
Virus-Induced Tumorigenesis and IFN System


 , ,
, ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. IFN System
3. IFN Signaling and MicroRNA
4. IFN System and Oncoviruses
4.1. HPV
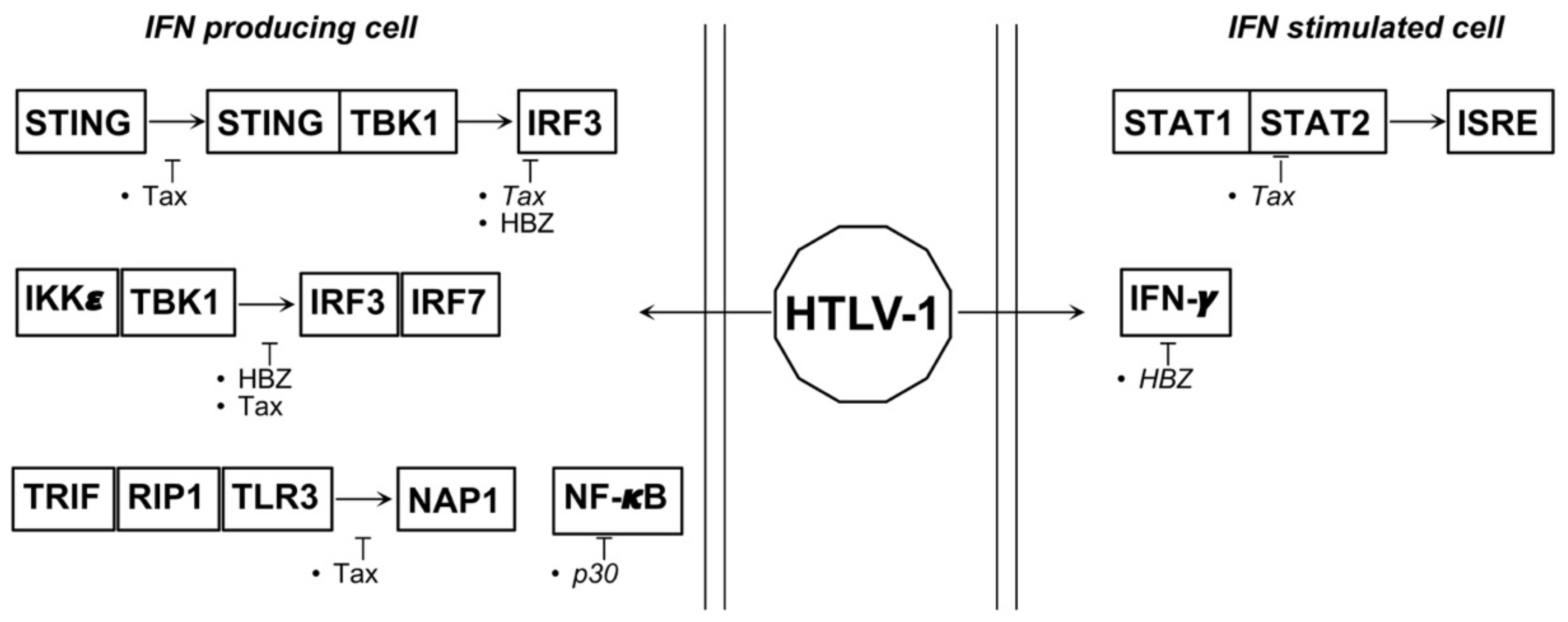
4.2. HTLV-1
4.3. MCPyV and JCPyV
4.4. Oncogenic Herpesviruses
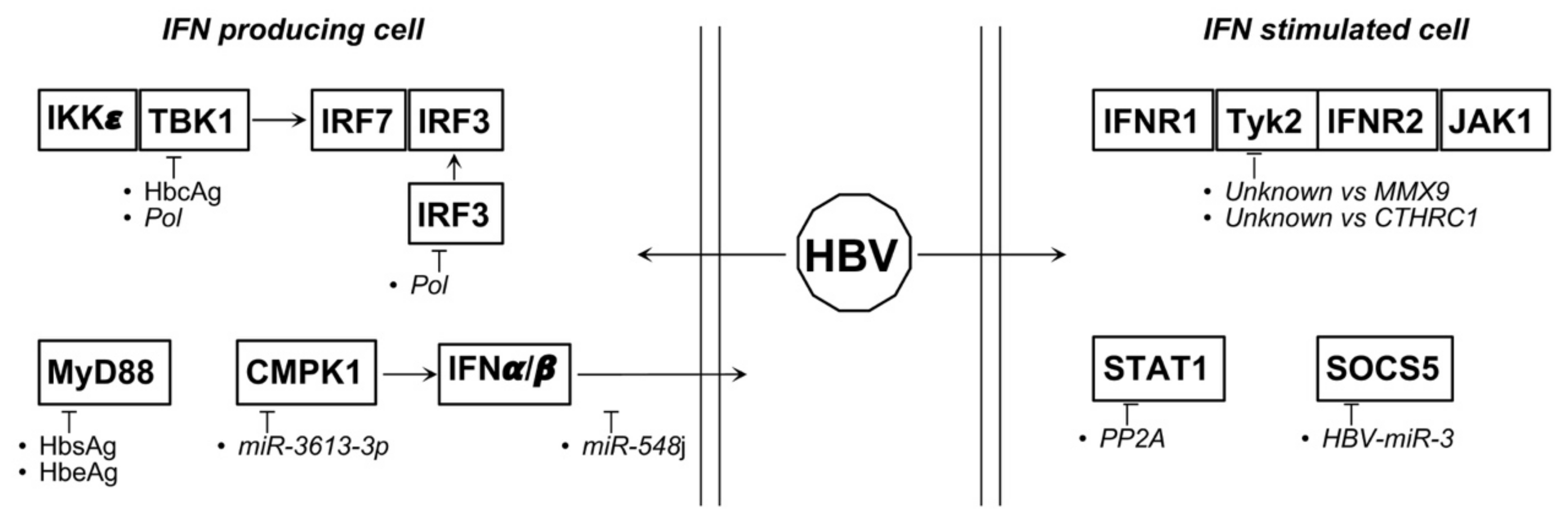
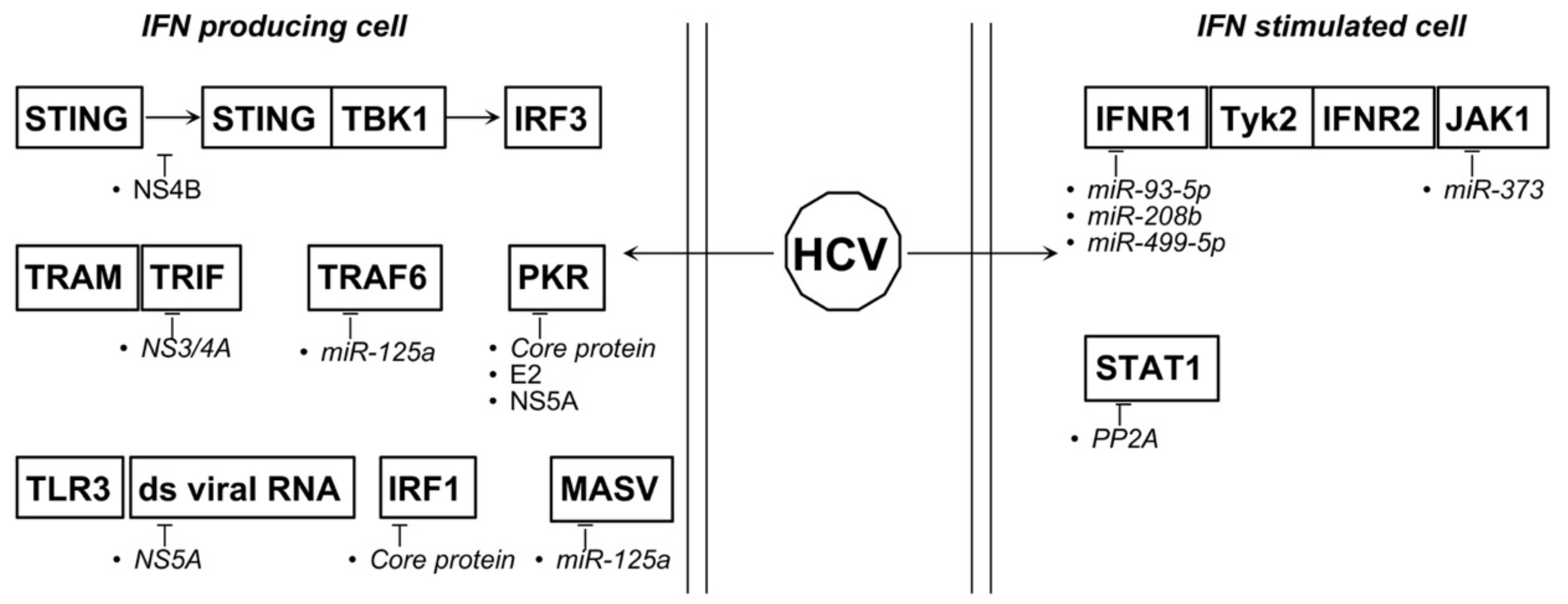
4.5. HBV and HCV

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oncoviruses | MicroRNAs 1 | Targets/Function 2 | Refs. |
|---|---|---|---|
| HPV | miR-122 ↑ | E6 ↓ | [64] |
| miR-129-5P ↑ | HPV18 E6 and E7 ↓ | [63] | |
| miR-221 ↑ | SOCS ↓ | [65] | |
| miR-10a, -18a, -19a, -21, -34a, -98, -182, -194, -590-5p | Exploitation of some E6 and E7 activities | [62] | |
| miR-34a ↑ | p53 ↓ | [37] | |
| HTLV-1 | miR-155 ↑ | IFNγ upregulation | [30] |
| EBV | miR-BART92 | FOXO3 ↓ | [113] |
| miR-141 ↑ | ZCCHC3 ↓ | [113] | |
| miR-494-3P ↑ | AKT activation | [114] | |
| miR-142-3P ↑ | AKT suppression | [114] | |
| miR-BART20-5P2 | TBX21/T-bet ↓ | [115] | |
| EBV-miR-82 | STAT1 ↓ | [114] | |
| miR-BART6-3p2 | RIG-I ↓ | [115] | |
| EBERs2 | induce IFN I response | [115] | |
| miR-BART162 | CREB-binding protein ↓ | [116] | |
| miR-BART82 | STAT1 ↓ | [117] | |
| miR-let7a ↓ | progression of NNL | [117] | |
| HBV | miR-548j ↑ | less IFNα/β release | [144] |
| miR-3613-3p ↑ | CMPK1 ↓ | [145] | |
| miR1287-5p ↓ | FSRT1 ↑ | [146] | |
| HBV-miR-32 | SOCS-5 ↓ | [147] | |
| HCV | miR-93-5p ↑ | IFNAR1 ↓ | [148] |
| miR-208b ↑ | IFNAR1, IFNλ ↓ | [149] | |
| miR-499a-5p ↑ | IFNAR1, IFNλ ↓ | [149] | |
| mir-373 ↑ | JAK1, IRF5 and -9 ↓ | [150,151] | |
| mir-125a ↑ | MASV, TRAF6 ↓ | [152] | |
| miR181a-2-3p, -374a-3p, -374a-5p, -204-5p ↓ | [153] | ||
| miR-146b-5p ↓ | activation NF-κB signaling | [153] |
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bekisz, J.; Baron, S.; Balinsky, C.; Morrow, A.; Zoon, K.C. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals 2010, 3, 994–1015. [Google Scholar] [CrossRef]
- Chiantore, M.V.; Vannucchi, S.; Mangino, G.; Percario, Z.A.; Affabris, E.; Fiorucci, G.; Romeo, G. Senescence and cell death pathways and their role in cancer therapeutic outcome. Curr. Med. Chem. 2009, 16, 287–300. [Google Scholar] [CrossRef]
- Fiorucci, G.; Chiantore, M.V.; Mangino, G.; Romeo, G. MicroRNAs in virus-induced tumorigenesis and IFN system. Cytokine Growth Factor Rev. 2015, 26, 183–194. [Google Scholar] [CrossRef]
- Fiorucci, G.; Chiantore, M.V.; Mangino, G.; Percario, Z.A.; Affabris, E.; Romeo, G. Cancer regulator microRNA: Potential relevance in diagnosis, prognosis and treatment of cancer. Curr. Med. Chem. 2012, 19, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Crosse, K.M.; Monson, E.A.; Beard, M.R.; Helbig, K.J. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J. Innate Immun. 2018, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef]
- Makris, S.; Paulsen, M.; Johansson, C. Type I Interferons as Regulators of Lung Inflammation. Front. Immunol. 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Das, N.; Mukherjee, P. Picking up a Fight: Fine Tuning Mitochondrial Innate Immune Defenses Against RNA Viruses. Front. Microbiol. 2020, 11, 1990. [Google Scholar] [CrossRef]
- Duic, I.; Tadakuma, H.; Harada, Y.; Yamaue, R.; Deguchi, K.; Suzuki, Y.; Yoshimura, S.H.; Kato, H.; Takeyasu, K.; Fujita, T. Viral RNA recognition by LGP2 and MDA5, and activation of signaling through step-by-step conformational changes. Nucleic Acids Res. 2020, 48, 11664–11674. [Google Scholar] [CrossRef]
- Lee, S.; Channappanavar, R.; Kanneganti, T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Louis, C.; Burns, C.; Wicks, I. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 2018, 9, 434. [Google Scholar] [CrossRef]
