The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy


Abstract
Simple Summary
Abstract
1. Introduction
2. Stress-Induced Signaling Directs Cell Fate Decisions
2.1. Mitochondria Are Highly Dynamic Organelles
2.2. The Crosstalk between Mitochondrial Shape Changes and Reactive Oxygen Species
2.3. Damaged Mitochondrial Fragments Are Cleared by Mitophagy
2.4. The Complex Regulation of Intrinsic Apoptosis
2.5. Mitochondrial Fission Is the First Step in Intrinsic Apoptosis
3. Reactive Oxygen Species and Cancer—A Dangerous Liaison
3.1. The Good and the Bad of Mitochondrial Reactive Oxygen Species

3.2. The Endogenous Antioxidant Defense Machinery




3.3. Apoptosis Is a Redox-Dependent Process
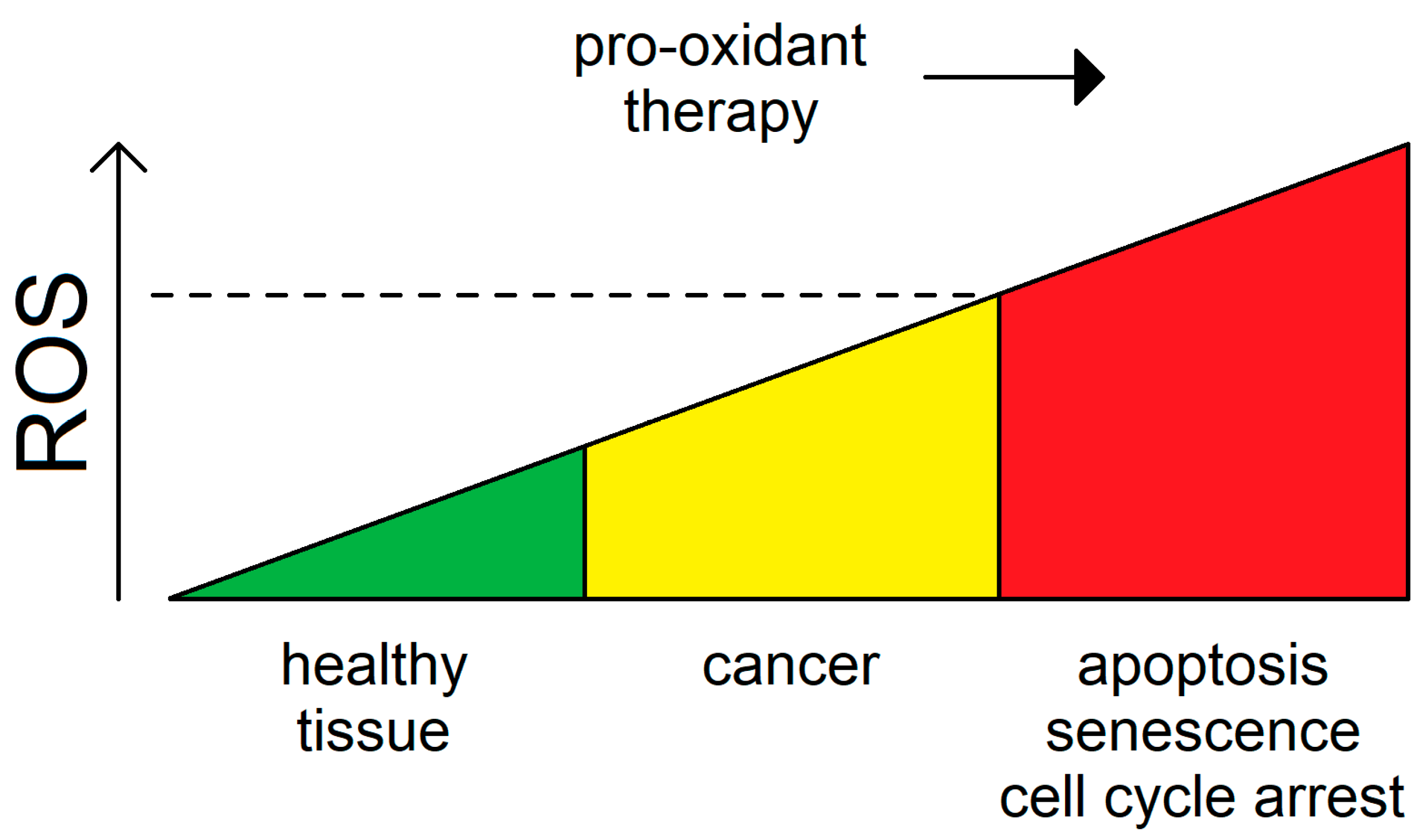
3.4. Reactive Oxygen Species as a Double-Edged Sword for Cancer
4. Surpassing the Redox Threshold for Apoptosis in Tumors by Inducing Mitochondrial Fission
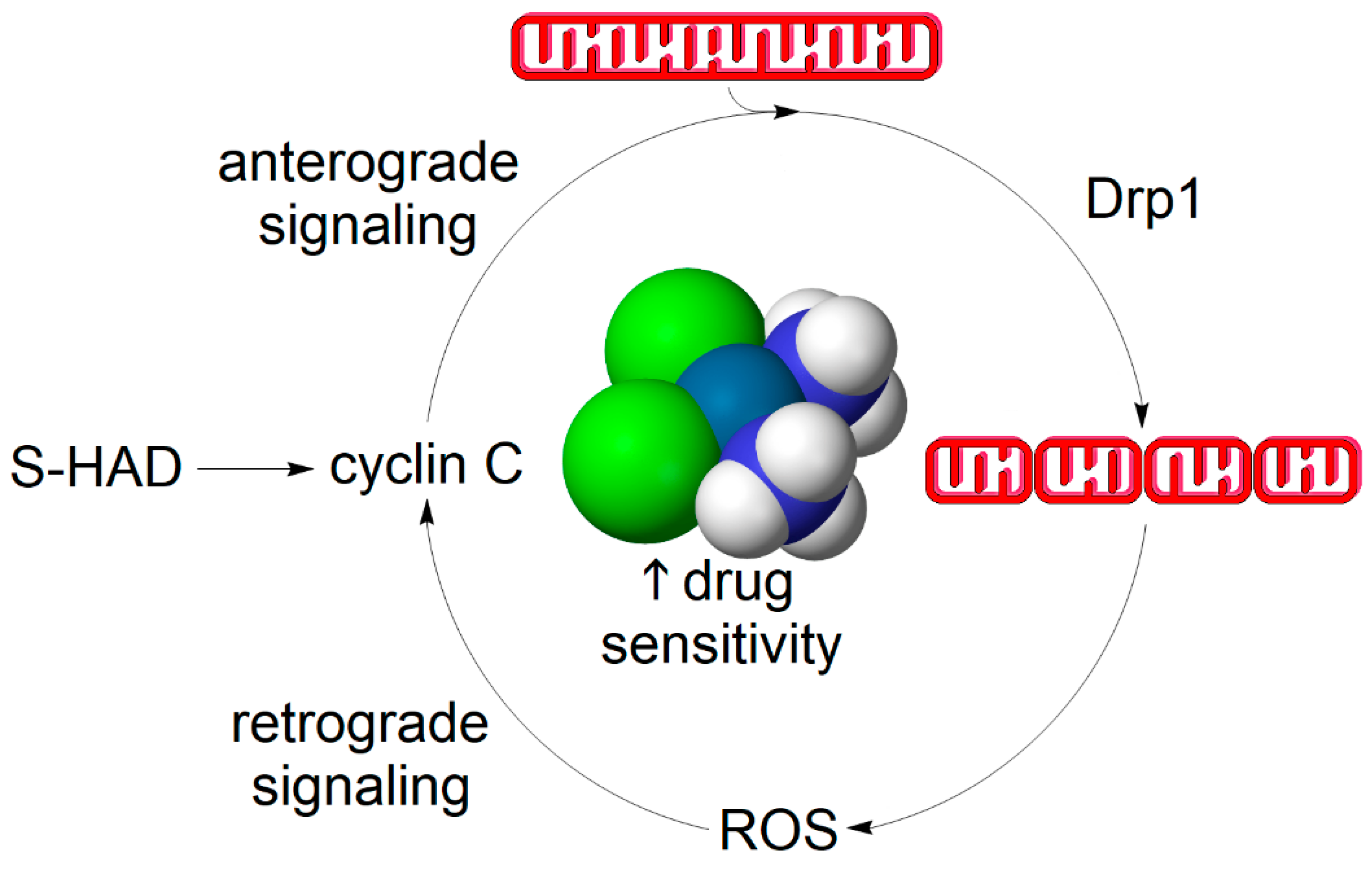
4.1. Cyclin C Acts as a Bridge between Stress-Induced Mitochondrial Fission and Apoptosis
4.2. Does a Self-Perpetuating Cycle of Mitochondrial Fission and Reactive Oxygen Species Production Underpin the Mechanism of Apoptotic Sensitization?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pospisil, P.; Prasad, A.; Rac, M. Mechanism of the Formation of Electronically Excited Species by Oxidative Metabolic Processes: Role of Reactive Oxygen Species. Biomolecules 2019, 9, 258. [Google Scholar] [CrossRef]
- Matschke, V.; Theiss, C.; Matschke, J. Oxidative stress: The lowest common denominator of multiple diseases. Neural Regen Res. 2019, 14, 238–241. [Google Scholar] [CrossRef]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef]
- Jezek, J.; Smethurst, D.G.J.; Stieg, D.C.; Kiss, Z.A.C.; Hanley, S.E.; Ganesan, V.; Chang, K.T.; Cooper, K.F.; Strich, R. Cyclin C: The Story of a Non-Cycling Cyclin. Biology 2019, 8, 3. [Google Scholar] [CrossRef]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev. 2018, 2018, 7857251. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Spurgers, K.B.; Chari, N.S.; Bohnenstiehl, N.L.; McDonnell, T.J. Molecular mediators of cell death in multistep carcinogenesis: A path to targeted therapy. Cell Death Differ. 2006, 13, 1360–1370. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Cernigliaro, C.; Giuliano, M.; Lauricella, M. The Double-Edged Sword Profile of Redox Signaling: Oxidative Events as Molecular Switches in the Balance between Cell Physiology and Cancer. Chem. Res. Toxicol. 2018, 31, 201–210. [Google Scholar] [CrossRef]
- Sprenger, H.G.; Langer, T. The Good and the Bad of Mitochondrial Breakups. Trends Cell Biol. 2019, 29, 888–900. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta Bioenergy 2018, 1859, 263–269. [Google Scholar] [CrossRef]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef]
- Atkins, K.; Dasgupta, A.; Chen, K.H.; Mewburn, J.; Archer, S.L. The role of Drp1 adaptor proteins MiD49 and MiD51 in mitochondrial fission: Implications for human disease. Clin. Sci. 2016, 130, 1861–1874. [Google Scholar] [CrossRef]
