ecosystem services: A framework for empirical analysis. Ecosyst. Serv. 2015, 16, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.A.; Ramankutty, N.; Brauman, K.A.; Cassidy, E.S.; Gerber, J.S.; Johnston, M.; Mueller, N.D.; O’Connell, C.; Ray, D.K.; West, P.C.; et al. Solutions for a cultivated planet. Nature 2011, 478, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wei, W.; Fu, B.; Lü, Y. Soil and water conservation on the Loess Plateau in China: Review and perspective. Prog. Phys. Geogr. Earth Environ. 2007, 31, 389–403. [Google Scholar] [CrossRef]
- Fu, B.; Liu, Y.; Lü, Y.; He, C.; Zeng, Y.; Wu, B. Assessing the soil erosion control service of ecosystems change in the Loess Plateau of China. Ecol. Complex. 2011, 8, 284–293. [Google Scholar] [CrossRef]
- Zhao, F.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Policy-guided nationwide ecological recovery: Assessment of the Grain-to-Green Program of China. J. Food Agric. Environ. 2013, 11, 1882–1890. [Google Scholar] [CrossRef]
- Gong, H.; Meng, D.; Li, X.; Zhu, F. Soil degradation and food security coupled with global climate change in northeastern China. Chin. Geogr. Sci. 2013, 23, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Ilunga wa Ilunga, E.; Mahy, G.; Piqueray, J.; Séleck, M.; Shutcha, M.N.; Meerts, P.; Faucon, M.P. Plant functional traits as a promising tool for the ecological restoration of degraded tropical metal-rich habitats and revegetation of metal-rich bare soils: A case study in copper vegetation of Katanga, DRC. Ecol. Eng. 2015, 82, 214–221. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, X.; Wu, Y.; Zhang, L.; Cheng, J.; Wei, G.; Lin, Y. Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci. Total Environ. 2018, 635, 598–606. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, Y.; Tang, Z.; Shangguan, Z.; Chang, F.; Jia, F.; Chen, Y.; He, X.; Shi, W.; Deng, L. Effects of grassland afforestation on structure and function of soil bacterial and fungal communities. Sci. Total Environ. 2019, 676, 396–406. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Y.; Sun, H.; An, S. The restoration age of Robinia pseudoacacia plantation impacts soil microbial biomass and microbial community structure in the Loess Plateau. Catena 2018, 165, 192–200. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, S.; Sun, Q.; Li, N.; Jiang, J.; Wang, R.; Zhang, Y.; Liu, Q.; Wu, D.; Li, R.; et al. Soil organic carbon sequestration potential of artificial and natural vegetation in the hilly regions of Loess Plateau. Ecol. Eng. 2015, 82, 547–554. [Google Scholar] [CrossRef]
- Yang, Y.; Dou, Y.; Huang, Y.; An, S. Links between soil fungal diversity and plant and soil properties on the Loess Plateau. Front. Microbiol. 2017, 8, 2198. [Google Scholar] [CrossRef]
- Wang, B.; Xue, S.; Liu, G.B.; Zhang, G.H.; Li, G.; Ren, Z.P. Changes in soil nutrient and enzyme activities under different vegetations in the Loess Plateau area, Northwest China. Catena 2012, 92, 186–195. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, L.; Zhang, Z.; Zhang, J.; Cheng, J.; Wei, G.; Lin, Y. Soil microbial diversity during 30 years of grassland restoration on the Loess Plateau, China: Tight linkages with plant diversity. Land Degrad. Dev. 2019, 30, 1172–1182. [Google Scholar] [CrossRef]
