
Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors
 , , , ,
, , , , 
Simple Summary
Abstract
1. Introduction
2. Hypertension and Endothelial Dysfunction
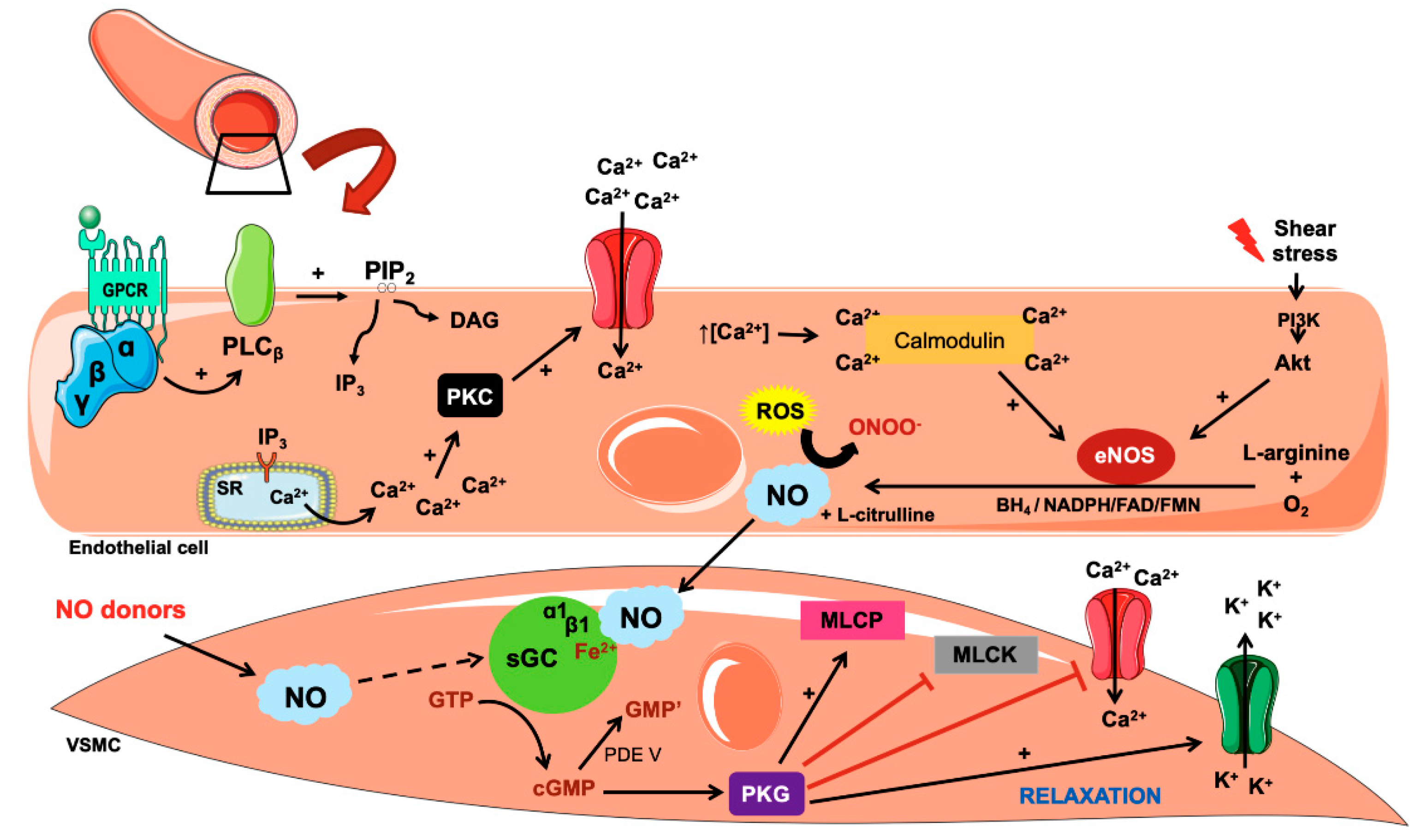
3. Biosynthesis and Action of Nitric Oxide
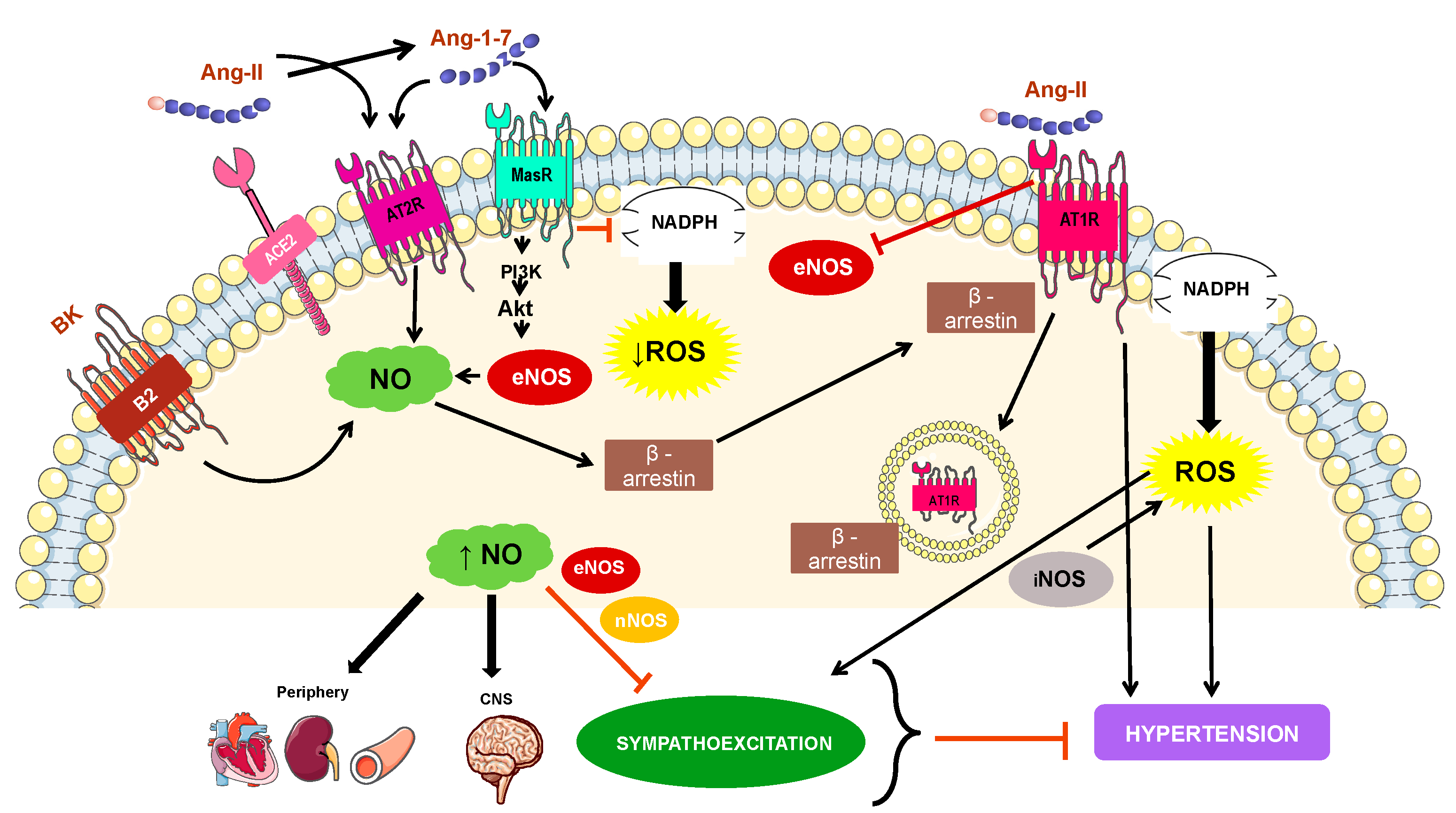
4. Mechanisms Involved in NO-Related Hypertension
5. Nitric Oxide Donors
5.1. Sodium Nitroprusside (SNP)
5.2. Organic Nitrates
5.3. Clinical Use and Limitations of Nitric Oxide Donors
5.4. Metal-Based Drugs as NO Donors
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pardali, E.; Dimmerler, S.; Zeiher, M.A.; Rieger, A.M. Clonal hematopoiesis, aging, and cardiovascular diseases. Exp. Hematol. 2020, 83, 95–104. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Risk Factor Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar]
- James, A.P.; Oparil, S.; Carter, L.B.; Cushman, C.W.; Dennison-Himmelfarb, C.; Handler, J.; Lackland, T.D.; LeFevre, L.M.; MacKenzie, D.T.; Ogedegbe, O.; et al. Evidence-based guideline for the management of high blood pressure in adults: Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014, 311, 507–520. [Google Scholar] [CrossRef]
- Herringto, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Oprail, S.; Schmieder, E.R. New Approaches in the Treatment of Hypertension. Circ. Res. 2015, 116, 1074–1095. [Google Scholar] [CrossRef] [PubMed]
- Pintévorá, M.; Kuneš, J.; Zicha, J. Altered neural and vascular mechanisms in hypertension. Physiol. Res. 2011, 60, 381–402. [Google Scholar]
- Ledoux, J.; Werner, E.M.; Brayden, E.J.; Nelson, T.M. Calcium-activated potassium channels and the regulation of vascular tone. Physiology 2006, 21, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–140. [Google Scholar] [PubMed]
- Davel, A.P.; Wenceslau, C.F.; Akamine, E.H.; Akamine, E.H.; Xavier, F.E.; Couto, G.K.; Oliveira, H.T.; Rossoni, L.V. Endothelial dysfunction in cardiovascular and endocrine-metabolic diseases: An update. Braz. J. Med. Biol. Res. 2011, 44, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.I.; Duncan, B.B.; Silva, G.A.; Menezes, A.M.; Monteiro, C.M.; Barreto, S.M.; Chor, D.; Menezes, P.R. Doenças crônicas não transmissíveis no Brasil: Carga e desafios atuais. Lancet 2011, 377, 61–74. [Google Scholar]
- Rahimi, K.; Emdin, C.A.; MacMahon, S. The epidemiology of blood pressure and its worldwide management. Circ. Res. 2015, 116, 925–936. [Google Scholar] [CrossRef]
- Rossier, B.C.; Bochud, M.; Devuyst, O. The Hypertension Pandemic: An Evolutionary Perspective. Physiology 2017, 32, 112–125. [Google Scholar] [CrossRef]
- Praxedes, J.N.; Santello, J.L.; Amodeo, C. Encontro multicêntrico sobre crises hipertensivas: Relatório e recomendações. Hipertensão 2001, 4, 23–41. [Google Scholar]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in blood pressure from 1975 to 2015: A pooled analysis of 1479 population-based measurement studies with 19·1 million participants. Lancet 2017, 389, 37–55. [Google Scholar] [CrossRef]
- World Health Organization. Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Ajayi, I.O.; Sowemimo, I.O.; Akpa, O.M.; Ossai, N.E. Prevalence of hypertension and associated factors among residents of Ibadan-North Local Government Area of Nigeria. Niger. J. Cardiol. 2016, 13, 67. [Google Scholar] [CrossRef]
- Hasan, M.; Akter, I.S.T.; Gupta, R.J.; Joshi, H.; Haider, M.R.; Sarker, M. Prevalence and determinants of hypertension among adult population in Nepal: Data from Nepal Demographic and Health Survey 2016. PLoS ONE 2018, 13, e0198028. [Google Scholar] [CrossRef] [PubMed]
- Olié, V.; Perrine, A.L.; Lecoffre, C.; Blacher, J. National prevalence of hypertension, treatment and control, in France in 2015 and temporal trends since 2006. Arch. Cardiovasc. Dis. Suppl. 2018, 11, 96–103. [Google Scholar] [CrossRef]
- Picon, R.V.; Fuchs, F.D.; Moreira, L.B.; Riegel, G.; Fuchs, S.C. Trends in Prevalence of Hypertension in Brazil: A Systematic Review with Meta-Analysis. PLoS ONE 2021, 7, e48255. [Google Scholar] [CrossRef] [PubMed]
- Malta, D.C.; Bernal, R.T.I.; Andrade, S.S.C.A.; Silva, M.M.A.; Velasquez-Melendez, G. Prevalência e fatores associados com hipertensão arterial autorreferida em adultos brasileiros. Rev. Saúde Pública 2017, 51, 1–11. [Google Scholar]
- Tortorella, C.C.S.; Corso, A.C.T.; Gonzáles-Chica, D.A.; Melhen, A.R.F. Time trends of hypertension and diabetes mellitus prevalence among adults registered in the Brazilian National Health System, in Florianópolis, Santa Catarina State, Brazil, 2004–2011. Epidemiol. Serv. Saúde 2017, 26, 469–480. [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Himmelfarb, C.D.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary. J. Am. Soc. Hypertens. 2018, 12, 579.e1–579.e73. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeck, G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes 1999, 48, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Buchanan, M.R.; Anderson, T.J. Endothelial function testing as a biomarker of vascular disease. Circulation 2003, 108, 2054–2059. [Google Scholar] [CrossRef]
- Le Brocq, M.; Leslie, S.J.; Milliken, P.; Megson, I.L. Endothelial Dysfunction: From molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1631–1674. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Oh, H.; Li, X. Ethanol extract of seeds of Oenothera odorata induces vasorelaxation via endothelium dependente NO-Cgmp signaling through activation of Akt-Enos-sGC pathaway. J. Ethnopharmacol. 2011, 133, 315–523. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Feletou, M.; Taddei, S. Endothelium-dependent contractions in hypertension. Br. J. Pharmacol. 2010, 144, 449–458. [Google Scholar] [CrossRef]
- Bernatova, I. Endothelial Dysfunction in Experimental Models of Arterial Hypertension: Cause or Consequence? BioMed Res. Int. 2014, 2014, 598271. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and Its Alterations in Cardiovascular Diseases: Life Style Intervention. BioMed Res. Int. 2014, 2014, 801896. [Google Scholar] [CrossRef]
- Sun, S.; Yang, F.; Tan, G.; Costanzo, M.; Oughtred, R.; Hirschman, J.; Theesfeld, C.L.; Bansal, P.; Sahni, N.; Yi, S.; et al. An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res. 2016, 26, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 15, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Köhler, R.; Vanhoutte, P.M. Endothelium-derived vasoactive factors and hypertension: Possible roles in pathogenesis and as treatment targets. Curr. Hypertens. Rep. 2010, 12, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Chopp, M.; Bailey, F.; Malinski, T. Nitric oxide changes in the rat brain after trasient middle cerebral artery occlusion. J. Neurol. Sci. 1995, 14, 22–27. [Google Scholar] [CrossRef]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Giardina, B. Pharmacological modulation of nitric oxide release: New pharmacological perspectives, potential benefits and risks. Curr. Med. Chem. 2010, 17, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Moncada, V.; Higgs, E.A. Prostaglandins in the pathogenesis and prevention of vascular disease. Blood Rev. 1987, 1, 141–145. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Napoli, C.; Loscalzo, J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: An overview. Circ. Res. 2002, 90, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef]
