Methods, Challenges and Potentials of Single Cell RNA-seq

{kind=link}
Abstract
:1. Introduction—the Importance of Transcriptional Profiling at Single Cell Resolution
2. RNA-seq—Basic Experimental Procedures
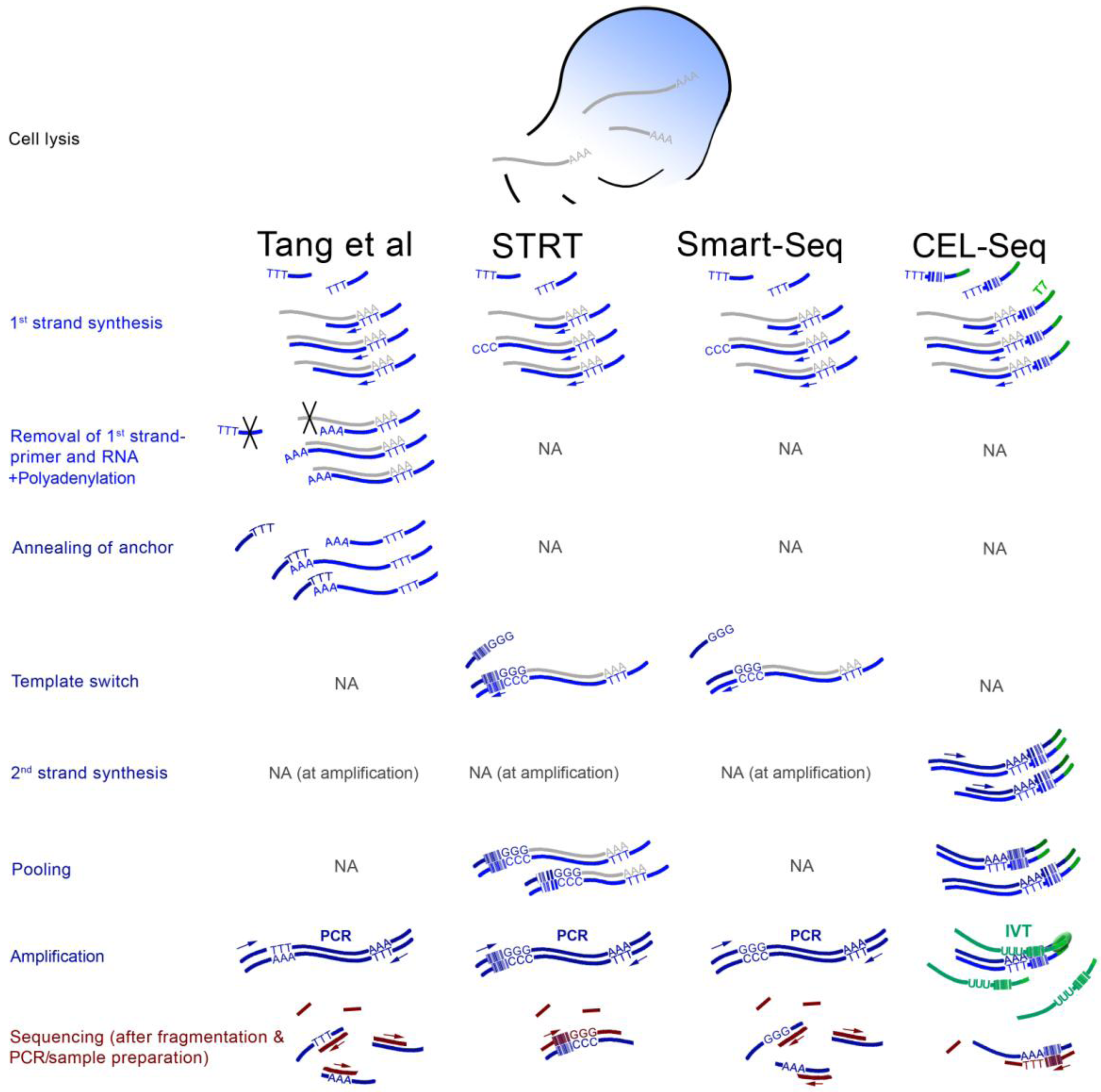
3. ScRNA-seq Strategies
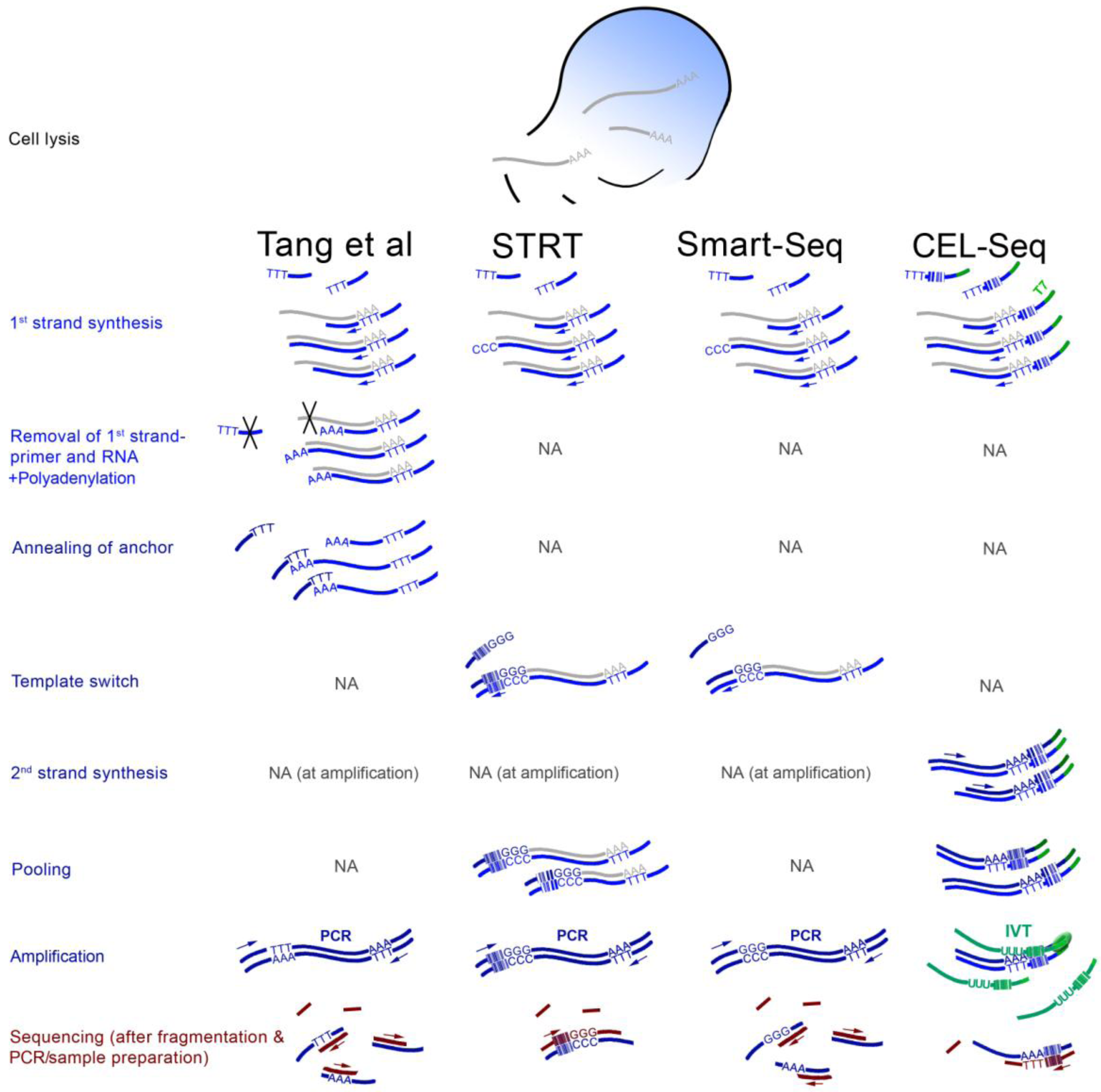
4. Findings from the First scRNA-seq Studies
5. Outlook—Challenges, Future Issues and Emerging Technologies
Acknowledgments
References and Notes
- McKinney, E.F.; Lyons, P.A.; Carr, E.J.; Hollis, J.L.; Jayne, D.R.; Willcocks, L.C.; Koukoulaki, M.; Brazma, A.; Jovanovic, V.; Kemeny, D.M.; et al. A cd8+ t cell transcription signature predicts prognosis in autoimmune disease. Nat. Med. 2010, 16, 586–591, 581 following 591. [Google Scholar]
- Wei, G.; Wei, L.; Zhu, J.; Zang, C.; Hu-Li, J.; Yao, Z.; Cui, K.; Kanno, Y.; Roh, T.Y.; Watford, W.T.; et al. Global mapping of h3k4me3 and h3k27me3 reveals specificity and plasticity in lineage fate determination of differentiating cd4+ t cells. Immunity 2009, 30, 155–167. [Google Scholar] [CrossRef]
- Morin, R.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.; McDonald, H.; Varhol, R.; Jones, S.; Marra, M. Profiling the hela s3 transcriptome using randomly primed cdna and massively parallel short-read sequencing. Biotechniques 2008, 45, 81–94. [Google Scholar]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar]
- Ozsolak, F.; Milos, P.M. Rna sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir el, A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011, 332, 687–696. [Google Scholar]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar]