- Liu, R.; Chan, D.C. The mitochondrial fission receptor Mff selectively recruits oligomerized Drp1. Mol. Biol. Cell 2015, 26, 4466–4477. [Google Scholar] [CrossRef]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Ganesan, V.; Willis, S.D.; Chang, K.T.; Beluch, S.; Cooper, K.F.; Strich, R. Cyclin C directly stimulates Drp1 GTP affinity to mediate stress-induced mitochondrial hyperfission. Mol. Biol. Cell 2019, 30, 302–311. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Breitzig, M.T.; Alleyn, M.D.; Lockey, R.F.; Kolliputi, N. A mitochondrial delicacy: Dynamin-related protein 1 and mitochondrial dynamics. Am. J. Physiol. Cell Physiol. 2018, 315, C80–C90. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Q.; Zhang, P.; Sun, L.; Peng, C.; Yuan, Z.; Cheng, J. c-Abl-mediated Drp1 phosphorylation promotes oxidative stress-induced mitochondrial fragmentation and neuronal cell death. Cell Death Dis. 2017, 8, e3117. [Google Scholar] [CrossRef]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Zaja, I.; Bai, X.; Liu, Y.; Kikuchi, C.; Dosenovic, S.; Yan, Y.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys. Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef]
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; Del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat. Commun. 2016, 7, 13189. [Google Scholar] [CrossRef]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef]
- Wang, K.; Yan, R.; Cooper, K.F.; Strich, R. Cyclin C mediates stress-induced mitochondrial fission and apoptosis. Mol. Biol. Cell 2015, 26, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.F.; Khakhina, S.; Kim, S.K.; Strich, R. Stress-induced nuclear-to-cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev. Cell 2014, 28, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.R.; Thomenius, M.J.; Johnson, E.S.; Freel, C.D.; Wu, J.Q.; Coloff, J.L.; Yang, C.S.; Tang, W.; An, J.; Ilkayeva, O.R.; et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol. Biol. Cell 2011, 22, 1207–1216. [Google Scholar] [CrossRef]
- Rong, R.; Xia, X.; Peng, H.; Li, H.; You, M.; Liang, Z.; Yao, F.; Yao, X.; Xiong, K.; Huang, J.; et al. Cdk5-mediated Drp1 phosphorylation drives mitochondrial defects and neuronal apoptosis in radiation-induced optic neuropathy. Cell Death Dis. 2020, 11, 720. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5: Potential implications in amyloid-beta-related neurotoxicity in Alzheimer disease. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Gan, X.; Chen, R.; Ren, Y.; Yu, H.; You, C. The cyclophilin D/Drp1 axis regulates mitochondrial fission contributing to oxidative stress-induced mitochondrial dysfunctions in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2017, 483, 765–771. [Google Scholar] [CrossRef]
- Xiao, L.; Xian, H.; Lee, K.Y.; Xiao, B.; Wang, H.; Yu, F.; Shen, H.M.; Liou, Y.C. Death-associated Protein 3 Regulates Mitochondrial-encoded Protein Synthesis and Mitochondrial Dynamics. J. Biol. Chem. 2015, 290, 24961–24974. [Google Scholar] [CrossRef]