- Oshiumi, H.; Kouwaki, T.; Seya, T. Accessory Factors of Cytoplasmic Viral RNA Sensors Required for Antiviral Innate Immune Response. Front. Immunol. 2016, 7, 200. [Google Scholar] [CrossRef]
- Zanin, N.; Viaris de Lesegno, C.; Lamaze, C.; Blouin, C.M. Interferon Receptor Trafficking and Signaling: Journey to the Cross Roads. Front. Immunol. 2020, 11, 615603. [Google Scholar] [CrossRef]
- Stark, G.R.; Cheon, H.; Wang, Y. Responses to Cytokines and Interferons that Depend upon JAKs and STATs. Cold Spring Harb. Perspect. Biol. 2018, 10, a028555. [Google Scholar] [CrossRef]
- Raftery, N.; Stevenson, N.J. Advances in anti-viral immune defence: Revealing the importance of the IFN JAK/STAT pathway. Cell. Mol. Life Sci. 2017, 74, 2525–2535. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus. J. Virol. 2019, 93, e00238-19. [Google Scholar] [CrossRef]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef]
- Lever, A.M.; Jeang, K.T. Insights into cellular factors that regulate HIV-1 replication in human cells. Biochemistry 2011, 50, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Houzet, L.; Jeang, K.T. MicroRNAs and human retroviruses. Biochim. Biophys. Acta 2011, 1809, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Okada, H. MicroRNAs and STAT interplay. Semin. Cancer Biol. 2012, 22, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Song, W.; Qian, J.; Li, Y.; He, J.; Zhang, Q.; Li, W.; Zhai, A.; Kao, W.; Hu, Y.; et al. MiR-122 modulates type I interferon expression through blocking suppressor of cytokine signaling 1. Int. J. Biochem. Cell Biol. 2013, 45, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells. Virol. J. 2011, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.S.; McCoy, C.E.; Lloyd, A.T.; O’Farrelly, C.; Stevenson, N.J. miR-19a: An effective regulator of SOCS3 and enhancer of JAK-STAT signalling. PLoS ONE 2013, 8, e69090. [Google Scholar] [CrossRef]
- Wiesen, J.L.; Tomasi, T.B. Dicer is regulated by cellular stresses and interferons. Mol. Immunol. 2009, 46, 1222–1228. [Google Scholar] [CrossRef]
- Ostermann, E.; Tuddenham, L.; Macquin, C.; Alsaleh, G.; Schreiber-Becker, J.; Tanguy, M.; Bahram, S.; Pfeffer, S.; Georgel, P. Deregulation of type I IFN-dependent genes correlates with increased susceptibility to cytomegalovirus acute infection of dicer mutant mice. PLoS ONE 2012, 7, e43744. [Google Scholar] [CrossRef]
- Trotta, R.; Chen, L.; Ciarlariello, D.; Josyula, S.; Mao, C.; Costinean, S.; Yu, L.; Butchar, J.P.; Tridandapani, S.; Croce, C.M.; et al. miR-155 regulates IFN-γ production in natural killer cells. Blood 2012, 119, 3478–3485. [Google Scholar] [CrossRef]
- Noguchi, S.; Yamada, N.; Kumazaki, M.; Yasui, Y.; Iwasaki, J.; Naito, S.; Akao, Y. socs7, a target gene of microRNA-145, regulates interferon-β induction through STAT3 nuclear translocation in bladder cancer cells. Cell Death Dis. 2013, 4, e482. [Google Scholar] [CrossRef]
- Polioudakis, D.; Bhinge, A.A.; Killion, P.J.; Lee, B.K.; Abell, N.S.; Iyer, V.R. A Myc-microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013, 41, 2239–2254. [Google Scholar] [CrossRef]
- Buggele, W.A.; Horvath, C.M. MicroRNA profiling of Sendai virus-infected A549 cells identifies miR-203 as an interferon-inducible regulator of IFIT1/ISG56. J. Virol. 2013, 87, 9260–9270. [Google Scholar] [CrossRef]
- Li, Y.; Xie, J.; Xu, X.; Wang, J.; Ao, F.; Wan, Y.; Zhu, Y. MicroRNA-548 down-regulates host antiviral response via direct targeting of IFN-λ1. Protein Cell 2013, 4, 130–141. [Google Scholar] [CrossRef]
- Honegger, A.; Schilling, D.; Bastian, S.; Sponagel, J.; Kuryshev, V.; Sültmann, H.; Scheffner, M.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS Pathog. 2015, 11, e1004712. [Google Scholar] [CrossRef]