- Bonaventura, D.; Oliveira, F.S.; Silva, R.S.; Bendhack, L.M. Decreased vasodilation induced by a new nitric oxide donor in two kidney one clip hypertensive rats is due to impaired k channel activation. Clin. Exp. Pharmacol. Physiol. 2005, 32, 478–481. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. Molecular mechanisms and therapeutic strategies related to nitric oxide. J. Fed. Am. Soc. Exp. Biol. 1995, 9, 1319–1330. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharm. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.V.; Andrade, F.A.; Paulo, M.; Paula, T.P.; Potje, S.R.; Pereira, A.C.; Bendhack, L.M. NO donors induce vascular relaxation by different cellular mechanisms in hypertensive and normotensive rats. Nitric Oxide 2019, 86, 12–20. [Google Scholar] [CrossRef] [PubMed]
- McCall, T.B.; Boughton-Smith, N.K.; Palmer, R.J.M.; Whittle, B.J.R.; Moncada, S. Synthesis of nitric oxide from L-arginine by neutrophil release and interaction with superoxide anion. Biochem. J. 1989, 261, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Chaudhuri, G. Inhibition of constitutive and inducible nitric oxide synthase: Potencial selective inhibition. Annu. Rev. Pharm. Toxicol. 1995, 35, 165–194. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Giroud, J.P.; Willoughby, D.A. Studies of the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J. Pathol. 1996, 104, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Watkins, S.C.; Stefanovic-Racic, M. Nitric oxide and cartilage metabolism. Methods Enzymol. 1996, 269, 75–88. [Google Scholar] [PubMed]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef]
- Schmidt, H.H.H.W.; Murad, F. Purification and characterization of human NOS. Biochem. Biophys. Res. Commun. 1991, 181, 1372–1377. [Google Scholar] [CrossRef]
- Pollock, J.S.; Forstermann, V.; Mitchell, J.A.; Warner, T.D.; Schmidt, H.H.; Nakane, M.; Murad, F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10480–10484. [Google Scholar] [CrossRef] [PubMed]
- Kirkeboen, K.A.; Strand, O.A. The role of nitric oxide in sepsis: An overview. Acta Anaesthesiol. Scand. 1999, 43, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.P. Nitric oxide, inducible nitric oxide synthase and inflammation in veterinary medicine. Anim. Health Res. Rev. 2002, 3, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Förstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 2000, 190, 244–254. [Google Scholar] [CrossRef]
- Li, H.; Poulos, T.L. Structure-functions studies on nitric oxide synthases. J. Inorg. Biochem. 2005, 99, 293–305. [Google Scholar] [CrossRef]
- Hemmens, B.; Goessler, W.; Schmidt, K.; Mayer, B. Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J. Biol. Chem. 2000, 275, 35786–35791. [Google Scholar] [CrossRef]
- Wever, R.M.; Van Dam, T.; Van Rijn, H.J.M.; Groot, F.; Rabelink, T.J. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 1997, 237, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Chalupsky, K.; Cai, H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA 2005, 102, 9056–9061. [Google Scholar] [CrossRef]
- Siu, K.L.; Lotz, C.; Ping, P.; Cai, H. Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunction via nitric oxide-dependent attenuation of NOX4 activation and recoupling of NOS. J. Mol. Cell. Cardiol. 2015, 78, 174–185. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Rubbo, H.; Radi, R.; Trujillo, M.; Telleri, R.; Kalyanaraman, B.; Barnes, S.; Kirk, M.; Freeman, B.A. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994, 269, 26066–26075. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cardena, G.; Oh, P.; Liu, J.; Schnitzer, J.E.; Sessa, W.C. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implications for nitric oxide signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Xie, Q.W.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 1992, 176, 599–604. [Google Scholar] [CrossRef]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Xie, Q.W.; Cho, H.J.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Ding, A.; Troso, T.; Nathan, C. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 1992, 256, 225–228. [Google Scholar] [CrossRef]
- Xi, L.; Jarrett, N.C.; Hess, M.L.; Kukreja, R.C. Essential role of inducible nitric oxide synthase in monophosphoryl lipid A-induced late cardioprotection: Evidence from pharmacological inhibition and gene knockout mice. Circulation 1999, 99, 2157–2163. [Google Scholar] [CrossRef]
- Kuhl, S.J.; Rosen, H. Nitric oxide and septic shock-from bench to bedside. West. J. Med. 1998, 168, 176–181. [Google Scholar]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [CrossRef] [PubMed]
- Salerno, J.C.; Harris, D.E.; Irizarry, K.; Patel, B.; Morales, A.J.; Smith, S.M.; Martasek, P.; Roman, L.J.; Masters, B.S.; Jones, C.L.; et al. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J. Biol. Chem. 1997, 272, 29769–29777. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 7, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Nussler, A.K.; Di Silvoi, M.; Lowenstein, C.J.; Shapiro, R.A.; Wang, S.C.; Simmons, R.L.; Billiar, T.R. Cytokines, endotoxin, and glucocorticoids regulate the expression of inducible nitric oxide synthase in hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Spink, J.; Cohen, J.; Evans, J.T.L. The cytokine responsive vascular smooth muscle cell enhancer of inducible nitric oxide synthases activation by nuclear factor-kappa B. J. Biol. Chem. 1995, 270, 29541–29547. [Google Scholar] [CrossRef] [PubMed]
- Kobzik, L.; Bredt, D.S.; Lowenstein, C.J.; Drazen, J.; Gaston, B.; Sugarbaker, D.; Stamler, J.S. Nitric oxide synthase in human and rat lung: Immunocytochemical and histochemical localization. Am. J. Respir. Cell Mol. Biol. 1993, 9, 371–377. [Google Scholar] [CrossRef]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol Educ. 2003, 27, 201–206. [Google Scholar] [CrossRef]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [PubMed]
- Ford, P.C.; Lorkovic, I.M. Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes. Chem. Rev. 2002, 102, 993–1018. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Kraehling, J.R.; Eitner, F.; Bénardeau, A.; Sandner, P. The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective. Int. J. Mol. Sci. 2018, 19, 1712. [Google Scholar] [CrossRef]
- Friebe, A.; Koesling, D. The function of NO-sensitive guanylyl cyclase: What we can learn from genetic mouse models. Nitric Oxide 2009, 21, 149–156. [Google Scholar] [CrossRef]
- Kots, A.Y.; Martin, E.; Sharina, I.G.; Murad, F. A short history of cGMP, guanylyl cyclases, and cGMP-dependent protein kinases. Handb. Exp. Pharmacol. 2009, 191, 1–14. [Google Scholar]
- Fernhoff, A.N.B.; Derbyshirea, E.R.; Marletta, M.A. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc. Natl. Acad. Sci. USA 2009, 106, 21602–21607. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl Cyclases and signaling by cyclic GMP. Pharm. Rev. 2000, 52, 375–414. [Google Scholar] [PubMed]
- Gibb, B.J.; Wykes, V.; Garthwaite, J. Properties of NO-activated guanylyl cyclases expressed in cells. Br. J. Pharm. 2003, 139, 1032–1040. [Google Scholar] [CrossRef]
- Sager, G. Cyclic GMP transporters. Neurochem. Int. 2004, 45, 865–873. [Google Scholar] [CrossRef]
- Taguchi, K.; Ueda, M.; Kubo, T. Effects of cAMP and cGMP on L-type calcium channel currents in rat mesenteric artery cells. Jpn. J. Pharm. 1997, 74, 179–186. [Google Scholar] [CrossRef]
- Heydrick, S. Nitric Oxide and the Cardiovascular System; Humana Press: Totowa, NJ, USA, 2000; pp. 33–49. [Google Scholar]
- Busse, R.; Fleming, I. Nitric Oxide, Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2000; p. 179206. [Google Scholar]
- Waldman, S.A.; Murad, F. Cyclic GMP synthesis and function. Pharm. Rev. 1987, 39, 163–196. [Google Scholar] [PubMed]
- Wang, G.R.; Zhu, Y.; Halushka, P.V.; Lincoln, M.T.; Mendelsohn, M.E. Mechanism of platelet inhibition by nitric oxide: In vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 4888–4893. [Google Scholar] [CrossRef] [PubMed]