- Karbowski, M.; Jeong, S.Y.; Youle, R.J. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 2004, 166, 1027–1039. [Google Scholar] [CrossRef]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017, 284, 4177–4195. [Google Scholar] [CrossRef]
- Niemann, A.; Ruegg, M.; La Padula, V.; Schenone, A.; Suter, U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 2005, 170, 1067–1078. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935 e925. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Rodrigues, V.; Silvestre, R.; Gautier, C.; Weil, R.; Corti, O.; Estaquier, J. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Sabouny, R.; Fraunberger, E.; Geoffrion, M.; Ng, A.C.; Baird, S.D.; Screaton, R.A.; Milne, R.; McBride, H.M.; Shutt, T.E. The Keap1-Nrf2 Stress Response Pathway Promotes Mitochondrial Hyperfusion Through Degradation of the Mitochondrial Fission Protein Drp1. Antioxid. Redox Signal 2017, 27, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Lampe, P.A.; Stojanovski, D.; Korwitz, A.; Anand, R.; Tatsuta, T.; Langer, T. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014, 33, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef]
- Yu, B.; Ma, J.; Li, J.; Wang, D.; Wang, Z.; Wang, S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat. Commun. 2020, 11, 2549. [Google Scholar] [CrossRef]
- Von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef]
- Gao, Q.; Frohman, M.A. Roles for the lipid-signaling enzyme MitoPLD in mitochondrial dynamics, piRNA biogenesis, and spermatogenesis. BMB Rep. 2012, 45, 7–13. [Google Scholar] [CrossRef]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Fan, S.; Wang, D.; Huyan, T.; Chen, J.; Chen, J.; Su, J.; Li, X.; Wang, Z.; Xie, S.; et al. FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2481–2494. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.; Jung, J.H.; Park, J.; Lee, H.; Hong, Y.; Cho, H.; Kim, M.; Park, S.; Kwon, S.H.; Kim, S.H.; et al. S6 kinase 1 plays a key role in mitochondrial morphology and cellular energy flow. Cell. Signal. 2018, 48, 13–24. [Google Scholar] [CrossRef]
- Qin, N.; Cai, T.; Ke, Q.; Yuan, Q.; Luo, J.; Mao, X.; Jiang, L.; Cao, H.; Wen, P.; Zen, K.; et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol. 2019, 247, 392–405. [Google Scholar] [CrossRef]
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 2016, 164, 872–883. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Watanabe, T.; Saotome, M.; Nobuhara, M.; Sakamoto, A.; Urushida, T.; Katoh, H.; Satoh, H.; Funaki, M.; Hayashi, H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp. Cell Res. 2014, 323, 314–325. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Kobashigawa, S.; Suzuki, K.; Yamashita, S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun. 2011, 414, 795–800. [Google Scholar] [CrossRef]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef]