- An, S.S.; Cheng, Y.; Huang, Y.M.; Liu, D. Effects of revegetation on soil microbial biomass, enzyme activities, and nutrient cycling on the Loess Plateau in China. Restor. Ecol. 2013, 21, 600–607. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Y.; Yan, H.; Jiang, Y.; Zhao, T.; An, S. Dynamics of soil nitrogen fractions and their relationship with soil microbial communities in two forest species of northern China. PLoS ONE 2018, 13, e0196567. [Google Scholar] [CrossRef]
- Zeng, Q.; An, S.; Liu, Y.; Wang, H.; Wang, Y. Biogeography and the driving factors affecting forest soil bacteria in an arid area. Sci. Total Environ. 2019, 680, 124–131. [Google Scholar] [CrossRef]
- Liu, D.; Keiblinger, K.M.; Leitner, S.; Wegner, U.; Zimmermann, M.; Fuchs, S.; Lassek, C.; Riedel, K.; Zechmeister-Boltenstern, S. Response of microbial communities and their metabolic functions to drying–rewetting stress in a temperate forest soil. Microorganisms 2019, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Zhang, C.; Xue, S.; Liu, G.-B.; Song, Z.-L. A comparison of soil qualities of different revegetation types in the Loess Plateau, China. Plant Soil 2011, 347, 163–178. [Google Scholar] [CrossRef]
- Powell, M.J. Chytridiomycota. In Handbook of the Protists; Springer International Publishing: Cham, Germany, 2017; pp. 1523–1558. ISBN 978-331-928-149-0. [Google Scholar]
- Longcore, J.E.; Simmons, D.R. Chytridiomycota. In eLS; John Wiley & Sons Ltd.: Chichester, UK, 2012. [Google Scholar]
- Deng, J.; Bai, X.; Zhou, Y.; Zhu, W.; Yin, Y. Variations of soil microbial communities accompanied by different vegetation restoration in an open-cut iron mining area. Sci. Total Environ. 2020, 704, 135243. [Google Scholar] [CrossRef] [PubMed]
- Su, J.Q.; Ding, L.J.; Xue, K.; Yao, H.Y.; Quensen, J.; Bai, S.J.; Wei, W.X.; Wu, J.S.; Zhou, J.; Tiedje, J.M.; et al. Long-term balanced fertilization increases the soil microbial functional diversity in a phosphorus-limited paddy soil. Mol. Ecol. 2015, 24, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jiang, Y.; Liang, C.; Luo, Y.; Xu, Q.; Han, C.; Zhao, Q.; Sun, B. Competitive interaction with keystone taxa induced negative priming under biochar amendments. Microbiome 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Xiao, E.; Wang, Y.; Xiao, T.; Sun, W.; Deng, J.; Jiang, S.; Fan, W.; Tang, J.; Ning, Z. Microbial community responses to land-use types and its ecological roles in mining area. Sci. Total Environ. 2021, 775, 145753. [Google Scholar] [CrossRef]
- Liu, D.; Huang, Y.; An, S.; Sun, H.; Bhople, P.; Chen, Z. Soil physicochemical and microbial characteristics of contrasting land-use types along soil depth gradients. Catena 2018, 162, 345–353. [Google Scholar] [CrossRef]
- Herren, C.M.; McMahon, K.D. Keystone taxa predict compositional change in microbial communities. Environ. Microbiol. 2018, 20, 2207–2217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Han, X.; Yan, J.; Zou, W.; Wang, E.; Lu, X.; Chen, X. Keystone microbiomes revealed by 14 years of field restoration of the degraded agricultural soil under distinct vegetation scenarios. Front. Microbiol. 2020, 11, 1915. [Google Scholar] [CrossRef]
- Cheng, X.; Yun, Y.; Wang, H.; Ma, L.; Tian, W.; Man, B.; Liu, C. Contrasting bacterial communities and their assembly processes in karst soils under different land use. Sci. Total Environ. 2021, 751, 142263. [Google Scholar] [CrossRef]