- Bolotina, V.M.; Najibi, S.; Palacino, J.J.; Pagano, P.J.; Cohen, R.A. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368, 850–853. [Google Scholar] [CrossRef]
- Leo, M.D.; Bannister, J.P.; Narayanan, D.; Nair, A.; Grubbs, J.E.; Gabrick, K.S.; Boop, F.A.; Jaggar, J.H. Dynamic regulation of beta1 subunit trafficking controls vascular contractility. Proc. Natl. Acad. Sci. USA 2014, 111, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy-Natarajan, G.; Koide, M. BK Channels in the Vascular System. Int. Rev. Neurobiol. 2016, 128, 401–438. [Google Scholar]
- Thippeswamy, T.; McKay, J.S.; Quinn, J.P.; Morris, R. Nitric Oxide, a biological double-faced janus—Is this good or bad? Histol. Histopathol. 2006, 21, 445–458. [Google Scholar] [PubMed]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.L.; Ritter, J.K. Role of Nitric Oxide in the Cardiovascular and Renal Systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef] [PubMed]
- Mirabito Colafella, K.M.; Bovee, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp. Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef] [PubMed]
- Kellici, T.F.; Tzakos, A.G.; Mavromoustakos, T. Rational drug design and synthesis of molecules targeting the angiotensin II type 1 and type 2 receptors. Molecules 2015, 20, 3868–3897. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, J.; Cui, G.; Wei, Y.; Lu, C.; Wang, L.; Diao, H. Pathophysiological role of osteopontin and angiotensin II in atherosclerosis. Biochem. Biophys. Res. Commun. 2016, 471, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Abdel Rahman, F.; Jurisch, V.; Kupatt, C. Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells 2019, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef]
- Assersen, K.B.; Sumners, C.; Steckelings, U.M. The Renin-Angiotensin System in Hypertension, a Constantly Renewing Classic: Focus on the Angiotensin AT(2)-Receptor. Can. J. Cardiol. 2020, 36, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.D.; Gyombolai, P.; Szalai, B.; Várnai, P.; Turu, G.; Hunyady, L. Angiotensin type 1A receptor regulates β-arrestin binding of the β(2)-adrenergic receptor via heterodimerization. Mol. Cell Endocrinol. 2017, 442, 113–124. [Google Scholar] [CrossRef]
- Ichiki, T.; Usui, M.; Kato, M.; Funakoshi, Y.; Ito, K.; Egashira, K.; Takeshita, A. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension 1998, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sattar, M.A.; Azam, M.; Khan, S.A.; Bhatt, O.; Johns, E.J. Interaction between nitric oxide and renal α1-adrenoreceptors mediated vasoconstriction in rats with left ventricular hypertrophyin Wistar Kyoto rats. PLoS ONE 2018, 13, e0189386. [Google Scholar] [CrossRef]
- Yang, J.; Sun, Y.; Dong, M.; Yang, X.; Meng, X.; Niu, R.; Guan, J.; Zhang, Y.; Zhang, C. Comparison of angiotensin-(1-7), losartan and their combination on atherosclerotic plaque formation in apolipoprotein E knockout mice. Atherosclerosis 2015, 240, 544–549. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef]
- Walters, P.E.; Gaspari, T.A.; Widdop, R.E. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 2005, 45, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Villela, D.; Leonhardt, J.; Patel, N.; Joseph, J.; Kirsch, S.; Hallberg, A.; Unger, T.; Bader, M.; Santos, R.A.; Sumners, C.; et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: A complex liaison. Clin. Sci. 2015, 128, 227–234. [Google Scholar] [CrossRef]
- Silva, G.M.; Franca-Falcao, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of Renin-Angiotensin System Components in Atherosclerosis: Focus on Ang-II, ACE2, and Ang-1-7. Front. Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Hamid, S.; Rhaleb, I.A.; Kassem, K.M.; Rhaleb, N.E. Role of Kinins in Hypertension and Heart Failure. Pharmaceuticals 2020, 13, 347. [Google Scholar] [CrossRef]
- Garthwaite, J. NO as a multimodal transmitter in the brain: Discovery and current status. Br. J. Pharm. 2019, 176, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Hirooka, Y.; Matsuo, I.; Eshima, K.; Shigematsu, H.; Shimokawa, H.; Takeshita, A. Overexpression of eNOS in NTS causes hypotension and bradycardia in vivo. Hypertension 2000, 36, 1023–1028. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kishi, T.; Hirooka, Y.; Sakai, K.; Shigematsu, H.; Shimokawa, H.; Takeshita, A. Overexpression of eNOS in the RVLM causes hypotension and bradycardia via GABA release. Hypertension 2001, 38, 896–901. [Google Scholar] [CrossRef]
- McBryde, F.D.; Liu, B.H.; Roloff, E.V.; Kasparov, S.; Paton, J.F.R. Hypothalamic paraventricular nucleus neuronal nitric oxide synthase activity is a major determinant of renal sympathetic discharge in conscious Wistar rats. Exp. Physiol. 2018, 103, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, Y. Sympathetic Activation in Hypertension: Importance of the Central Nervous System. Am. J. Hypertens. 2020, 33, 914–926. [Google Scholar] [CrossRef]
- Waki, H.; Kasparov, S.; Wong, L.F.; Murphy, D.; Shimizu, T.; Paton, J.F. Chronic inhibition of endothelial nitric oxide synthase activity in nucleus tractus solitarii enhances baroreceptor reflex in conscious rats. J. Physiol. 2003, 546, 233–242. [Google Scholar] [CrossRef]
- Kimura, Y.; Hirooka, Y.; Sagara, Y.; Ito, K.; Kishi, T.; Shimokawa, H.; Takeshita, A.; Sunagawa, K. Overexpression of inducible nitric oxide synthase in rostral ventrolateral medulla causes hypertension and sympathoexcitation via an increase in oxidative stress. Circ. Res. 2005, 96, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, P.; Diem, R.; Dun, N.J.; Förstermann, U. Endogenous and exogenous nitric oxide inhibits norepinephrine release from rat heart sympathetic nerves. Circ. Res. 1995, 77, 841–848. [Google Scholar] [CrossRef]
- Krob, H.A.; Vinsant, S.L.; Ferrario, C.M.; Friedman, D.P. Angiotensin-(1-7) immunoreactivity in the hypothalamus of the (mRen-2d)27 transgenic rat. Brain Res. 1998, 798, 36–45. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, X.; Patel, K.P. Angiotensin-converting enzyme 2 overexpression improves central nitric oxide-mediated sympathetic outflow in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2402–H2412. [Google Scholar] [CrossRef][Green Version]
- Patel, K.P.; Schultz, H.D. Angiotensin peptides and nitric oxide in cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1121–1132. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.; Corbin, D.J. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Pharm. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Feelisch, M. The use of nitric oxide donors in pharmacological studies. Naunyn-Schmiedebergs Arch. Pharmacol. 1998, 358, 113–122. [Google Scholar] [CrossRef]
- Janero, D.R.; Bryan, N.S.; Fumito, S.; Dhawan, V.; Schwalb, D.J.; Warren, M.C.; Feelisch, M. Differential nitrosylation of blood and tissue constituents during glyceryl trinitrate biotransformation in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 16958–16963. [Google Scholar] [CrossRef]
- Pereira, A.C.; Araújo, A.V.; Paulo, M.; Andrade, F.A.; Silva, B.R.; Vercesi, J.A.; Silva, R.S.; Bendhack, L.M. Hypotensive effect and vascular relaxation in different arteries induced by the nitric oxide donor RuBPY. Nitric Oxide 2017, 30, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Machado, N.T.; Maciel, P.M.; Alustau, M.C.; Queiroz, T.M.; Furtado, F.F.; Assis, V.L.; Veras, R.C.; Araújo, I.G.; Athayde-Filho, P.F.; Medeiros, I.A. Nitric oxide as a target for the hypotensive and vasorelaxing effects induced by (Z)-ethyl 12-nitrooxy-octadec-9-enoate in rats. Eur. J. Pharm. Sci. 2014, 62, 317–325. [Google Scholar] [CrossRef]