- Zachari, M.; Ktistakis, N.T. Mammalian Mitophagosome Formation: A Focus on the Early Signals and Steps. Front. Cell Dev. Biol. 2020, 8, 171. [Google Scholar] [CrossRef]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Joaquim, M.; Escobar-Henriques, M. Role of Mitofusins and Mitophagy in Life or Death Decisions. Front. Cell Dev. Biol. 2020, 8, 572182. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, A. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Gavathiotis, E. BAX unleashed: The biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci. 2011, 36, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef]
- Pena-Blanco, A.; Garcia-Saez, A.J. Bax, Bak and beyond—Mitochondrial performance in apoptosis. FEBS J. 2017, 285, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Gahl, R.F.; He, Y.; Yu, S.; Tjandra, N. Conformational rearrangements in the pro-apoptotic protein, Bax, as it inserts into mitochondria: A cellular death switch. J. Biol. Chem. 2014, 289, 32871–32882. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2019, 21, 85–100. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An extreme multifunctional protein with a key role in cell fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Chen, X.; Yao, J.M.; Fang, X.; Zhang, C.; Yang, Y.S.; Hu, C.P.; Chen, Q.; Zhong, G.W. Hypoxia promotes pulmonary vascular remodeling via HIF-1alpha to regulate mitochondrial dynamics. J. Geriatr. Cardiol. 2019, 16, 855–871. [Google Scholar]
- Jezek, J.; Chang, K.T.; Joshi, A.M.; Strich, R. Mitochondrial translocation of cyclin C stimulates intrinsic apoptosis through Bax recruitment. EMBO Rep. 2019, 20, e47425. [Google Scholar] [CrossRef]
- Tanaka, H.; Okazaki, T.; Aoyama, S.; Yokota, M.; Koike, M.; Okada, Y.; Fujiki, Y.; Gotoh, Y. Peroxisomes control mitochondrial dynamics and the mitochondrion-dependent apoptosis pathway. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef]
- Fischer, T.D.; Hylin, M.J.; Zhao, J.; Moore, A.N.; Waxham, M.N.; Dash, P.K. Altered Mitochondrial Dynamics and TBI Pathophysiology. Front. Syst. Neurosci. 2016, 10, 29. [Google Scholar] [CrossRef]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Cho, S.G.; Wang, C.Y.; Yang, T.; Dong, Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol. 2011, 300, C447–C455. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Cheung, E.C.; Neuspiel, M.; MacLaurin, J.G.; Fortin, A.; Park, D.S.; McBride, H.M.; Slack, R.S. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J. Biol. Chem. 2007, 282, 23788–23798. [Google Scholar] [CrossRef] [PubMed]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Georgieva, E.; Ivanova, D.; Zhelev, Z.; Bakalova, R.; Gulubova, M.; Aoki, I. Mitochondrial Dysfunction and Redox Imbalance as a Diagnostic Marker of “Free Radical Diseases”. Anticancer Res. 2017, 37, 5373–5381. [Google Scholar]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Larosa, V.; Remacle, C. Insights into the respiratory chain and oxidative stress. Biosci. Rep. 2018, 38, BSR20171492. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly 2012, 142, w13659. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Koppenol, W.H. The Haber-Weiss reaction—The latest revival. Free Radic. Biol. Med. 2019, 145, 221–222. [Google Scholar] [CrossRef]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.; Cheng, K.; Chen, Y.; Yang, R.; Chen, D.Z. Enhancement of tumor lethality of ROS in photodynamic therapy. Cancer Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Haag-Liautard, C.; Coffey, N.; Houle, D.; Lynch, M.; Charlesworth, B.; Keightley, P.D. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 2008, 6, 1706–1714. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer Iv, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef]
- Mari, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Sheeja, A.A.; Janardanan, D. Hydroxyl radical scavenging activity of melatonin and its related indolamines. Free Radic Res. 2020, 54, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Mujika, J.I.; Uranga, J.; Matxain, J.M. Computational study on the attack of .OH radicals on aromatic amino acids. Chemistry 2013, 19, 6862–6873. [Google Scholar] [CrossRef]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid. Med. Cell. Longev. 2011, 2011, 809696. [Google Scholar] [CrossRef] [PubMed]
- Fatima, K.; Masood, N.; Luqman, S. Quenching of singlet oxygen by natural and synthetic antioxidants and assessment of electronic UV/Visible absorption spectra for alleviating or enhancing the efficacy of photodynamic therapy. Biomed. Res. Ther. 2016, 3, 514–527. [Google Scholar] [CrossRef]
- To, T.L.; Fadul, M.J.; Shu, X. Singlet oxygen triplet energy transfer-based imaging technology for mapping protein-protein proximity in intact cells. Nat. Commun. 2014, 5, 4072. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Sakurai, Y.; Tajima, K. Reactive oxygen species inhibitory diagrams and their usability for the evaluation of antioxidant ability. Oxid. Antioxid. Med. Sci. 2016, 5, 1–7. [Google Scholar] [CrossRef]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite with uric acid in the presence of ascorbate and thiols: Implications for uncoupling endothelial nitric oxide synthase. Biochem. Pharmacol. 2005, 70, 343–354. [Google Scholar] [CrossRef]
- Pieri, C.; Marra, M.; Moroni, F.; Recchioni, R.; Marcheselli, F. Melatonin: A peroxyl radical scavenger more effective than vitamin E. Life Sci. 1994, 55, PL271–PL276. [Google Scholar] [CrossRef]
- Bhat, T.A.; Kumar, S.; Chaudhary, A.K.; Yadav, N.; Chandra, D. Restoration of mitochondria function as a target for cancer therapy. Drug Discov. Today 2015, 20, 635–643. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed]
- Kardeh, S.; Ashkani-Esfahani, S.; Alizadeh, A.M. Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur. J. Pharmacol. 2014, 735, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Iskandar, K.B.; Hirpara, J.L.; Clement, M.V.; Pervaiz, S. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 2004, 64, 7867–7878. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Tian, C.; Zhao, L.; Petit, P.X.; Mehrpour, M.; Chen, Q. Cysteine 62 of Bax is critical for its conformational activation and its proapoptotic activity in response to H2O2-induced apoptosis. J. Biol. Chem. 2008, 283, 15359–15369. [Google Scholar] [CrossRef]