- Zeng, Q.; Liu, D.; An, S. Decoupled diversity patterns in microbial geographic distributions on the arid area (the Loess Plateau). Catena 2021, 196, 104922. [Google Scholar] [CrossRef]
- Watson, R.T.; Zakri, A.H. Ecosystems and Human Well-Being: Desertification Synthesis: Millenium Ecosystem Assessment; WHO: Geneva, Switzerland, 2015; ISBN 156-973-590-5. [Google Scholar]
- Ostwald, M.; Chen, D. Land-use change: Impacts of climate variations and policies among small-scale farmers in the Loess Plateau, China. Land Use Policy 2006, 23, 361–371. [Google Scholar] [CrossRef]
- Tian, Q.; Taniguchi, T.; Shi, W.-Y.; Li, G.; Yamanaka, N.; Du, S. Land-use types and soil chemical properties influence soil microbial communities in the semiarid Loess Plateau region in China. Sci. Rep. 2017, 7, 45289. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Frey, B.; Yang, L.; Li, M.-H.; Ni, H. Land use change effects on diversity of soil bacterial, Acidobacterial and fungal communities in wetlands of the Sanjiang Plain, northeastern China. Sci. Rep. 2019, 9, 18535. [Google Scholar] [CrossRef]
- Liu, T.; Wu, X.; Li, H.; Alharbi, H.; Wang, J.; Dang, P.; Chen, X.; Kuzyakov, Y.; Yan, W. Soil organic matter, nitrogen and pH driven change in bacterial community following forest conversion. For. Ecol. Manag. 2020, 477, 118473. [Google Scholar] [CrossRef]
- Hou, L.; Hoag, D.; Keske, C.M.H.; Lu, C. Sustainable value of degraded soils in China’s Loess Plateau: An updated approach. Ecol. Econ. 2014, 97, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wang, B.; Bhople, P.; Davlatbekov, F.; Yu, F. Land rehabilitation improves edaphic conditions and increases soil microbial biomass and abundance. Soil Ecol. Lett. 2020, 2, 145–156. [Google Scholar] [CrossRef]
- Rinnan, R.; Bååth, E. Differential utilization of carbon substrates by bacteria and fungi in tundra soil. Appl. Environ. Microbiol. 2009, 75, 3611–3620. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Li, S.; Avera, B.N.; Strahm, B.D.; Badgley, B.D. Soil bacterial and fungal communities show distinct recovery patterns during forest ecosystem restoration. Appl. Environ. Microbiol. 2017, 83, e00966-17. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, C.; Kou, Y.; Yao, M.; He, Z.; Li, X. Distinct mechanisms shape soil bacterial and fungal co-occurrence networks in a mountain ecosystem. FEMS Microbiol. Ecol. 2020, 96, fiaa030. [Google Scholar] [CrossRef]
- Deng, L.; Shangguan, Z.P.; Li, R. Effects of the grain-for-green program on soil erosion in China. Int. J. Sediment Res. 2012, 27, 120–127. [Google Scholar] [CrossRef]
- Zhao, G.; Mu, X.; Wen, Z.; Wang, F.; Gao, P. Soil erosion, conservation, and eco-environment changes in the loess plateau of china. Land Degrad. Dev. 2013, 24, 499–510. [Google Scholar] [CrossRef]
- Zhao, F.; Jiao, S.; Chengjie, R.; Di, K.; Jian, D.; Xinhui, H.; Gaihe, Y.; Yongzhong, F.; Guangxin, R. Land use change influences soil C, N and P stoichiometry under ‘Grain-to-Green Program’ in China. Sci. Rep. 2015, 5, 10195. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.B.; Yang, X.M.; Li, Y. Effect of apple base on regional water cycle in Weibei upland of the Loess Plateau. Acta Geogr. A Sin. 2001, 56, 7–12. [Google Scholar] [CrossRef]