- Paulsson, J. Models of stochastic gene expression. Phys. Life Rev. 2005, 2, 157–175. [Google Scholar] [CrossRef]
- Maheshri, N.; O'Shea, E.K. Living with noisy genes: How cells function reliably with inherent variability in gene expression. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 413–434. [Google Scholar] [CrossRef]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using gene expression noise to understand gene regulation. Science 2012, 336, 183–187. [Google Scholar]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mrnas: Life without a poly(a) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef]
- Tariq, M.A.; Kim, H.J.; Jejelowo, O.; Pourmand, N. Whole-transcriptome rnaseq analysis from minute amount of total rna. Nucleic Acids Res. 2011, 39, e120. [Google Scholar] [CrossRef]
- Ortin, J.; Parra, F. Structure and function of rna replication. Annu. Rev. Microbiol. 2006, 60, 305–326. [Google Scholar] [CrossRef]
- Von Hippel, P.H.; Fairfield, F.R.; Dolejsi, M.K. On the processivity of polymerases. Ann. NY Acad. Sci. 1994, 726, 118–131. [Google Scholar] [CrossRef]
- Fox-Walsh, K.; Davis-Turak, J.; Zhou, Y.; Li, H.; Fu, X.D. A multiplex rna-seq strategy to profile poly(a+) rna: Application to analysis of transcription response and 3' end formation. Genomics 2011, 98, 266–271. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Mamanova, L.; Andrews, R.M.; James, K.D.; Sheridan, E.M.; Ellis, P.D.; Langford, C.F.; Ost, T.W.; Collins, J.E.; Turner, D.J. Frt-seq: Amplification-free, strand-specific transcriptome sequencing. Nat. Methods 2010, 7, 130–132. [Google Scholar]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. Mrna-seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A smart approach for full-length cdna library construction. Biotechniques 2001, 30, 892–897. [Google Scholar]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Highly multiplexed and strand-specific single-cell rna 5' end sequencing. Nat. Protoc. 2012, 7, 813–828. [Google Scholar] [CrossRef]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex rna-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar]
- Tang, F.; Barbacioru, C.; Nordman, E.; Li, B.; Xu, N.; Bashkirov, V.I.; Lao, K.; Surani, M.A. Rna-seq analysis to capture the transcriptome landscape of a single cell. Nat. Protoc. 2010, 5, 516–535. [Google Scholar]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. Cel-seq: Single-cell rna-seq by multiplexed linear amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Ramskold, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mrna-seq from single-cell levels of rna and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Qiu, S.; Luo, S.; Evgrafov, O.; Li, R.; Schroth, G.P.; Levitt, P.; Knowles, J.A.; Wang, K. Single-neuron rna-seq: Technical feasibility and reproducibility. Frontiers Genet. 2012, 3, 124. [Google Scholar]
- RNA Sample Amplification. Available online: http://www.ipc.nxgenomics.org/newsletter/no7.htm (accessed on 11 October 2012).