- Chiantore, M.V.; Mangino, G.; Iuliano, M.; Zangrillo, M.S.; De Lillis, I.; Vaccari, G.; Accardi, R.; Tommasino, M.; Columba Cabezas, S.; Federico, M.; et al. Human papillomavirus E6 and E7 oncoproteins affect the expression of cancer-related microRNAs: Additional evidence in HPV-induced tumorigenesis. J. Cancer Res. Clin. Oncol. 2016, 142, 1751–1763. [Google Scholar] [CrossRef]
- Iuliano, M.; Mangino, G.; Chiantore, M.V.; Zangrillo, M.S.; Accardi, R.; Tommasino, M.; Fiorucci, G.; Romeo, G. Human Papillomavirus E6 and E7 oncoproteins affect the cell microenvironment by classical secretion and extracellular vesicles delivery of inflammatory mediators. Cytokine 2018, 106, 182–189. [Google Scholar] [CrossRef]
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef]
- Scott, M.; Nakagawa, M.; Moscicki, A.B. Cell-mediated immune response to human papillomavirus infection. Clin. Diagn. Lab. Immunol. 2001, 8, 209–220. [Google Scholar] [CrossRef]
- Karim, R.; Meyers, C.; Backendorf, C.; Ludigs, K.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; van der Burg, S.H.; Boer, J.M. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes. PLoS ONE 2011, 6, e17848. [Google Scholar] [CrossRef]
- Chang, Y.E.; Laimins, L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [PubMed]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Bortnik, V.; McMillan, N.A.; Idris, A. cGAS-STING responses are dampened in high-risk HPV type 16 positive head and neck squamous cell carcinoma cells. Microb. Pathog. 2019, 132, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. [Google Scholar] [CrossRef]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2020, 94, e01582-19. [Google Scholar] [CrossRef]
- Guo, H.; König, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Yu, Y.; Taxman, D.J.; Zhang, L.; Widman, D.G.; Swanson, K.V.; Wen, K.W.; Damania, B.; Moore, C.B.; et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 2012, 36, 933–946. [Google Scholar] [CrossRef]
- Lei, Y.; Wen, H.; Ting, J.P. The NLR protein, NLRX1, and its partner, TUFM, reduce type I interferon, and enhance autophagy. Autophagy 2013, 9, 432–433. [Google Scholar] [CrossRef]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.J.; Melief, C.J.; et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef]
- He, C.; Lv, X.; Huang, C.; Angeletti, P.C.; Hua, G.; Dong, J.; Zhou, J.; Wang, Z.; Ma, B.; Chen, X.; et al. A Human Papillomavirus-Independent Cervical Cancer Animal Model Reveals Unconventional Mechanisms of Cervical Carcinogenesis. Cell Rep. 2019, 26, 2636–2650. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef]
- Chiantore, M.V.; Mangino, G.; Iuliano, M.; Zangrillo, M.S.; De Lillis, I.; Vaccari, G.; Accardi, R.; Tommasino, M.; Fiorucci, G.; Romeo, G. IFN-β antiproliferative effect and miRNA regulation in Human Papilloma Virus E6- and E7-transformed keratinocytes. Cytokine 2017, 89, 235–238. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Yan, Q.; Chen, X.; Yang, Y.; Liu, X.; Wan, X. Interferon-β induced microRNA-129-5p down-regulates HPV-18 E6 and E7 viral gene expression by targeting SP1 in cervical cancer cells. PLoS ONE 2013, 8, e81366. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ji, Y.; Li, A.; Zhang, Q.; Song, W.; Li, Y.; Huang, H.; Qian, J.; Zhai, A.; Yu, X.; et al. MiR-122 directly inhibits human papillomavirus E6 gene and enhances interferon signaling through blocking suppressor of cytokine signaling 1 in SiHa cells. PLoS ONE 2014, 9, e108410. [Google Scholar] [CrossRef]
- Lu, H.; Gu, X. MicroRNA-221 inhibits human papillomavirus 16 E1-E2 mediated DNA replication through activating SOCS1/Type I IFN signaling pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 1518–1528. [Google Scholar] [PubMed]