- Jin, J.; Su, J.; Bai, Y.; Jing, Z.; Jing, G.; Cheng, G.; Cheng, J. Response of soil quality to artificial vegetation restoration patterns in the arid area of northern Weihe river basin. Acta Agrestia Sin. 2014, 22, 737–742. [Google Scholar] [CrossRef]
- Liu, C.A.; Li, F.R.; Zhou, L.M.; Zhang, R.H.; Yu, J.; Lin, S.L.; Wang, L.J.; Siddique, K.H.M.; Li, F.M. Effect of organic manure and fertilizer on soil water and crop yields in newly-built terraces with loess soils in a semi-arid environment. Agric. Water Manag. 2013, 117, 123–132. [Google Scholar] [CrossRef]
- Wang, L.; Yang, F.; Yuan, J.; Raza, W.; Huang, Q.; Shen, Q. Long-term application of bioorganic fertilizers improved soil biochemical properties and microbial communities of an apple orchard soil. Front. Microbiol. 2016, 7, 1893. [Google Scholar] [CrossRef]
- Zhao, W.; Liang, B.; Yang, X.; Zhou, J. Fate of residual 15 N-labeled fertilizer in dryland farming systems on soils of contrasting fertility. Soil Sci. Plant Nutr. 2015, 61, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Yang, Y.; An, S.; Wang, H.; Wang, Y. The biogeographical distribution of soil bacterial communities in the Loess Plateau as revealed by high-throughput sequencing. Front. Microbiol. 2018, 9, 2456. [Google Scholar] [CrossRef] [Green Version]
- Fujita, S.I.; Senda, Y.; Nakaguchi, S.; Hashimoto, T. Multiplex PCR using internal transcribed spacer 1 and 2 regions for rapid detection and identification of yeast strains. J. Clin. Microbiol. 2001, 39, 3617–3622. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, 1–22. [Google Scholar] [CrossRef]
- Kemp, P.F.; Aller, J.Y. Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 2004, 47, 161–177. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, S.; Liu, X.; Yao, P.; Ge, T.; Zhang, X.-H. Spatiotemporal dynamics of the archaeal community in coastal sediments: Assembly process and co-occurrence relationship. ISME J. 2020, 14, 1463–1478. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. MBio 2010, 1, 113. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Sul, W.J.; Asuming-Brempong, S.; Wang, Q.; Tourlousse, D.M.; Penton, C.R.; Deng, Y.; Rodrigues, J.L.M.; Adiku, S.G.K.; Jones, J.W.; Zhou, J.; et al. Tropical agricultural land management influences on soil microbial communities through its effect on soil organic carbon. Soil Biol. Biochem. 2013, 65, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Kim, Y.; Kim, J.M.; Chu, B.; Joa, J.-H.; Sang, M.K.; Song, J.; Weon, H.-Y. A preliminary examination of bacterial, archaeal, and fungal communities inhabiting different rhizocompartments of tomato plants under real-world environments. Sci. Rep. 2019, 9, 9300. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wang, S.P.; Lin, Q.Y.; Zhou, J.Z. Molecular ecological network analyses revealing the effects of livestock grazing on soil microbial community in the Tibetan grassland. Microbiol. China 2015, 42, 1818–1831. [Google Scholar]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B. Package vegan. In Community Ecology Package; Version 2; CRAN: Vienna, Austria, 2013; pp. 1–295. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Sterkenburg, E.; Bahr, A.; Brandström Durling, M.; Clemmensen, K.E.; Lindahl, B.D. Changes in fungal communities along a boreal forest soil fertility gradient. New Phytol. 2015, 207, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Kirk, P.; Cannon, P.; Minter, D.; Stalpers, J. Dictionary of the Fungi, 10th ed.; CABI: London, UK, 2008; ISBN 085-199-826-7. [Google Scholar]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, C.; Luo, Y. Meta-analysis of the impacts of global change factors on soil microbial diversity and functionality. Nat. Commun. 2020, 11, 3072. [Google Scholar] [CrossRef]