- Miller, M.R.; Megson, I.L. Recent developments in nitric oxide donor drug. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Ulker, S.; Mcmaster, D.; Mckeown, P.P.; Bayraktutan, U. Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovasc. Res. 2003, 59, 488–500. [Google Scholar] [CrossRef]
- Floryszak-Wieczorek, J.; Milczarek, G.; Arasimowicz, M.; Ciszewski, A. Do nitric oxide donors mimic endogenous NO-related response in plants? Planta 2006, 224, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Priviero, F.B.; Webb, R.C. Heme-dependent and independent soluble guanylate cyclase activators and vasodilation. J. Cardiovasc. Pharm. 2010, 56, 229–233. [Google Scholar] [CrossRef]
- Kodja, G.; Feelisch, M.; Noack, E. Sulfhydryl-containing nitrate esters: A new class of nitric oxide donors. Cardiovasc Drug Rev. 1995, 13, 275–288. [Google Scholar]
- Friederich, J.A.; Butterworth, J.F. Sodium nitroprusside: Twenty years and couting. Anesth. Analg. 1995, 81, 152–162. [Google Scholar] [PubMed]
- Cobb, A.; Thornton, L. Sodium Nitroprusside as a Hyperinflation Drug and Therapeutic Alternatives. J. Pharm. Pract. 2018, 31, 374–381. [Google Scholar] [CrossRef]
- Hottinger, D.G.; Beebe, D.S.; Kozhimannil, T.; Prielipp, R.C.; Belani, K.G. Sodium nitroprusside in 2014: A clinical concepts review. J. Anaesthesiol. Clin. Pharm. 2014, 30, 462–471. [Google Scholar]
- Fukatsu, A.; Hayashi, T.; Miyazaki-Akita, A.; Matsui-Hirai, H.; Furutate, Y.; Ishitsuka, A.; Hattori, Y.; Iguchi, A. Possible Usefulness of apocynun, na NADPH oxidase inibitor, for nitrate tolerance: Prevention of NO donor-induced endotelial cell abnormalities. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 790–797. [Google Scholar] [CrossRef]
- Yakazu, Y.; Iwasawa, K.; Narita, H.; Kindscher, J.D.; Benson, K.T.; Goto, H. Hemodynamic and sympathetic effects of feoldopam and sodium nitroprusside. Acta Anaesthesiol. A Scand. 2001, 45, 1176–1180. [Google Scholar] [CrossRef]
- Brunton, T. On the use of nitrite of amyl in angina pectoris. Lancet 1867, 2, 97–98. [Google Scholar] [CrossRef]
- Lindenfeld, J.; Albert, N.M.; Boehmer, J.P.; Collins, S.P.; Ezekowitz, J.A.; Givertz, M.M.; Katz, S.D.; Klapholz, M.; Moser, D.K.; Rogers, J.G.; et al. Comprehensive heart failure practice guideline. J. Card. Fail. 2010, 16, 194. [Google Scholar]
- Fung, H.L.; Chung, S.J.; Bauer, J.A.; Chong, S.; Kowaluk, E.A. Biochemical mechanism of organic nitrate action. AJC 1992, 70, 4B–10B. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; McDonald, K.M.; Hastie, T.; Fadel, B.; Hagan, V.; Lee, B.K.; Hlatky, M.A. Meta-analysis of trials comparing β-blockers, calcium antagonists, and nitrates for stable angina. JAMA Am. Med. Assoc. 1999, 281, 1927–1936. [Google Scholar] [CrossRef]
- Münzel, T.; Steven, S.; Daiber, A. Organic nitrates: Update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vasc. Pharm. 2014, 63, 105–113. [Google Scholar] [CrossRef]
- Tarkin, J.M.; Kaski, J.C. Vasodilator Therapy: Nitrates and Nicorandil. Cardiovasc. Drugs 2017, 31, 367–378. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; Primi, M.C.; Trossini, G.H.G.; Ferreira, E.I. Nitric Oxide: State of the Art in Drug Design. Curr. Med. Chem. 2012, 19, 386–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.H.; Frishman, W.H. The antiplatelet effects of nitrates: Is it of clinical significance in patients with cardiovascular disease? Cardiol. Rev. 2010, 18, 198–203. [Google Scholar] [CrossRef]
- Omar, S.A.; Artime, E.; Webb, A.J. A comparison of organic and inorganic nitrates/nitrites. Nitric Oxide 2012, 26, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Abshagen, U.W. Pharmacokinetics of isosorbide mononitrate. Am. J. Cardiol. 1992, 27, G61–G66. [Google Scholar] [CrossRef]
- Chen, Z.; Stamler, J.S. Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc. Med. 2006, 16, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.G.; Xian, M.; Tang, X.; Wu, X.; Wen, Z.; Cai, T.; Janczuk, A.J. Nitric oxide donors: Chemical activities and biological applications. Chem. Rev. 2002, 102, 1091–1134. [Google Scholar] [CrossRef]
- Miller, M.R.; Wadsworth, R.M. Understanding organic nitrates—A vein hope? Br. J. Pharm. 2009, 157, 565–567. [Google Scholar] [CrossRef] [PubMed]
- França-Silva, M.S.; Balarini, C.M.; Cruz, J.C.; Khan, B.A.; Rampelotto, P.H.; Braga, V.A. Organic nitrates: Past, present and future. Molecules 2014, 19, 15314–15323. [Google Scholar] [CrossRef]
- Lee, S.R.; Nilius, B.; Han, J. Gaseous Signaling Molecules in Cardiovascular Function: From Mechanisms to Clinical Translation. Ver. Physiol. Biochem. Pharmacol. 2018, 174, 81–156. [Google Scholar]
- Marczin, N.; Riedel, B.; Royston, D.; Yacoub, M. Intravenous nitrate vasodilators and exhaled nitric oxide. Lancet 1997, 349, 1742. [Google Scholar] [CrossRef]
- Kövesi, T.; Royston, D.; Yacoub, M.; Marczin, N. Basal and nitroglycerin-induced exhaled nitric oxide before and after cardiac surgery with cardiopulmonary bypass. Br. J. Anaesth. 2003, 90, 608–616. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boshier, P.R.; Hanna, G.B.; Marczin, N. Exhaled nitric oxide as biomarker of acute lung injury: An unfulfilled promise? J. Breath. Res. 2013, 7, 017118. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 28, 7617–7635. [Google Scholar] [CrossRef]
- Yasuda, H.; Yamaya, M.; Nakayama, K.; Sasaki, T.; Ebihara, S.; Kanda, A.; Asada, M.; Inoue, D.; Suzuki, T.; Okazaki, T.; et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.K.; Giesing, D.H.; Williams, R.L.; Benet, L.Z.; Lin, E.T. Pharmacokinetics of nitroglycerin and metabolites in humans following oral dosing. Biopharm. Drug Dipos. 1988, 9, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Assinder, D.F.; Chasseaud, L.F.; Taylor, T. Plasma Isosorbide Dinitrate Concentrations in Human Subjects after Administration of Standard and Sustained-Release Formulations. J. Pharm. Sci. 1977, 66, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Chowdhury, Y.S. Isosorbide; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hashimoto, S.; Kobayashi, A. Clinical Pharmacokinetics and Pharmacodynamics of Glyceryl Trinitrate and its Metabolites. Clin. Pharm. 2003, 42, 205–221. [Google Scholar] [CrossRef]
- Pannala, A.S.; Mani, A.R.; Spencer, J.P.E.; Skinner, V.; Bruckdorfer, K.R.; Moore, K.P.; Rice-Evans, C.A. The effect of dietary nitrate on salivary, plasma, and urinary nitrate metabolism in humans. Free Radic. Biol. Med. 2003, 34, 576–584. [Google Scholar] [CrossRef]
- Hunault, C.C.; van Velzen, A.G.; Sips, A.J.A.M.; Schothorst, R.C.; Meulenbelt, J. Bioavailability of sodium nitrite from an aqueous solution in healthy adults. Toxicol. Lett. 2009, 190, 48–53. [Google Scholar] [CrossRef]
- Wagner, A.D.; Schultz, D.S.; Deen, W.M.; Young, V.R.; Tannernbaum, S.R. Metabolic fate of an oral dose of 15N-labeled nitrate in humans: Effect of diet supplementation with ascorbic acid. Cancer Res. 1983, 43, 1921–1925. [Google Scholar]
- Munzel, T. Explaining the Phenomenon of Nitrate Tolerance. Circ. Res. 2005, 97, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Wenzel, P.; Oelze, M.; Münzel, T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin. Res. Cardiol. 2008, 97, 12–20. [Google Scholar] [CrossRef]
- Fung, H.L. biochemical mechanism of nitroglycerin action and tolerance: Is this old mystery solved? Annu. Rev. Pharmacol. Toxicol. 2004, 44, 67–85. [Google Scholar] [CrossRef]
- Klemenska, E.; Beresewicz, A. Bioactivation of organic nitrates and the mechanism of nitrate tolerance. Cardiol. J. 2009, 16, 11–19. [Google Scholar]