- Zheng, Y.; Yamaguchi, H.; Tian, C.; Lee, M.W.; Tang, H.; Wang, H.G.; Chen, Q. Arsenic trioxide (As(2)O(3)) induces apoptosis through activation of Bax in hematopoietic cells. Oncogene 2005, 24, 3339–3347. [Google Scholar] [CrossRef]
- Kirkland, R.A.; Saavedra, G.M.; Franklin, J.L. Rapid activation of antioxidant defenses by nerve growth factor suppresses reactive oxygen species during neuronal apoptosis: Evidence for a role in cytochrome c redistribution. J. Neurosci. 2007, 27, 11315–11326. [Google Scholar] [CrossRef]
- Kirkland, R.A.; Windelborn, J.A.; Kasprzak, J.M.; Franklin, J.L. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J. Neurosci. 2002, 22, 6480–6490. [Google Scholar] [CrossRef]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J. Natl. Cancer Inst. 2006, 98, 1462–1473. [Google Scholar] [CrossRef]
- Ruckenstuhl, C.; Buttner, S.; Carmona-Gutierrez, D.; Eisenberg, T.; Kroemer, G.; Sigrist, S.J.; Frohlich, K.U.; Madeo, F. The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer. PLoS ONE 2009, 4, e4592. [Google Scholar] [CrossRef]
- Hahm, E.R.; Moura, M.B.; Kelley, E.E.; Van Houten, B.; Shiva, S.; Singh, S.V. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS ONE 2011, 6, e23354. [Google Scholar] [CrossRef]
- Sanz, A.; Soikkeli, M.; Portero-Otin, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellila, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Nicco, C.; Chereau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Levy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [PubMed]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist Update 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent Oxidative Stress in Cancer. Febs Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Konstantinov, A.A.; Peskin, A.V.; Popova, E.; Khomutov, G.B.; Ruuge, E.K. Superoxide generation by the respiratory chain of tumor mitochondria. Biochim. Biophys. Acta 1987, 894, 1–10. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Payen, V.L.; Zampieri, L.X.; Porporato, P.E.; Sonveaux, P. Pro- and antitumor effects of mitochondrial reactive oxygen species. Cancer Metastasis Rev. 2019, 38, 189–203. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Kong, Q.; Beel, J.A.; Lillehei, K.O. A threshold concept for cancer therapy. Med. Hypotheses 2000, 55, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Liu, W.; Zhang, Q.; Huang, J.; Hu, C.; Liu, Y.; Wang, Q.; Zhou, M.; Lai, W.; Sheng, F.; et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to IR-783-induced apoptosis in human breast cancer cells. J. Cell Mol. Med. 2018, 22, 4474–4485. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Wang, J.; Van Houten, B. The role of dynamin-related protein 1 in cancer growth: A promising therapeutic target? Expert Opin. Ther. Targets 2013, 17, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Khakhina, S.; Cooper, K.F.; Strich, R. Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol. Biol. Cell 2014, 25, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Stieg, D.C.; Willis, S.D.; Ganesan, V.; Ong, K.L.; Scuorzo, J.; Song, M.; Grose, J.; Strich, R.; Cooper, K.F. A complex molecular switch directs stress-induced cyclin C nuclear release through SCF(Grr1)-mediated degradation of Med13. Mol. Biol. Cell 2018, 29, 363–375. [Google Scholar] [CrossRef]