- Schmidt, T.M. Encyclopedia of Microbiology; Academic Press: Cambridge, MA, USA, 2019; ISBN 978-012-811-737-8. [Google Scholar]
- Schaechter, M. Encyclopedia of microbiology. Choice Rev. Online 2011, 49, 49–1215. [Google Scholar] [CrossRef]
- Xiao, L.; Liu, G.; Zhang, J.; Xue, S. Long-term effects of vegetational restoration on soil microbial communities on the Loess Plateau of China. Restor. Ecol. 2016, 24, 794–804. [Google Scholar] [CrossRef]
- Wang, B.; Guo, B.L.; Xue, S.; Zhu, B. Changes in soil physico-chemical and microbiological properties during natural succession on abandoned farmland in the Loess Plateau. Environ. Earth Sci. 2011, 62, 915–925. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, S.; Li, C.; Zhao, L.; Feng, H.; Yue, G.; Ren, Z.; Cheng, G. The soil carbon/nitrogen ratio and moisture affect microbial community structures in alkaline permafrost-affected soils with different vegetation types on the Tibetan plateau. Res. Microbiol. 2014, 165, 128–139. [Google Scholar] [CrossRef]
- Fan, T.; Stewart, B.A.; Yong, W.; Junjie, L.; Guangye, Z. Long-term fertilization effects on grain yield, water-use efficiency and soil fertility in the dryland of Loess Plateau in China. Agric. Ecosyst. Environ. 2005, 106, 313–329. [Google Scholar] [CrossRef]
- Wu, T.; Schoenau, J.J.; Li, F.; Qian, P.; Malhi, S.S.; Shi, Y.; Xu, F. Influence of cultivation and fertilization on total organic carbon and carbon fractions in soils from the Loess Plateau of China. Soil Tillage Res. 2004, 77, 59–68. [Google Scholar] [CrossRef]
- Neutel, A.M.; Heesterbeek, J.A.P.; Van De Koppel, J.; Hoenderboom, G.; Vos, A.; Kaldeway, C.; Berendse, F.; De Ruiter, P.C. Reconciling complexity with stability in naturally assembling food webs. Nature 2007, 449, 599–602. [Google Scholar] [CrossRef] [Green Version]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Button, D.K. Kinetics of nutrient-limited transport and microbial growth. Microbiol. Rev. 1985, 49, 270–297. [Google Scholar] [CrossRef]
- Cheng, M.; Xiang, Y.; Xue, Z.; An, S.; Darboux, F. Soil aggregation and intra-aggregate carbon fractions in relation to vegetation succession on the Loess Plateau, China. Catena 2015, 124, 77–84. [Google Scholar] [CrossRef]
- Banning, N.C.; Murphy, D.V. Effect of heat-induced disturbance on microbial biomass and activity in forest soil and the relationship between disturbance effects and microbial community structure. Appl. Soil Ecol. 2008, 40, 109–119. [Google Scholar] [CrossRef]
- Allison, S.D.; Wallenstein, M.D.; Bradford, M.A. Soil-carbon response to warming dependent on microbial physiology. Nat. Geosci. 2010, 3, 336–340. [Google Scholar] [CrossRef]
- Mitchell, R.J.; Guo, D.; Mou, P.; Jones, R.H. Spatio-temporal patterns of soil available nutrients following experimental disturbance in a pine forest. Oecologia 2004, 138, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wardle, D.A.; Lindahl, B.D. Ecology. Disentangling global soil fungal diversity. Science 2014, 346, 1052–1053. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Johnson, D.; Burslem, D.F.R.P.; Yu, S.; Fang, M.; Taylor, J.D.; Taylor, A.F.S.; Helgason, T.; Liu, X. Soil fungal networks maintain local dominance of ectomycorrhizal trees. Nat. Commun. 2020, 11, 2636. [Google Scholar] [CrossRef]