- Lopez, M.; Malacarne, P.F.; Gajos-Draus, A.; Ding, X.; Daiber, A.; Lundberg, J.O.; Offermanns, S.; Brandes, R.P.; Rezende, F. Vascular biotransformation of organic nitrates is independent of cytochrome P450 monooxygenases. Br. J. Pharmacol. 2021, 178, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003, 9, 1498–1505. [Google Scholar] [CrossRef]
- Rassaf, T.; Flogel, U.; Drexhage, C.; Hendgen-Cotta, U.; Kelm, M.; Schrader, J. Nitrite reductase function of deoxymyoglobin: Oxygen sensor and regulator of cardiac energetics and function. Circ. Res. 2007, 100, 1749–1754. [Google Scholar] [CrossRef]
- Shiva, S.; Huang, Z.; Grubina, R.; Sun, J.; Ringwood, L.A.; MacArthur, P.H.; Xu, X.; Murphy, E.; Darley-Usmar, V.M.; Gladwin, M.T. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ. Res. 2007, 100, 654–661. [Google Scholar] [CrossRef]
- Webb, A.J.; Milsom, A.B.; Rathod, K.S.; Chu, W.L.; Qureshi, S.; Lovell, M.J.; Lecomte, F.M.; Perrett, D.; Raimondo, C.; Khoshbin, E.; et al. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: Role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ. Res. 2008, 103, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cui, H.; Kundu, T.K.; Alzawahra, W.; Zweier, J.L. Nitric Oxide Production from Nitrite Occurs Primarily in Tissues Not in the Blood. J. Biol. Chem. 2008, 283, 17855–17863. [Google Scholar] [CrossRef]
- Aamand, R.; Dalsgaard, T.; Jensen, F.B.; Simonsen, U.; Roepstorff, A.; Fago, A. Generation of nitric oxide from nitrite by carbonic anhydrase: A possible link between metabolic activity and vasodilation. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H2068–H2074. [Google Scholar] [CrossRef]
- Kapil, V.; Haydar, S.M.A.; Pearl, V.; Lundberg, J.O.; Weitzberg, E.; Ahluwalia, A. Physiological role for nitrate-reducing oral bacteria in blood pressure control. Free Radic. Biol. Med. 2013, 55, 93–100. [Google Scholar] [CrossRef]
- Moretti, C.; Zhuge, Z.; Zhang, G.; Haworth, S.M.; Paulo, L.L.; Guimarães, D.D.; Lundberg, J.O. The obligatory role of host microbiota in bioactivation of dietary nitrate. Free Radic. Biol. Med. 2019, 145, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Dejam, A.; Hunter, C.J.; Tremonti, C.; Pluta, R.M.; Hon, Y.Y.; Grimes, G.; Gladwin, M.T. Nitrite Infusion in Humans and Nonhuman Primates: Endocrine Effects, Pharmacokinetics, and Tolerance Formation. Circulation 2007, 116, 1821–1831. [Google Scholar] [CrossRef]
- Magee, P.N.; Barnes, J.M. The Production of Malignant Primary Hepatic Tumours in the Rat by Feeding Dimethylnitrosamine. Br. J. Cancer 1956, 10, 114–122. [Google Scholar] [CrossRef]
- Oelze, M.; Knorr, M.; Kröller-Schön, S.; Kossmann, S.; Gottschlich, A.; Rümmler, R.; Schuff, A.; Daub, S.; Doppler, C.; Kleinert, H.; et al. Chronic therapy with isosorbide-5-mononitrate causes endothelial dysfunction, oxidative stress, and a marked increase in vascular endothelin-1 expression. Eur. Heart J. 2013, 34, 3206–3216. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T. Effects of long-term nitroglycerin treatment on endothelial nitric oxide synthase (NOS III) gene expression, NOS III-mediated superoxide production, and vascular NO bioavailability. Circ. Res. 2000, 86, E7–E12. [Google Scholar] [CrossRef]
- Preik, M.; Kelm, M.; Feelisch, M.; Strauer, B.E. Impaired effectiveness of nitric oxide-donors in resistance arteries of patients with arterial hypertension. J. Hypertens. 1996, 14, 903–908. [Google Scholar] [CrossRef]
- Evora, P.R.; Evora, P.M.; Celotto, A.C.; Rodrigues, A.J.; Joviliano, E.E. Cardiovascular therapeutics targets on the NO-sGC-cGMP signaling pathway: A critical overview. Curr. Drug Targets 2012, 13, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Giaid, A.; Kurz, S.; Stewart, D.J.; Harrison, D.G. Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc. Natl. Acad. Sci. USA 1995, 92, 5244–5248. [Google Scholar] [CrossRef]
- Divakaran, S.; Loscalzo, J. The Role of Nitroglycerin and Other Nitrogen Oxides in Cardiovascular Therapeutics. J. Am. Coll. Cardiol. 2017, 70, 2393–2410. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Burstein, J.M.; Ahmed, S.; Miner, S.E.S.; Al-Hesayen, A.; Kelly, S.; Parker, J.D. Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: A human in vivo study. Circulation 2001, 104, 1119–1123. [Google Scholar] [CrossRef]
- Rapoport, R.M.; Waldman, S.A.; Ginsburg, R.; Molina, C.R.; Murad, F. Effects of glyceryl trinitrate on endothelium-dependent and -independent relaxation and cyclic GMP levels in rat aorta and human coronary artery. J. Cardiovasc. Pharm. 1988, 1987, 82–89. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z.; Cobb, F.R.; Stamler, J.S. Role of mitochondrial aldehyde dehydrogenase in nitroglycerin-induced vasodilation of coronary and systemic vessels: An intact canine model. Circulation 2004, 110, 750–755. [Google Scholar] [CrossRef]
- Parker, J.D.; Farrell, B.; Fenton, T.; Cohanim, M.; Parker, J.O. Counter-regulatory responses to continuous and intermittent therapy with nitroglycerin. Circulation 1991, 84, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Al-Hesayen, A.; Courtney, J.; Parker, J.D. Comparison of the effects of pentaerythritoltetranitrate and nitroglycerin on endothelium-dependent vasorelaxation in male volunteers. Am. J. Cardiol. 2003, 91, 1392–1394. [Google Scholar] [CrossRef]
- Jurt, U.; Goti, T.; Ravandi, A.; Babaei, S.; Zeman, P.; Parker, J.D. Differential effects of pentaerythritoltetranitrate and nitroglycerin on the development of tolerance and evidence of lipid peroxidation: A human in vivo study. J. Am. Coll. Cardiol. 2001, 38, 854–859. [Google Scholar] [CrossRef]
- Müllenheim, J.; Müller, S.; Laber, U.; Thämer, V.; Meyer, W.; Bassenge, E.; Fink, B.; Kojda, G. The effect of high-dose pentaerythritoltetranitrate on the development of nitrate tolerance in rabbits. Naunyn-Schmiedebergs Arch. Pharmacol. 2001, 364, 269–275. [Google Scholar]
- Dikalov, S.; Fink, B.; Skatchkov, M.; Bassenge, E. Comparison of glyceryl trinitrate-induced with pentaerythrityl tetranitrate-induced in vivo formation of superoxide radicals: Effect of vitamin C. Free Radic. Biol. Med. 1999, 27, 170–176. [Google Scholar] [CrossRef]
- Daiber, A.; Oelze, M.; Coldewey, M.; Bachschmid, M.; Wenzel, P.; Sydow, K.; Wendt, M.; Kleschyov, A.L.; Stalleicken, D.; Ullrich, V.; et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: A comparison of pentaerythritol tetranitrate with other organic nitrates. Mol. Pharm. 2004, 66, 1372–1382. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Brandt, M.; Ullmann, E.; Kröller-Schön, S.; Heeren, T.; Daiber, A. Pentaerythritol Tetranitrate In Vivo Treatment Improves Oxidative Stress and Vascular Dysfunction by Suppression of Endothelin-1 Signaling in Monocrotaline-Induced Pulmonary Hypertension. Oxidative Med. Cell. Longev. 2017, 2017, 4353462. [Google Scholar] [CrossRef]
- Fraccarollo, D.; Galuppo, P.; Neuser, J.; Bauersachs, J.; Widder, J.D. Pentaerythritol Tetranitrate Targeting Myocardial Reactive Oxygen Species Production Improves Left Ventricular Remodeling and Function in Rats with Ischemic Heart Failure Novelty and Significance. Hypertension 2015, 66, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Münzel, T.; Li, H. Maternal Treatment of Spontaneously Hypertensive Rats With Pentaerythritol Tetranitrate Reduces Blood Pressure in Female off spring Novelty and Significance. Hypertension 2014, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Man, A.W.C.; Chen, M.; Wu, Z.; Reifenberg, G.; Daiber, A.; Münzel, T.; Li, H. Renal Effects of Fetal Reprogramming with Pentaerythritol Tetranitrate in Spontaneously Hypertensive Rats. Front. Pharmacol. 2020, 11, 454. [Google Scholar] [CrossRef]