- Honegger, A.; Leitz, J.; Bulkescher, J.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Silencing of human papillomavirus (HPV) E6/E7 oncogene expression affects both the contents and the amounts of extracellular microvesicles released from HPV-positive cancer cells. Int. J. Cancer 2013, 133, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de Thé, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Igata, A.; Matsumoto, M.; Tara, M. HTLV-I associated myelopathy, a new clinical entity. Lancet 1986, 1, 1031–1032. [Google Scholar] [CrossRef]
- Zhang, L.L.; Wei, J.Y.; Wang, L.; Huang, S.L.; Chen, J.L. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharm. Sin. 2017, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.K.; Chan, C.P.; Fung, S.Y.; Wang, P.H.; Wong, W.M.; Tang, H.V.; Yuen, K.S.; Jin, D.Y.; Kok, K.H. Suppression of Type I Interferon Production by Human T-Cell Leukemia Virus Type 1 Oncoprotein Tax through Inhibition of IRF3 Phosphorylation. J. Virol. 2016, 90, 3902–3912. [Google Scholar] [CrossRef]
- Hyun, J.; Ramos, J.C.; Toomey, N.; Balachandran, S.; Lavorgna, A.; Harhaj, E.; Barber, G.N. Oncogenic human T-cell lymphotropic virus type 1 tax suppression of primary innate immune signaling pathways. J. Virol. 2015, 89, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Charoenthongtrakul, S.; Zhou, Q.; Shembade, N.; Harhaj, N.S.; Harhaj, E.W. Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF-kappaB-dependent induction of SOCS1. J. Virol. 2011, 85, 6955–6962. [Google Scholar] [CrossRef]
- Olière, S.; Hernandez, E.; Lézin, A.; Arguello, M.; Douville, R.; Nguyen, T.L.; Olindo, S.; Panelatti, G.; Kazanji, M.; Wilkinson, P.; et al. HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PLoS Pathog. 2010, 6, e1001177. [Google Scholar] [CrossRef]
- Zhang, J.; Yamada, O.; Kawagishi, K.; Araki, H.; Yamaoka, S.; Hattori, T.; Shimotohno, K. Human T-cell leukemia virus type 1 Tax modulates interferon-alpha signal transduction through competitive usage of the coactivator CBP/p300. Virology 2008, 379, 306–313. [Google Scholar] [CrossRef]
- Narulla, M.S.; Alsairi, A.; Charmier, L.; Noonan, S.; Conroy, D.; Hall, W.W.; Sheehy, N. Positive and Negative Regulation of Type I Interferons by the Human T Cell Leukemia Virus Antisense Protein HBZ. J. Virol. 2017, 91, e00853-17. [Google Scholar] [CrossRef]
- Sugata, K.; Satou, Y.; Yasunaga, J.; Hara, H.; Ohshima, K.; Utsunomiya, A.; Mitsuyama, M.; Matsuoka, M. HTLV-1 bZIP factor impairs cell-mediated immunity by suppressing production of Th1 cytokines. Blood 2012, 119, 434–444. [Google Scholar] [CrossRef]
- Koralnik, I.J.; Lemp, J.F.; Gallo, R.C.; Franchini, G. In vitro infection of human macrophages by human T-cell leukemia/lymphotropic virus type I (HTLV-I). AIDS Res. Hum. Retrovir. 1992, 8, 1845–1849. [Google Scholar] [CrossRef]
- Macatonia, S.E.; Cruickshank, J.K.; Rudge, P.; Knight, S.C. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res. Hum. Retrovir. 1992, 8, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Hishizawa, M.; Imada, K.; Kitawaki, T.; Ueda, M.; Kadowaki, N.; Uchiyama, T. Depletion and impaired interferon-alpha-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br. J. Haematol. 2004, 125, 568–575. [Google Scholar] [CrossRef]
- Datta, A.; Sinha-Datta, U.; Dhillon, N.K.; Buch, S.; Nicot, C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J. Biol. Chem. 2006, 281, 23414–23424. [Google Scholar] [CrossRef]