- Mamet, S.D.; Redlick, E.; Brabant, M.; Lamb, E.G.; Helgason, B.L.; Stanley, K.; Siciliano, S.D. Structural equation modeling of a winnowed soil microbiome identifies how invasive plants re-structure microbial networks. ISME J. 2019, 13, 1988–1996. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLOS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [Green Version]
- Achat, D.L.; Augusto, L.; Bakker, M.R.; Gallet-Budynek, A.; Morel, C. Microbial processes controlling P availability in forest spodosols as affected by soil depth and soil properties. Soil Biol. Biochem. 2012, 44, 39–48. [Google Scholar] [CrossRef]
- Cong, J.; Yang, Y.; Liu, X.; Lu, H.; Liu, X.; Zhou, J.; Li, D.; Yin, H.; Ding, J.; Zhang, Y. Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 2015, 5, 10007. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.R.; Wu, H.Y.; Song, X.D.; Yang, S.H.; Dong, Y.; Yang, J.L.; Zhang, G.L. Intra-horizon differentiation of the bacterial community and its co-occurrence network in a typical Plinthic horizon. Sci. Total Environ. 2019, 678, 692–701. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Nobre, M.F.; Moore, E.; Rainey, F.A.; Battista, J.R.; da Costa, M.S. Characterization and radiation resistance of new isolates of Rubrobacter radiotolerans and Rubrobacter xylanophilus. Extremophiles 1999, 3, 235–238. [Google Scholar] [CrossRef]
- Xu, L.; He, Y. Comparision of bacterial diversity between rhizpsphere and non-rhizophere soil of maize based on 16S rDNA high-throughput sequencing. J. Shanxi Agric. Sci. 2019, 47, 1212–1216. [Google Scholar] [CrossRef]
- Yoon, M.H.; Im, W.T. Flavisolibacter ginsengiterrae gen. nov., sp. nov. and Flavisolibacter ginsengisoli sp. nov., isolated from ginseng cultivating soil. Int. J. Syst. Evol. Microbiol. 2007, 57, 1834–1839. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Lee, C.H.; Oh, T.K. Aeromicrobium alkaliterrae sp. nov., isolated from an alkaline soil, and emended description of the genus Aeromicrobium. Int. J. Syst. Evol. Microbiol. 2005, 55, 2171–2175. [Google Scholar] [CrossRef]
- Ozdemir-Kocak, F.; Isik, K.; Saricaoglu, S.; Saygin, H.; Inan-Bektas, K.; Cetin, D.; Guven, K.; Sahin, N. Kribbella sindirgiensis sp. nov. isolated from soil. Arch. Microbiol. 2017, 199, 1399–1407. [Google Scholar] [CrossRef]
- Sandoval-Denis, M.; Guarro, J.; Cano-Lira, J.F.; Sutton, D.A.; Wiederhold, N.P.; de Hoog, G.S.; Abbott, S.P.; Decock, C.; Sigler, L.; Gené, J. Phylogeny and taxonomic revision of Microascaceae with emphasis on synnematous fungi. Stud. Mycol. 2016, 83, 193–233. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, N.; Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of the genus Talaromyces. Stud. Mycol. 2014, 78, 175–341. [Google Scholar] [CrossRef] [Green Version]
- Gardeli, C.; Athenaki, M.; Xenopoulos, E.; Mallouchos, A.; Koutinas, A.A.; Aggelis, G.; Papanikolaou, S. Lipid production and characterization by Mortierella (Umbelopsis) isabellina cultivated on lignocellulosic sugars. J. Appl. Microbiol. 2017, 123, 1461–1477. [Google Scholar] [CrossRef]