- Man, A.W.C.; Chen, M.; Zhou, Y.; Wu, Z.; Reifenberg, G.; Daiber, A.; Li, H. Fetal programming effects of pentaerythritol tetranitrate in a rat model of superimposed preeclampsia. J. Mol. Med. 2020, 98, 1287–1299. [Google Scholar] [CrossRef]
- Bohn, H.; Schönafinger, K. Oxygen and oxidation promote the release of nitric oxide from sydnonimines. J. Cardiovasc. Pharm. 1989, 14 (Suppl. 11), S6–S12. [Google Scholar] [CrossRef]
- Noack, E.; Feelisch, M. Molecular aspects underlying the vasodilator action of molsidomine. J. Cardiovasc. Pharm. 1989, 14 (Suppl. 11), S1–S5. [Google Scholar] [CrossRef]
- Kristek, F.; Fiberov, V.; Varga, I. Long-Term Effect of Molsidomine and Pentaerythrityl Tetranitrate on Cardiovascular System of Spontaneously Hypertensive Rats. Physiol. Res. 2003, 52, 709–717. [Google Scholar] [PubMed]
- Benigni, A.; Zoja, C.; Noris, M.; Corna, D.; Benedetti, G.; Bruzzi, I.; Todeschini, M.; Remuzzi, G. Renoprotection by nitric oxide donor and lisinopril in the remnant kidney model. Am. J. Kidney Dis. 1999, 33, 746–753. [Google Scholar] [CrossRef]
- Oosterhuis, N.R.; Bongartz, L.G.; Verhaar, M.C.; Cheng, C.; Xu, Y.J.; van Koppen, A.; Cramer, M.J.; Goldsschmeding, R.; Gaillard, C.A.; Doevendans, P.P.; et al. Targeting multiple pathways reduces renal and cardiac fibrosis in rats with subtotal nephrectomy followed by coronary ligation. Acta Physiol. 2017, 220, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Reinero, M.; Beghetti, M.; Tozzi, P.; von Segesser, L.K.; Samaja, M.; Milano, G. Nitric Oxide–cGMP Pathway Modulation in an Experimental Model of Hypoxic Pulmonary Hypertension. J. Cardiovasc. Pharmacol. Ther. 2021, 107424842110141. [Google Scholar] [CrossRef]
- Koeners, M.P.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Joles, J.A. A perinatal nitric oxide donor increases renal vascular resistance and ameliorates hypertension and glomerular injury in adult fawn-hooded hypertensive rats. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 294, R1847–R1855. [Google Scholar] [CrossRef]
- Lablanche, J.M.; Grollier, G.; Lusson, J.R.; Bassand, J.P.; Drobinski, G.; Bertrand, B.; Battaglia, S.; Desveaux, B.; Juillière, Y.; Juliard, J.M.; et al. Effect of the direct nitric oxide donors linsidomine and molsidomine on angiographic restenosis after coronary balloon angioplasty. The ACCORD Study. Circulation 1997, 95, 83–89. [Google Scholar] [CrossRef]
- Belhassen, L.; Carville, C.; Pelle, G.; Sediame, S.; Benacerraf, S.; Dubois-Rande, J.L.; Adnot, S. Molsidomine improves flow-dependent vasodilation in brachial arteries of patients with coronary artery disease. J. Cardiovasc. Pharmacol. 2000, 35, 560–563. [Google Scholar] [CrossRef]
- Van Hove, C.; Carreer-Bruhwyler, F.; Geczy, J.; Herman, A.G. Long-term treatment with the NO-donor molsidomine reduces circulating ICAM-1 levels in patients with stable angina. Atherosclerosis 2005, 180, 399–405. [Google Scholar] [CrossRef]
- Cirino, G.; Distrutti, E.; Wallace, J.L. Nitric oxide and inflammation. Inflamm. Allergy Drug Targets 2006, 5, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Wattanapitayakul, S.K.; Weinstein, D.M.; Holycross, B.J.; Bauer, J.A. Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J. 2000, 14, 271–278. [Google Scholar] [CrossRef]
- Szabó, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Bachschmid, M.; Schildknecht, S.; Ullrich, V. Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem. Biophys. Res. Commun. 2005, 338, 536–542. [Google Scholar] [CrossRef]
- Schildknecht, S.; Ullrich, V. Peroxynitrite as regulator of vascular prostanoid synthesis. Arch. Biochem. Biophys. 2009, 484, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Colli, S.; Habib, A.; Aldini, G.; Altomare, A.; Banfi, C. Cyclooxygenase-2 Glycosylation Is Affected by Peroxynitrite in Endothelial Cells: Impact on Enzyme Activity and Degradation. Antioxidants 2021, 10, 496. [Google Scholar] [CrossRef]
- Hogg, N. The biochemistry and physiology of S-nitrosothiols. Annu Rev. Pharm. Toxicol. 2002, 42, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Darley-Usmar, V.M.; Wilson, M.T.; Moncada, S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochem. J. 1992, 281, 419–424. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M.; London, E.D.; Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 6368–6371. [Google Scholar] [CrossRef]
- Yamada, M.; Momose, K.; Richelson, E.; Yamada, M. Sodium nitroprusside-induced apoptotic cellular death via production of hydrogen peroxide in murine neuroblastoma N1E-115 cells. J. Pharm. Toxicol. Methods 1996, 35, 11–17. [Google Scholar] [CrossRef]
- Terwel, D.; Nieland, L.J.; Schutte, B.; Reutelingsperger, C.P.; Ramaekers, F.C.; Steinbusch, H.W. S-nitroso-N-acetylpenicillamine and nitroprusside induce apoptosis in a neuronal cell line by the production of different reactive molecules. Eur. J. Pharm. 2000, 400, 19–33. [Google Scholar] [CrossRef][Green Version]
- Godínez-Rubí, M.; Rojas-Mayorquín, A.E.; Ortuño-Sahagún, D. Nitric oxide donors as neuroprotective agents after an ischemic stroke-related inflammatory reaction. Oxid. Med. Cell Longev. 2013, 2013, 297357. [Google Scholar] [CrossRef]
- Katsuyama, K.; Shichiri, M.; Marumo, F.; Hirata, Y. NO inhibits cytokine-induced iNOS expression and NF-kappaB activation by interfering with phosphorylation and degradation of IkappaB-alpha. Arter. Thromb. Vasc. Biol. 1998, 18, 1796–1802. [Google Scholar] [CrossRef]
- Blais, V.; Rivest, S. Inhibitory action of nitric oxide on circulating tumor necrosis factor-induced NF-kappaB activity and COX-2 transcription in the endothelium of the brain capillaries. J. Neuropathol. Exp. Neurol. 2001, 60, 893–905. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Walley, K.R.; McDonald, T.E.; Higashimoto, Y.; Hayashi, S. Modulation of proinflammatory cytokines by nitric oxide in murine acute lung injury. Am. J. Respir. Crit. Care Med. 1999, 160, 698–704. [Google Scholar] [CrossRef]
- Park, S.W.; Huq, M.D.; Hu, X.; Wei, L.N. Tyrosine nitration on p65: A novel mechanism to rapidly inactivate nuclear factor-kappaB. Mol. Cell Proteom. 2005, 4, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Gasco, A.; Fruttero, R.; Sorba, G.; Stilo, A.D.; Calvino, R. NO donors: Focus on furoxan derivatives. Pure Appl. Chem. 2004, 76, 973–981. [Google Scholar] [CrossRef]
- França-Silva, M.S.; Monteiro, M.M.; Queiroz, T.M.; Santos, A.F.; Athayde-Filho, P.F.; Braga, V.A. The new nitric oxide donor 2-nitrate-1,3-dibuthoxypropan alters autonomic function in spontaneously hypertensive rats. Auton. Neurosci. 2012, 171, 28–35. [Google Scholar] [CrossRef]
- Queiroz, T.M.; Mendes-Júnior, L.G.; Guimarães, D.D.; França-Silva, M.S.; Nalivaiko, E.; Braga, V.A. Cardiorespiratory effects induced by 2-nitrate-1,3-dibuthoxypropan are reduced by nitric oxide scavenger in rats. Auton. Neurosci. 2014, 181, 31–36. [Google Scholar] [CrossRef]
- Porpino, S.K.; Zollbrecht, C.; Peleli, M.; Montenegro, M.F.; Brandão, M.C.; Athayde-Filho, P.F.; França-Silva, M.S.; Larsson, E.; Lundberg, J.O.; Weitzberg, E.; et al. Nitric oxide generation by the organic nitrate NDBP attenuates oxidative stress and angiotensin II-mediated hypertension. Br. J. Pharmacol. 2016, 173, 2290–2302. [Google Scholar] [CrossRef] [PubMed]
- De Gaitani, C.M.; de Melo, M.C.; Lunardi, C.N.; de Oliveira, F.S.; da Silva, R.S.; Bendhack, L.M. Hypotensive effect of the nitrosyl ruthenium complex nitric oxide donor in renal hypertensive rats. Nitric Oxide 2009, 20, 195–199. [Google Scholar] [CrossRef]