- Jezek, J.; Wang, K.; Yan, R.; Di Cristofano, A.; Cooper, K.F.; Strich, R. Synergistic repression of thyroid hyperplasia by cyclin C and Pten. J. Cell Sci. 2019, 132, jcs230029. [Google Scholar] [CrossRef]
- Jezek, J.; Plecita-Hlavata, L.; Jezek, P. Aglycemic HepG2 Cells Switch from Aminotransferase Glutaminolytic Pathway of Pyruvate Utilization to Complete Krebs Cycle at Hypoxia. Front. Endocrinol. 2018, 9, 637. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name. | Abbreviation | References |
|---|---|---|
| Abelson tyrosine kinase | c-Abl | [21] |
| AMP-activated protein kinase | AMPK | [22,23] |
| calcineurin | – | [24,25,26,27] |
| Ca2+/calmodulin-dependent protein kinase | CaMK | [28,29] |
| c-Jun N-terminal kinase | JNK | [30] |
| cyclin C | – | [31,32] |
| cell division cycle 20 related 1 | Cdh1 | [33] |
| cyclin-dependent kinase 1 | Cdk1 | [19,24] |
| cyclin-dependent kinase 5 | Cdk5 | [34,35] |
| cyclophilin D | – | [36] |
| death-associated protein 3 | DAP3 | [37] |
| endophilin B1 | – | [38] |
| extracellular signal-regulated kinase | Erk | [39] |
| ganglioside-induced differentiation associated protein 1 | GDAP1 | [40] |
| hypoxia-inducible factor 1α | HIF1α | [41] |
| mitochondrial fission process 1 | MTFP1 | [42] |
| nuclear factor κB | NF-κB | [43] |
| nuclear factor (erythroid-derived 2)-like 2 | Nrf2 | [44] |
| overlapping activity with m-AAA protease 1 | Oma1 | [45] |
| mitofilin | – | [46] |
| p38 mitogen-activated protein kinase | p38 MAPK | [47] |
| PGAM5 | – | [48] |
| phospholipase D | PLD | [49,50] |
| protein disulfide isomerase A1 | PDIA1 | [51] |
| protein kinase A | PKA | [27,52] |
| protein kinase C, isoform δ | PKCδ | [24] |
| Rho-associated coiled-coil containing protein kinase 1 | ROCK1 | [53] |
| S6 kinase 1 | S6K1 | [54] |
| uncoupling protein 2 | UCP2 | [55,56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ježek, J.; Cooper, K.F.; Strich, R. The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy. Biology 2021, 10, 33. https://doi.org/10.3390/biology10010033
Ježek J, Cooper KF, Strich R. The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy. Biology. 2021; 10(1):33. https://doi.org/10.3390/biology10010033
Chicago/Turabian StyleJežek, Jan, Katrina F. Cooper, and Randy Strich. 2021. "The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy" Biology 10, no. 1: 33. https://doi.org/10.3390/biology10010033
APA StyleJežek, J., Cooper, K. F., & Strich, R. (2021). The Impact of Mitochondrial Fission-Stimulated ROS Production on Pro-Apoptotic Chemotherapy. Biology, 10(1), 33. https://doi.org/10.3390/biology10010033