- Middelburg, J.J.; Barranguet, C.; Boschker, H.T.S.; Herman, P.M.J.; Moens, T.; Heip, C.H.R. The fate of intertidal microphytobenthos carbon: An in situ 13C-labeling study. Limnol. Oceanogr. 2000, 45, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, N.P.; Riordan, S.M.; Castaño-Rodríguez, N.; Wilkins, M.R.; Kaakoush, N.O. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome 2018, 6, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Fungi | |||
|---|---|---|---|---|
| AL | RL | AL | RL | |
| Threshold | 0.96 | 0.96 | 0.89 | 0.87 |
| avgCC a | 0.08 | 0.10 | 0.12 | 0.16 |
| avgK | 2.46 | 5.04 | 3.39 | 3.98 |
| Module | 246 | 204 | 39 | 25 |
| Modularity | 0.94 | 0.77 | 0.68 | 0.63 |
| GD | 9.58 | 6.33 | 5.14 | 4.35 |
| Bacteria | Fungi | |||||||
|---|---|---|---|---|---|---|---|---|
| AL | RL | AL | RL | |||||
| r | p | r | p | r | p | r | p | |
| pH | 0.010 | 0.269 | 0.015 | 0.125 | 0.017 | 0.278 | 0.071 | 0.121 |
| a TOC | 0.005 | 0.382 | 0.034 | 0.039 | 0.026 | 0.181 | 0.068 | 0.114 |
| TN | 0.006 | 0.371 | 0.082 | 0.001 | 0.005 | 0.388 | 0.080 | 0.120 |
| TP | −0.001 | 0.494 | 0.003 | 0.385 | −0.038 | 0.881 | 0.112 | 0.052 |
| Moisture | −0.015 | 0.775 | −0.016 | 0.831 | −0.011 | 0.610 | 0.047 | 0.146 |
| SC | 0.009 | 0.273 | 0.033 | 0.997 | 0.076 | 0.018 | 0.000 | 0.370 |
| NO3 | 0.103 | 0.001 | −0.033 | 0.994 | −0.016 | 0.631 | −0.049 | 0.903 |
| NH4 | 0.041 | 0.023 | 0.068 | 0.001 | −0.045 | 0.897 | 0.035 | 0.232 |
| AP | 0.052 | 0.004 | 0.047 | 0.005 | 0.023 | 0.230 | 0.017 | 0.264 |
| DOC | 0.006 | 0.301 | 0.007 | 0.002 | 0.005 | 0.381 | 0.052 | 0.142 |
| MBC | −0.027 | 0.937 | 0.081 | 0.001 | −0.035 | 0.850 | 0.021 | 0.241 |
| Altitude | 0.325 | 0.211 | 0.314 | 0.240 | 0.341 | 0.193 | 0.329 | 0.224 |
| Plant type | 0.355 | 0.153 | 0.345 | 0.194 | 0.369 | 0.124 | 0.355 | 0.172 |
| OTUs | Kingdom | Class | Order | Family | Genus | Species | |
|---|---|---|---|---|---|---|---|
| AL | OTU69876 | Bacteria | Saprospirae | [Saprospirales] | Chitinophagaceae | Flavisolibacter | Not assigned |
| AL | OTU72155 | Bacteria | Actinobacteria | Actinomycetales | Nocardioidaceae | Aeromicrobium | Not assigned |
| AL | OTU37190 | Bacteria | Actinobacteria | Actinomycetales | Nocardioidaceae | Kribbella | Not assigned |
| AL | OTU5872 | Bacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Devosia | Not assigned |
| AL | OTU1603 | Bacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | Kaistobacter | Not assigned |
| AL | OTU46084 | Bacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | Not assigned |
| AL | OTU53987 | Bacteria | Gammaproteobacteria | Pseudomonadales | Pseudomonadaceae | Pseudomonas | Not assigned |
| AL | OTU36507 | Bacteria | Gammaproteobacteria | Xanthomonadales | Sinobacteraceae | Steroidobacter | Not assigned |
| RL | OTU40252 | Bacteria | Alphaproteobacteria | Rhizobiales | Bradyrhizobiaceae | Balneimonas | Not assigned |
| RL | OTU46298 | Bacteria | Alphaproteobacteria | Rhizobiales | Brucellaceae | Ochrobactrum | Not assigned |
| RL | OTU65537 | Bacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | Not assigned |
| RL | OTU60443 | Bacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | Not assigned |
| RL | OTU6889 | Bacteria | Alphaproteobacteria | Rhizobiales | Hyphomicrobiaceae | Rhodoplanes | Not assigned |
| RL | OTU28903 | Bacteria | Planctomycetia | Pirellulales | Pirellulaceae | A17 | Not assigned |