- Fenizia, C.; Fiocchi, M.; Jones, K.; Parks, R.W.; Ceribelli, M.; Chevalier, S.A.; Edwards, D.; Ruscetti, F.; Pise-Masison, C.A.; Franchini, G. Human T-cell leukemia/lymphoma virus type 1 p30, but not p12/p8, counteracts toll-like receptor 3 (TLR3) and TLR4 signaling in human monocytes and dendritic cells. J. Virol. 2014, 88, 393–402. [Google Scholar] [CrossRef]
- Bellon, M.; Lepelletier, Y.; Hermine, O.; Nicot, C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood 2009, 113, 4914–4917. [Google Scholar] [CrossRef]
- Prado, J.C.M.; Monezi, T.A.; Amorim, A.T.; Lino, V.; Paladino, A.; Boccardo, E. Human polyomaviruses and cancer: An overview. Clinics 2018, 73, e558s. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; MacDonald, M.; You, J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 2016, 20, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Khalili, K.; White, M.K. Human demyelinating disease and the polyomavirus JCV. Mult. Scler. 2006, 12, 133–142. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Steiger, J. Polyomavirus BK. Lancet Infect. Dis. 2003, 3, 611–623. [Google Scholar] [CrossRef]
- Lemos, B.; Nghiem, P. Merkel cell carcinoma: More deaths but still no pathway to blame. J. Invest. Derm. 2007, 127, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Lunder, E.J.; Stern, R.S. Merkel-cell carcinomas in patients treated with methoxsalen and ultraviolet A radiation. N. Engl. J. Med. 1998, 339, 1247–1248. [Google Scholar] [CrossRef]
- Verma, S.; Ziegler, K.; Ananthula, P.; Co, J.K.; Frisque, R.J.; Yanagihara, R.; Nerurkar, V.R. JC virus induces altered patterns of cellular gene expression: Interferon-inducible genes as major transcriptional targets. Virology 2006, 345, 457–467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Co, J.K.; Verma, S.; Gurjav, U.; Sumibcay, L.; Nerurkar, V.R. Interferon- alpha and-beta restrict polyomavirus JC replication in primary human fetal glial cells: Implications for progressive multifocal leukoencephalopathy therapy. J. Infect. Dis. 2007, 196, 712–718. [Google Scholar] [CrossRef] [PubMed]
- De-Simone, F.I.; Sariyer, R.; Otalora, Y.L.; Yarandi, S.; Craigie, M.; Gordon, J.; Sariyer, I.K. IFN-Gamma Inhibits JC Virus Replication in Glial Cells by Suppressing T-Antigen Expression. PLoS ONE 2015, 10, e0129694. [Google Scholar] [CrossRef] [PubMed]
- Abend, J.R.; Low, J.A.; Imperiale, M.J. Inhibitory effect of gamma interferon on BK virus gene expression and replication. J. Virol. 2007, 81, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Willmes, C.; Adam, C.; Alb, M.; Völkert, L.; Houben, R.; Becker, J.C.; Schrama, D. Type I and II IFNs inhibit Merkel cell carcinoma via modulation of the Merkel cell polyomavirus T antigens. Cancer Res. 2012, 72, 2120–2128. [Google Scholar] [CrossRef]
- Liu, W.; Kim, G.B.; Krump, N.A.; Zhou, Y.; Riley, J.L.; You, J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 13730–13739. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Hjalgrim, H.; Smedby, K.E.; Rostgaard, K.; Molin, D.; Hamilton-Dutoit, S.; Chang, E.T.; Ralfkiaer, E.; Sundström, C.; Adami, H.O.; Glimelius, B.; et al. Infectious mononucleosis, childhood social environment, and risk of Hodgkin lymphoma. Cancer Res. 2007, 67, 2382–2388. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Hawkins, J.B.; Tracy, S.I.; Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef]
- Gupta, S.; Ylä-Anttila, P.; Callegari, S.; Tsai, M.H.; Delecluse, H.J.; Masucci, M.G. Herpesvirus deconjugases inhibit the IFN response by promoting TRIM25 autoubiquitination and functional inactivation of the RIG-I signalosome. PLoS Pathog. 2018, 14, e1006852. [Google Scholar] [CrossRef]