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific rna sequencing methods. Nat. Methods 2010, 7, 709–715. [Google Scholar]
- Tang, F.; Barbacioru, C.; Bao, S.; Lee, C.; Nordman, E.; Wang, X.; Lao, K.; Surani, M.A. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell rna-seq analysis. Cell Stem Cell 2010, 6, 468–478. [Google Scholar] [CrossRef]
- So, L.H.; Ghosh, A.; Zong, C.; Sepulveda, L.A.; Segev, R.; Golding, I. General properties of transcriptional time series in escherichia coli. Nat. Genet. 2011, 43, 554–560. [Google Scholar]
- Hebenstreit, D.; Fang, M.; Gu, M.; Charoensawan, V.; van Oudenaarden, A.; Teichmann, S.A. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 2011, 7, 497. [Google Scholar]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Fu, G.K.; Hu, J.; Wang, P.H.; Fodor, S.P. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc. Natl. Acad. Sci. USA 2011, 108, 9026–9031. [Google Scholar]
- Kivioja, T.; Vaharautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2012, 9, 72–74. [Google Scholar]
- Shiroguchi, K.; Jia, T.Z.; Sims, P.A.; Xie, X.S. Digital rna sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl. Acad. Sci. USA 2012, 109, 1347–1352. [Google Scholar]
- Lipson, D.; Raz, T.; Kieu, A.; Jones, D.R.; Giladi, E.; Thayer, E.; Thompson, J.F.; Letovsky, S.; Milos, P.; Causey, M. Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 2009, 27, 652–658. [Google Scholar] [CrossRef]
- Ozsolak, F.; Platt, A.R.; Jones, D.R.; Reifenberger, J.G.; Sass, L.E.; McInerney, P.; Thompson, J.F.; Bowers, J.; Jarosz, M.; Milos, P.M. Direct rna sequencing. Nature 2009, 461, 814–818. [Google Scholar]
- Ozsolak, F.; Milos, P.M. Single-molecule direct rna sequencing without cdna synthesis. Wiley Interdiscip. Rev. RNA 2011, 2, 565–570. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Uemura, S.; Aitken, C.E.; Korlach, J.; Flusberg, B.A.; Turner, S.W.; Puglisi, J.D. Real-time trna transit on single translating ribosomes at codon resolution. Nature 2010, 464, 1012–1017. [Google Scholar]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 83, 4327–4341. [Google Scholar]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hebenstreit, D. Methods, Challenges and Potentials of Single Cell RNA-seq. Biology 2012, 1, 658-667. https://doi.org/10.3390/biology1030658
Hebenstreit D. Methods, Challenges and Potentials of Single Cell RNA-seq. Biology. 2012; 1(3):658-667. https://doi.org/10.3390/biology1030658
Chicago/Turabian StyleHebenstreit, Daniel. 2012. "Methods, Challenges and Potentials of Single Cell RNA-seq" Biology 1, no. 3: 658-667. https://doi.org/10.3390/biology1030658
APA StyleHebenstreit, D. (2012). Methods, Challenges and Potentials of Single Cell RNA-seq. Biology, 1(3), 658-667. https://doi.org/10.3390/biology1030658