| RL | OTU3183 | Bacteria | Planctomycetia | Planctomycetales | Planctomycetaceae | Planctomyces | Not assigned |
| RL | OTU6858 | Bacteria | Rubrobacteria | Rubrobacterales | Rubrobacteraceae | Rubrobacter | Not assigned |
| RL | OTU14570 | Bacteria | Rubrobacteria | Rubrobacterales | Rubrobacteraceae | Rubrobacter | Not assigned |
| AL | OTU5193 | Fungi | Eurotiomycetes | Eurotiales | Trichocomaceae | Talaromyces | T. marneffei |
| AL | OTU13116 | Fungi | Incertae sedis | Mortierellales | Mortierellaceae | Mortierella | M. indohii |
| AL | OTU13053 | Fungi | Sordariomycetes | Microascales | Microascaceae | unidentified | unidentified |
| AL | OTU11811 | Fungi | unidentified | unidentified | unidentified | unidentified | unidentified |
| RL | OTU6627 | Fungi | Dothideomycetes | Pleosporales | Phaeosphaeriaceae | Phaeosphaeria | unidentified |
| RL | OTU10400 | Fungi | Dothideomycetes | Pleosporales | unidentified | unidentified | unidentified |
| RL | OTU9390 | Fungi | Eurotiomycetes | Eurotiales | Trichocomaceae | Penicillium | P. polonicum |
| RL | OTU9744 | Fungi | Eurotiomycetes | unidentified | unidentified | unidentified | unidentified. |
| RL | OTU7322 | Fungi | Incertae sedis | Mortierellales | Mortierellaceae | Mortierella | M. humilis |
| RL | OTU12611 | Fungi | Lecanoromycetes | Teloschistales | Physciaceae | Phaeophyscia | P. hispidula |
| RL | OTU4181 | Fungi | Pezizomycetes | Pezizales | Pyronemataceae | Geopora | unidentified |
| RL | OTU56 | Fungi | Pezizomycetes | Pezizales | Pyronemataceae | unidentified | unidentified |
| RL | OTU6753 | Fungi | Saccharomycetes | Saccharomycetales | Saccharomycetaceae | Kazachstania | K. telluris |
| RL | OTU2069 | Fungi | Sordariomycetes | Hypocreales | Incertae sedis | Acremonium | A. dichromosporum |
| RL | OTU3133 | Fungi | Sordariomycetes | Hypocreales | Nectriaceae | Fusarium | F. pseudensiforme |
| RL | OTU6835 | Fungi | unidentified | unidentified | unidentified | unidentified | unidentified |
| RL | OTU6835 | Fungi | unidentified | unidentified | unidentified | unidentified | unidentified |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Bhople, P.; Keiblinger, K.M.; Wang, B.; An, S.; Yang, N.; Chater, C.C.C.; Yu, F. Soil Rehabilitation Promotes Resilient Microbiome with Enriched Keystone Taxa than Agricultural Infestation in Barren Soils on the Loess Plateau. Biology 2021, 10, 1261. https://doi.org/10.3390/biology10121261
Liu D, Bhople P, Keiblinger KM, Wang B, An S, Yang N, Chater CCC, Yu F. Soil Rehabilitation Promotes Resilient Microbiome with Enriched Keystone Taxa than Agricultural Infestation in Barren Soils on the Loess Plateau. Biology. 2021; 10(12):1261. https://doi.org/10.3390/biology10121261
Chicago/Turabian StyleLiu, Dong, Parag Bhople, Katharina Maria Keiblinger, Baorong Wang, Shaoshan An, Nan Yang, Caspar C. C. Chater, and Fuqiang Yu. 2021. "Soil Rehabilitation Promotes Resilient Microbiome with Enriched Keystone Taxa than Agricultural Infestation in Barren Soils on the Loess Plateau" Biology 10, no. 12: 1261. https://doi.org/10.3390/biology10121261
APA StyleLiu, D., Bhople, P., Keiblinger, K. M., Wang, B., An, S., Yang, N., Chater, C. C. C., & Yu, F. (2021). Soil Rehabilitation Promotes Resilient Microbiome with Enriched Keystone Taxa than Agricultural Infestation in Barren Soils on the Loess Plateau. Biology, 10(12), 1261. https://doi.org/10.3390/biology10121261