- Gupta, S.; Ylä-Anttila, P.; Sandalova, T.; Sun, R.; Achour, A.; Masucci, M.G. 14-3-3 scaffold proteins mediate the inactivation of trim25 and inhibition of the type I interferon response by herpesvirus deconjugases. PLoS Pathog. 2019, 15, e1008146. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ylä-Anttila, P.; Sandalova, T.; Achour, A.; Masucci, M.G. Interaction With 14-3-3 Correlates With Inactivation of the RIG-I Signalosome by Herpesvirus Ubiquitin Deconjugases. Front. Immunol. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, L.; Liu, W.; Duan, Z. Latent Membrane Protein 1 of Epstein-Barr Virus Promotes RIG-I Degradation Mediated by Proteasome Pathway. Front. Immunol. 2018, 9, 1446. [Google Scholar] [CrossRef] [PubMed]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Poüs, C.; et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 2021, 17, 1296–1315. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Doong, S.L.; Teng, S.C.; Lee, C.P.; Tsai, C.H.; Chen, M.R. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 2009, 83, 1856–1869. [Google Scholar] [CrossRef]
- Song, Y.J.; Izumi, K.M.; Shinners, N.P.; Gewurz, B.E.; Kieff, E. IRF7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc. Natl. Acad. Sci. USA 2008, 105, 18448–18453. [Google Scholar] [CrossRef]
- Hahn, A.M.; Huye, L.E.; Ning, S.; Webster-Cyriaque, J.; Pagano, J.S. Interferon regulatory factor 7 is negatively regulated by the Epstein-Barr virus immediate-early gene, BZLF-1. J. Virol. 2005, 79, 10040–10052. [Google Scholar] [CrossRef]
- Wu, L.; Fossum, E.; Joo, C.H.; Inn, K.S.; Shin, Y.C.; Johannsen, E.; Hutt-Fletcher, L.M.; Hass, J.; Jung, J.U. Epstein-Barr virus LF2: An antagonist to type I interferon. J. Virol. 2009, 83, 1140–1146. [Google Scholar] [CrossRef]
- Geiger, T.R.; Martin, J.M. The Epstein-Barr virus-encoded LMP-1 oncoprotein negatively affects Tyk2 phosphorylation and interferon signaling in human B cells. J. Virol. 2006, 80, 11638–11650. [Google Scholar] [CrossRef]
- Liu, X.; Sadaoka, T.; Krogmann, T.; Cohen, J.I. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Suppresses Type I Interferon Signaling To Promote EBV Reactivation. J. Virol. 2020, 94, e00258-20. [Google Scholar] [CrossRef]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [PubMed]
- Michaud, F.; Coulombe, F.; Gaudreault, E.; Paquet-Bouchard, C.; Rola-Pleszczynski, M.; Gosselin, J. Epstein-Barr virus interferes with the amplification of IFNalpha secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PLoS ONE 2010, 5, e11908. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fachko, D.N.; Ivanov, N.S.; Skalsky, R.L. B Cell Receptor-Responsive miR-141 Enhances Epstein-Barr Virus Lytic Cycle via FOXO3 Inhibition. mSphere 2021, 6, e00093-21. [Google Scholar] [CrossRef] [PubMed]
- Soltani, S.; Zakeri, A.; Tabibzadeh, A.; Zakeri, A.M.; Zandi, M.; Siavoshi, S.; Seifpour, S.; Farahani, A. A review on EBV encoded and EBV-induced host microRNAs expression profile in different lymphoma types. Mol. Biol. Rep. 2021, 48, 1801–1817. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus miR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef]
- Hooykaas, M.J.G.; van Gent, M.; Soppe, J.A.; Kruse, E.; Boer, I.G.J.; van Leenen, D.; Groot Koerkamp, M.J.A.; Holstege, F.C.P.; Ressing, M.E.; Wiertz, E.J.H.J.; et al. EBV MicroRNA BART16 Suppresses Type I IFN Signaling. J. Immunol. 2017, 198, 4062–4073. [Google Scholar] [CrossRef]
- Huang, W.T.; Lin, C.W. EBV-encoded miR-BART20-5p and miR-BART8 inhibit the IFN-γ-STAT1 pathway associated with disease progression in nasal NK-cell lymphoma. Am. J. Pathol. 2014, 184, 1185–1197. [Google Scholar] [CrossRef]
- Meckes, D.G.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Tu, T.; Bühler, S.; Bartenschlager, R. Chronic viral hepatitis and its association with liver cancer. Biol. Chem. 2017, 398, 817–837. [Google Scholar] [CrossRef]