- Rodrigues, G.J.; Pereira, A.C.; Vercesi, J.A.; Lima, R.G.; Silva, R.S.; Bendhack, L.M. Long-lasting hypotensive effect in renal hypertensive rats induced by nitric oxide released from a ruthenium complex. J. Cardiovasc. Pharmacol. 2012, 60, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, F.C.; Potje, S.R.; Pereira, A.C.; Daruge, M.G.; da Silva, R.S.; Bendhack, L.M.; Antoniali, C. Hypotensive and vasorelaxing effects of the new NO donor [Ru(terpy)(bdq)NO+]3+ in spontaneously hypertensive rats. Nitric Oxide 2012, 26, 111–117. [Google Scholar] [CrossRef]
- Potje, S.R.; Troiano, J.A.; Grando, M.D.; Graton, M.E.; da Silva, R.S.; Bendhack, L.M.; Antoniali, C. Endothelial modulation of a nitric oxide donor complex-induced relaxation in normotensive and spontaneously hypertensive rats. Life Sci. 2018, 201, 130–140. [Google Scholar] [CrossRef]
- Das Mendes-Júnior, L.G.; Guimarães, D.D.; Gadelha, D.D.; Diniz, T.F.; Brandão, M.C.; Athayde-Filho, P.F.; Lemos, V.S.; França-do Silva, M.S.; Braga, V.A. The new nitric oxide donor cyclohexane nitrate induces vasorelaxation, hypotension, and antihypertensive effects via NO/cGMP/PKG pathway. Front. Physiol. 2015, 6, 243. [Google Scholar] [CrossRef]
- Paulo, L.L.; Cruz, J.C.; Zhuge, Z.; Carvalho-Galvão, A.; Brandão, M.C.R.; Diniz, T.F.; Haworth, S.M.; Athayde-Filho, P.F.; Lemos, V.S.; Lundberg, J.O.; et al. The novel organic mononitrate NDHP attenuates hypertension and endothelial dysfunction in hypertensive rats. Redox Biol. 2018, 15, 182–191. [Google Scholar] [CrossRef]
- Orvid, C.; Abrams, M.J. Medicinal inorganic chemistry: Introduction. Chem. Rev. 1999, 99, 2201–2204. [Google Scholar]
- Silva, D.O. Perspectives for movel mixed diruthenium-organic drug as metallopharmaceuticals in cancer therapy. Anticancer Agents Med. Chem. 2010, 10, 312–323. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef]
- Hambley, T.W. Developing new metal-based therapeutics: Challenges and opportunities. Dalton Trans. 2007, 21, 4929–4937. [Google Scholar] [CrossRef]
- Meggers, E. Targeting proteins with metal complexes. Chem. Commun. 2009, 7, 1001–1100. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, F.P.; Mayhew, E.; Roe, E.M.; Shulman, A. Inhibition of landschuetz ascites tumour growth by metal chelates derived from 3,4,7,8-Tetramethyl-1,10-phenanthroline. Br. J. Cancer 1965, 19, 195–199. [Google Scholar] [CrossRef]
- Bonaventura, D.; Oliveira, F.d.S.; Togniolo, V.; Tedesco, A.C.; da Silva, R.S.; Bendhack, L.M. A macrocyclic nitrosyl ruthenium complex is a NO donor that induces rat aorta relaxation. Nitric Oxide 2004, 10, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, D.; Oliveira, F.S.; Lunardi, C.N.; Vercesi, J.A.; da Silva, R.S.; Bendhack, L.M. Characterization of the mechanisms of action and nitric oxide species involved in the relaxation induced by the ruthenium complex. Nitric Oxide 2006, 15, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, D.; de Lima, R.G.; Vercesi, J.A.; da Silva, R.S.; Bendhack, L.M. Comparison of the mechanisms underlying the relaxation induced by two nitric oxide donors: Sodium nitroprusside and a new ruthenium complex. Vasc. Pharm. 2007, 46, 215–222. [Google Scholar] [CrossRef]
- Paulo, M.; Rodrigues, G.J.; da Silva, R.S.; Bendhack, L.M. A new NO donor failed to release NO and to induce relaxation in the rat basilar artery. Eur. J. Pharm. Sci. 2012, 45, 344–350. [Google Scholar] [CrossRef]
- Potje, S.R.; Munhoz, F.C.; Perassa, L.A.; Graton, M.E.; Pereira, A.A.; Nakamune, A.C.; da Silva, R.S.; Bendhack, L.M.; Sumida, D.H.; Antoniali, C. Mechanisms underlying the hypotensive and vasodilator effects of Ru(terpy)(bdq)NO]3+, a nitric oxide donor, differ between normotensive and spontaneously hypertensive rats. Eur. J. Pharm. Sci. 2014, 741, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.P.C.; Campos, R.; Cabral, P.H.B.; Gomes, V.M.; Santos, C.F.; Waller, S.B.; de Sousa, E.H.S.; Lopes, L.G.F.; Fonteles, M.C.; do Nascimento, N.R.F. Antihypertensive potential of cis-[Ru(bpy) 2(ImN)(NO)]3+, a ruthenium-based nitric oxide donor. Res. Vet. Sci. 2020, 130, 153–160. [Google Scholar] [CrossRef]
- Marcondes, F.G.; Ferro, A.A.; Souza-Torsoni, A.; Sumitani, M.; Clarke, M.J.; Franco, D.W.; Tfouni, E.; Krieger, M.H. In vivo effects of the controlled NO donor/scavenger ruthenium cyclam complexes on blood pressure. Life Sci. 2002, 70, 2735–2752. [Google Scholar] [CrossRef]
- Olesen, J.; Thomsen, L.L.; Iversen, H. Nitric oxide is a key molecule in migraine and other vascular headaches. Trends Pharm. Sci. 1994, 15, 149–153. [Google Scholar] [CrossRef]
- Araújo, A.V.; Pereira, A.C.; Grando, M.D.; da Silva, R.D.; Bendhack, L.M. The new NO donor Terpy in-duces similar relaxation in mesenteric resistance arteries of renal hyper-tensive and normotensive rats. Nitric Oxide 2013, 35, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, D.; de Lima, R.G.; da Silva, R.S.; Bendhack, L.M. NO donors-relaxation is impaired in aorta from hypertensive rats due to a reduced involvement of K(+) channels and sarcoplasmic reticulum Ca(2+)-ATPase. Life Sci. 2011, 89, 595–602. [Google Scholar] [CrossRef]
- Potje, S.R.; Chen, Z.; Oliveira, S.D.S.; Bendhack, L.M.; da Silva, R.S.; Bonini, M.G.; Antoniali, C.; Minshall, R.D. Nitric oxide donor [Ru(terpy)(bdq)NO]3+ induces uncoupling and phosphorylation of endothelial nitric oxide synthase promoting oxidant production. Free Radic. Biol. Med. 2017, 112, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Potje, S.R.; Hildebrand, M.C.; Munhoz, F.C.; Troiano, J.A.; Pereira, A.A.; Nakamune, A.C.; da Silva, R.S.; Bendhack, L.M.; Antoniali, C. The hypotensive effect of the ruthenium complex [Ru(terpy)(bdq)NO]3+ is higher in male than in female spontaneously hypertensive rats (SHR). Naunyn-Schmiedebergs Arch. Pharmacol. 2014, 387, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Ford, P.C.; da Silva, R.S.; Bendhack, L.M. Ruthenium-nitrite complex as pro-drug releases NO in a tissue and enzyme-dependent way. Nitric Oxide 2011, 24, 192–198. [Google Scholar] [CrossRef]
- Pereira, A.C.; Lunardi, C.N.; Paulo, M.; da Silva, R.S.; Bendhack, L.M. Nitric oxide generated by the compound RuBPY promotes the vascular smooth cell membrane hyperpolarization. Eur. J. Pharm. Sci. 2013, 48, 604–610. [Google Scholar] [CrossRef]
- Paulo, M.; Grando, M.D.; da Silva, R.S.; Minshall, R.D.; Bendhack, L.M. The nitric oxide donor RuBPY does not induce in vitro cross-tolerance with acetylcholine. Nitric Oxide 2017, 69, 69–77. [Google Scholar] [CrossRef]
- Crisalli, A.M.; Franco, L.P.; Silva, B.R.; Holanda, A.K.M.; Bendhack, L.M.; Da Silva, R.S.; Ford, P.C. Nitric oxide release from a photoactive water-soluble ruthenium nitrosyl. Biological effects. J. Coord. Chem. 2018, 71, 1690–1703. [Google Scholar] [CrossRef]
- Oishi, J.C.; Buzinnari, T.C.; Pestana, C.R.; De Moraes, T.F.; Vatanabe, I.P.; Wink, D.A., Jr.; da Silva, R.S.; Bendhack, L.M.; Rodrigues, G.J. In Vitro Treatment with cis-[Ru(H-dcbpy-)2(Cl)(NO)] Improves the Endothelial Function in Aortic Rings with Endothelial Dysfunction. J. Pharm. Pharm. Sci. 2015, 18, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Campelo, M.W.; Campelo, A.P.; Lopes, L.G.; Santos, A.A.; Guimarães, S.B.; Vasconcelos, P.R. Effects of Rut-bpy (Cis-[Ru(bpy)2(SO3)(NO)]PF6), a novel nitric oxide donor, in L-NAME-induced hypertension in rats. Acta Cir. Bras. 2011, 26, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, J.B.; Silva, L.F.; Lopes, L.G.; Moraes, M.E.; Nascimento, N.R. Relaxation of rabbit corpus cavernosum smooth muscle and aortic vascular endothelium induced by new nitric oxide donor substances of the nitrosyl-ruthenium complex. Int. Braz. J. Urol. 2008, 34, 638–647. [Google Scholar] [CrossRef]