- Taylor, P.E.; Zuckerman, A.J. Non-production of interfering substances by serum from patients with infectious hepatitis. J. Med. Microbiol. 1968, 1, 217–219. [Google Scholar] [CrossRef][Green Version]
- Blindenbacher, A.; Duong, F.H.; Hunziker, L.; Stutvoet, S.T.; Wang, X.; Terracciano, L.; Moradpour, D.; Blum, H.E.; Alonzi, T.; Tripodi, M.; et al. Expression of hepatitis c virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology 2003, 124, 1465–1475. [Google Scholar] [CrossRef]
- Christen, V.; Duong, F.; Bernsmeier, C.; Sun, D.; Nassal, M.; Heim, M.H. Inhibition of alpha interferon signaling by hepatitis B virus. J. Virol. 2007, 81, 159–165. [Google Scholar] [CrossRef]
- Twu, J.S.; Lee, C.H.; Lin, P.M.; Schloemer, R.H. Hepatitis B virus suppresses expression of human beta-interferon. Proc. Natl. Acad. Sci. USA 1988, 85, 252–256. [Google Scholar] [CrossRef]
- Tan, G.; Song, H.; Xu, F.; Cheng, G. When Hepatitis B Virus Meets Interferons. Front. Microbiol. 2018, 9, 1611. [Google Scholar] [CrossRef]
- Duong, F.H.; Filipowicz, M.; Tripodi, M.; La Monica, N.; Heim, M.H. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Tsunematsu, S.; Suda, G.; Yamasaki, K.; Kimura, M.; Izumi, T.; Umemura, M.; Ito, J.; Sato, F.; Nakai, M.; Sho, T.; et al. Hepatitis B virus X protein impairs α-interferon signaling via up-regulation of suppressor of cytokine signaling 3 and protein phosphatase 2A. J. Med. Virol. 2017, 89, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Samal, J.; Kandpal, M.; Vivekanandan, P. Molecular mechanisms underlying occult hepatitis B virus infection. Clin. Microbiol. Rev. 2012, 25, 142–163. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Liu, S.; Peng, N.; Xie, J.; Hao, Q.; Zhang, M.; Zhang, Y.; Xia, Z.; Xu, G.; Zhao, F.; Wang, Q.; et al. Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 2015, 62, 1015–1023. [Google Scholar] [CrossRef]
- Wang, J.; Liu, B.; Wang, N.; Lee, Y.M.; Liu, C.; Li, K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011, 85, 3733–3745. [Google Scholar] [CrossRef] [PubMed]
- Whitten, T.M.; Quets, A.T.; Schloemer, R.H. Identification of the hepatitis B virus factor that inhibits expression of the beta interferon gene. J. Virol. 1991, 65, 4699–4704. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, W.; Tan, L.; Yang, H.; Ge, M.; Zhu, C.; Zhang, R.; Cao, Y.; Chen, J.; Luo, Z.; et al. Hepatitis B virus hijacks CTHRC1 to evade host immunity and maintain replication. J. Mol. Cell Biol. 2015, 7, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, W.; Chen, Y.; Xie, X.; Zhang, Y.; Ma, C.; Yang, Q.; Han, Y.; Zhu, C.; Xiong, Y.; et al. Matrix Metalloproteinase 9 Facilitates Hepatitis B Virus Replication through Binding with Type I Interferon (IFN) Receptor 1 To Repress IFN/JAK/STAT Signaling. J. Virol. 2017, 91, e01824-16. [Google Scholar] [CrossRef] [PubMed]
- Ciccaglione, A.R.; Stellacci, E.; Marcantonio, C.; Muto, V.; Equestre, M.; Marsili, G.; Rapicetta, M.; Battistini, A. Repression of interferon regulatory factor 1 by hepatitis C virus core protein results in inhibition of antiviral and immunomodulatory genes. J. Virol. 2007, 81, 202–214. [Google Scholar] [CrossRef]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef]
- Gale, M.J.; Korth, M.J.; Tang, N.M.; Tan, S.L.; Hopkins, D.A.; Dever, T.E.; Polyak, S.J.; Gretch, D.R.; Katze, M.G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 1997, 230, 217–227. [Google Scholar] [CrossRef]