- Costa, P.P.C.; Waller, S.B.; Dos Santos, G.R.; Gondim, F.L.; Serra, D.S.; Cavalcante, F.S.A.; Gouveia, F.S., Jr.; de Paula, V.F., Jr.; Sousa, E.H.S.; Lopes, L.G.F.; et al. Anti-asthmatic effect of nitric oxide metallo-donor FOR811A [cis-[Ru(bpy)2(2-MIM)(NO)](PF6)3] in the respiratory mechanics of Swiss mice. PLoS ONE 2021, 16, e0248394. [Google Scholar] [CrossRef]
- Sasahara, G.L.; Gouveia Junior, F.S.; Rodrigues, R.O.; Zampieri, D.S.; Fonseca, S.; Goncalves, R.C.R.; Athaydes, B.R.; Kitagawa, R.R.; Santos, F.A.; Sousa, E.H.S.; et al. Nitro-imidazole-based ruthenium complexes with antioxidant and anti-inflammatory activities. J. Inorg. Biochem. 2020, 206, 111048. [Google Scholar] [CrossRef] [PubMed]
- Leitão Junior, A.S.; Campos, R.M.; Cerqueira, J.B.; Fonteles, M.C.; Santos, C.F.; de Nucci, G.; Sousa, E.H.; Lopes, L.G.; Gonzaga-Silva, L.F.; Nascimento, N.R. Relaxant effect of a metal-based drug in human corpora cavernosa and its mechanism of action. Int. J. Impot Res. 2016, 27, 181–187. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| No Donor | Class | Clinical Uses | Clinical Limitations | References |
|---|---|---|---|---|
| Sodium Nitroprusside (SNP) | Inorganic donor | -Vasodilation in hypertensive crisis and cardiovascular emergencies, such as angina pectoris and heart failure -Hypotensive control during surgery | -Formation of CN− -Reflex tachycardia -Endothelial dysfunction -Tolerance | [38,123,131,132,133,134,135,136] |
| Molsidomine | Sydnonimines | -Vasodilation in patients with artery disease -Antianginal effects | -Despite an improvement in the long-term angiographic result after angioplasty, it induced no effect on clinical outcome | [200,202,207,209] |
| Glyceryl trinitrate (GTN) | Organic nitrate | -Antianginal effect (reduction in the preload by peripheral vasodilation and dilation of the epicardial coronary artery) and reduction in systemic BP -Increase in oxygen supply due to dilation of both non- and atherosclerotic coronary arteries | -Small oral bioavailability -Endothelial dysfunction -Tolerance -Increases oxidative stress -Increases autocrine endothelin expression -Induces supersensitivity to vasoconstrictors | [143,144,145,146,167,181,182,183] |
| Isosorbide mononitrate (ISMN) | Organic nitrate | -Vasodilation for the treatment of angina pectoris -Vasodilation of coronary arteries | -Short effect, despite the oral bioavailability -Endothelial dysfunction -Therapy of post-infarct leads to an increased rate of coronary events -Increases oxidative stress -Increases autocrine endothelin expression; -Supersensitivity to vasoconstrictors | [143,144,145,146] |
| Isosorbide dinitrate (ISDN) | Organic nitrate | -Vasodilation for the treatment of angina pectoris -Vasodilation of coronary arteries | -Short effect, despite the oral bioavailability | [143,145,146] |
| Pentaerythrityl tetranitrate (PETN) | Organic nitrate | -Improvement in pulmonary hypertension beyond reduction in the preload -Treatment of ischemic heart diseases -Does not induce tolerance | -Little oral bioavailability | [143,144,145,146,195] |
| Nicorandil | -Vasodilation for chronic stable angina | -Short effect, despite the oral bioavailability | [142,143,144,145,146] |
| No Donor | Class | Effect | Species | Tolerance | References |
|---|---|---|---|---|---|
| (Z)-ethyl 12-nitrooxy-octadec-9-enoate (NCOE) | Organic nitrate | -Short-lasting hypotension and bradycardia -Vasorelaxation | Rat | Does not cause in vitro tolerance | [126] |
| 2-nitrate-1,3-dibuthoxypropan (NDBP) | Organic nitrate | -Hypotension, bradycardia, and bradypnea -Prevention of the progression of angiotensin II-mediated hypertension | Rat | Does not cause in vitro tolerance | [228,229,231] |
| Cyclohexane Nitrate (HEX) | Organic nitrate | -Reduction in blood pressure and heart rate -Antihypertensive effect in renovascular hypertension -Vasorelaxation in cranial artery | Rat | - | [235] |
| 1,3-bis (hexyloxy) propan-2-yl nitrate (NDHP) | Organic nitrate | - Reduction in blood pressure in hypertensive animals -Vasorelaxation -Prevention of the progression of hypertension and endothelial dysfunction | Rat | Does not cause in vitro tolerance | [236] |
| [Ru(terpy)(bdq)NO+]3+ (TERPY) | Metal-based drugs | -Vasorelaxation in aorta and mesenteric resistance arteries from Sham and two-kidney-one-clip hypertensive (2K1C) -Long-lasting hypotensive effect in 2K-1C, but not in normotensive -Similar vasorelaxation and released NO in aortas from Wistar and Spontaneously Hypertensive Rats (SHR) - Does not induce vasorelaxation in basilar arteries -Hypotensive effect in SHR | Rat | - | [231,232,233,234,245,251,252,254] |
| [Ru(bpy)2(py)(NO2)](PF6) (RuBPY) | Metal-based drugs | -Induced relaxation in aorta, mesenteric resistance arteries; coronary arteries between normotensive and 2K1C rats -Did not induce hypotensive effect in normotensive rats -Induced coronary artery relaxation (which may be useful for angina) and a minor effect in basilar artery (which may indicate that it does not induce headache). -NO· release that activates K+ channels in cultured VCMC aorta | Rat | Does not cause in vitro tolerance (self- or cross-tolerance) | [42,125,255,256,257] |
| trans-[Ru(Cl)NO(cyclam)2+ | Metal-based drugs | -Long-lasting hypotensive effect (20 times greater than SNP) in normotensive and hypertensive animals | Mouse | - | [249] |
| trans-[RuCl([15]aneN4)NO]2+ | Metal-based drugs | -Vasorelaxation in aorta (due to the release of NO· and NO-species) | Rat | - | [243,244] |
| Ru(NO)(salenCO2H)Cl | Metal-based drugs | -Vasorelaxation in aorta | Rat | - | [258] |
| Rut-bpy (Cis-[Ru(bpy)2(SO3)(NO)]PF6 | Metal-based drugs | -Stabilization of BP in anesthetized hypotensive animals | Rat | - | [260] |
| cis-[Ru(bpy)2(SO3)(NO)]PF-6-9 (FONO1) | Metal-based drugs | -Vasodilation in corpus cavernosum | Rabbit | - | [261] |
| trans-[Ru(NH3)4(caffeine)(NO)]C13 (LLNO1) | Metal-based drugs | -Vasodilation in corpus cavernosum | Rabbit | - | [261] |
| cis-[Ru(bpy)2(2-MIM)(NO)](PF6)3 (FOR811A) | Metal-based drugs | -Decrease in alveolar collapse and prevention of bronchoconstriction during asthma | Mouse | - | [262] |
| cis-[Ru(bpy)2(ImN)(NO)]3+ (FOR0811) | Metal-based drugs | -Decrease in BP (long-lasting) with no reflex tachycardia in L-NG-Nitro arginine methyl ester (L-NAME) hypertensive rats -Reduction in the low (LF) and very low (VLF) frequency bands in rats -Vasorelaxation in rat aorta -Vasorelaxation of human corpus cavernosum | Rat and human | - | [248] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Silva, G.M.; da Silva, M.C.; Nascimento, D.V.G.; Lima Silva, E.M.; Gouvêa, F.F.F.; de França Lopes, L.G.; Araújo, A.V.; Ferraz Pereira, K.N.; de Queiroz, T.M. Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors. Biology 2021, 10, 1041. https://doi.org/10.3390/biology10101041
da Silva GM, da Silva MC, Nascimento DVG, Lima Silva EM, Gouvêa FFF, de França Lopes LG, Araújo AV, Ferraz Pereira KN, de Queiroz TM. Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors. Biology. 2021; 10(10):1041. https://doi.org/10.3390/biology10101041
Chicago/Turabian Styleda Silva, Gabriela Maria, Mirelly Cunha da Silva, Déborah Victória Gomes Nascimento, Ellen Mayara Lima Silva, Fabíola Furtado Fialho Gouvêa, Luiz Gonzaga de França Lopes, Alice Valença Araújo, Kelli Nogueira Ferraz Pereira, and Thyago Moreira de Queiroz. 2021. "Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors" Biology 10, no. 10: 1041. https://doi.org/10.3390/biology10101041
APA Styleda Silva, G. M., da Silva, M. C., Nascimento, D. V. G., Lima Silva, E. M., Gouvêa, F. F. F., de França Lopes, L. G., Araújo, A. V., Ferraz Pereira, K. N., & de Queiroz, T. M. (2021). Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors. Biology, 10(10), 1041. https://doi.org/10.3390/biology10101041