- Taguchi, T.; Nagano-Fujii, M.; Akutsu, M.; Kadoya, H.; Ohgimoto, S.; Ishido, S.; Hotta, H. Hepatitis C virus NS5A protein interacts with 2’,5’-oligoadenylate synthetase and inhibits antiviral activity of IFN in an IFN sensitivity-determining region-independent manner. J. Gen. Virol. 2004, 85, 959–969. [Google Scholar] [CrossRef]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.; Ikeda, M.; Ray, S.C.; Gale, M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Nitta, S.; Sakamoto, N.; Nakagawa, M.; Kakinuma, S.; Mishima, K.; Kusano-Kitazume, A.; Kiyohashi, K.; Murakawa, M.; Nishimura-Sakurai, Y.; Azuma, S.; et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 2013, 57, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Li, Q.; Cheng, Q.; Huang, C.; Zheng, J.; Chen, S.; Ling, Q.; Zhu, M.; Chen, M.; Shi, G.; et al. MicroRNA-548j inhibits type I interferon production by targeting ZBTB11 in patients with chronic hepatitis B. Biochem. Biophys. Res. Commun. 2017, 488, 628–633. [Google Scholar] [CrossRef]
- Zhao, Y.; Yu, Y.; Ye, L. MiR-3613-3p impairs IFN-induced immune response by targeting CMPK1 in chronic hepatitis B. Infect. Genet. Evol. 2019, 74, 103919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z. Circular RNA hsa_circ_0004812 impairs IFN-induced immune response by sponging miR-1287-5p to regulate FSTL1 in chronic hepatitis B. Virol. J. 2020, 17, 40. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, L.; Mu, T.; Yi, J.; Ma, C.; Xie, H.; Liu, M.; Tang, H. An HBV-encoded miRNA activates innate immunity to restrict HBV replication. J. Mol. Cell Biol. 2020, 12, 263–276. [Google Scholar] [CrossRef]
- He, C.L.; Liu, M.; Tan, Z.X.; Hu, Y.J.; Zhang, Q.Y.; Kuang, X.M.; Kong, W.L.; Mao, Q. Hepatitis C virus core protein-induced miR-93-5p up-regulation inhibits interferon signaling pathway by targeting IFNAR1. World J. Gastroenterol. 2018, 24, 226–236. [Google Scholar] [CrossRef]
- Jarret, A.; McFarland, A.P.; Horner, S.M.; Kell, A.; Schwerk, J.; Hong, M.; Badil, S.; Joslyn, R.C.; Baker, D.P.; Carrington, M.; et al. Hepatitis-C-virus-induced microRNAs dampen interferon-mediated antiviral signaling. Nat. Med. 2016, 22, 1475–1481. [Google Scholar] [CrossRef]
- Mukherjee, A.; Di Bisceglie, A.M.; Ray, R.B. Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway. J. Virol. 2015, 89, 3356–3365. [Google Scholar] [CrossRef]
- Gong, W.; Guo, X.; Zhang, Y. Depletion of MicroRNA-373 Represses the Replication of Hepatitis C Virus via Activation of Type 1 Interferon Response by Targeting IRF5. Yonsei Med. J. 2018, 59, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Y.; Su, Y.; Tian, L.; Qin, P.; Xu, X.; Zhou, Y. microRNA-125a targets MAVS and TRAF6 to modulate interferon signaling and promote HCV infection. Virus Res. 2021, 296, 198336. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kogure, T.; Ninomiya, M.; Fukuda, R.; Monma, N.; Ikeo, K.; Tanaka, Y. The reduction of miR146b-5p in monocytes and T cells could contribute to the immunopathogenesis of hepatitis C virus infection. Sci. Rep. 2019, 9, 13393. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iuliano, M.; Mangino, G.; Chiantore, M.V.; Di Bonito, P.; Rosa, P.; Affabris, E.; Romeo, G. Virus-Induced Tumorigenesis and IFN System. Biology 2021, 10, 994. https://doi.org/10.3390/biology10100994
Iuliano M, Mangino G, Chiantore MV, Di Bonito P, Rosa P, Affabris E, Romeo G. Virus-Induced Tumorigenesis and IFN System. Biology. 2021; 10(10):994. https://doi.org/10.3390/biology10100994
Chicago/Turabian StyleIuliano, Marco, Giorgio Mangino, Maria Vincenza Chiantore, Paola Di Bonito, Paolo Rosa, Elisabetta Affabris, and Giovanna Romeo. 2021. "Virus-Induced Tumorigenesis and IFN System" Biology 10, no. 10: 994. https://doi.org/10.3390/biology10100994
APA StyleIuliano, M., Mangino, G., Chiantore, M. V., Di Bonito, P., Rosa, P., Affabris, E., & Romeo, G. (2021). Virus-Induced Tumorigenesis and IFN System. Biology, 10(10), 994. https://doi.org/10.3390/biology10100994