


pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria

Abstract
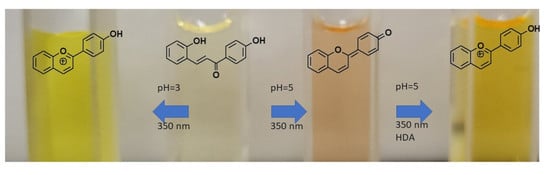
{kind=link}
{kind=link}
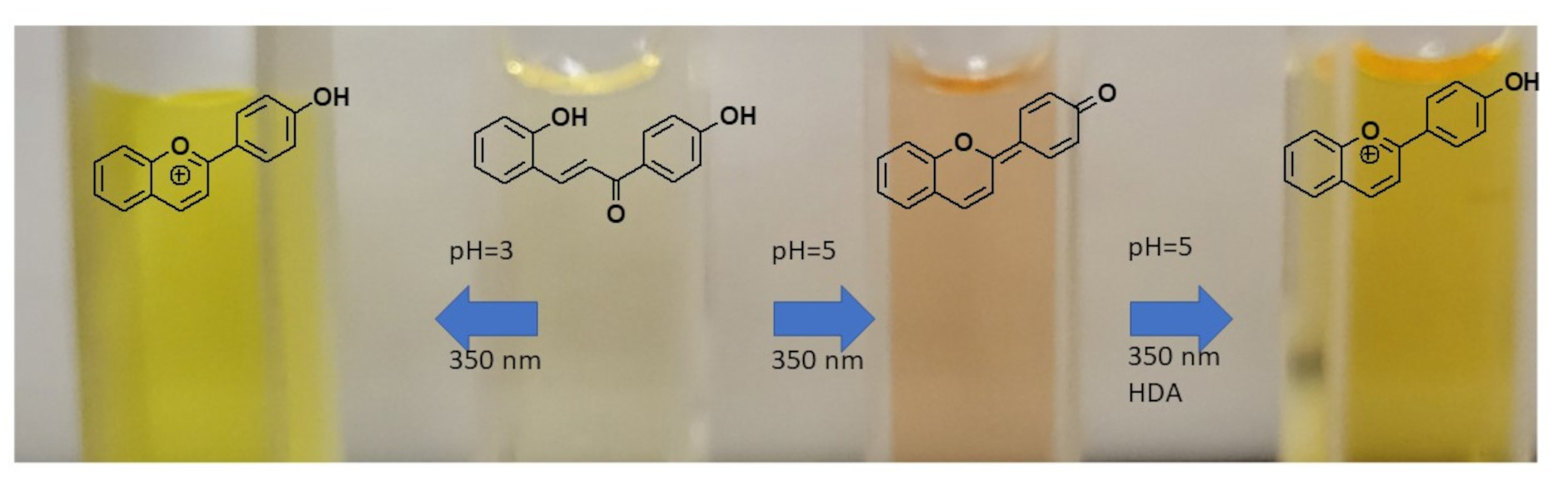
{kind=link}
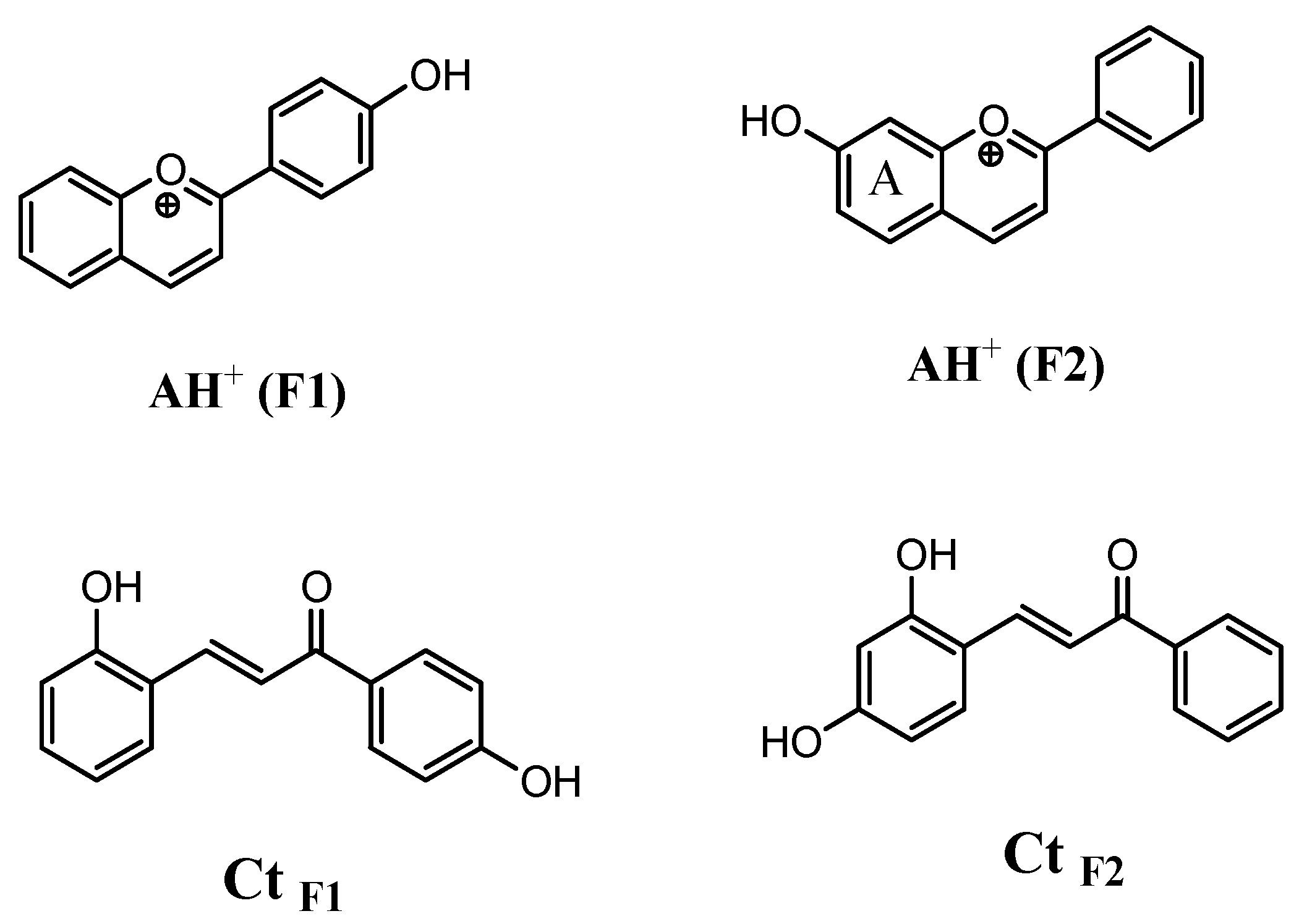
{kind=link}
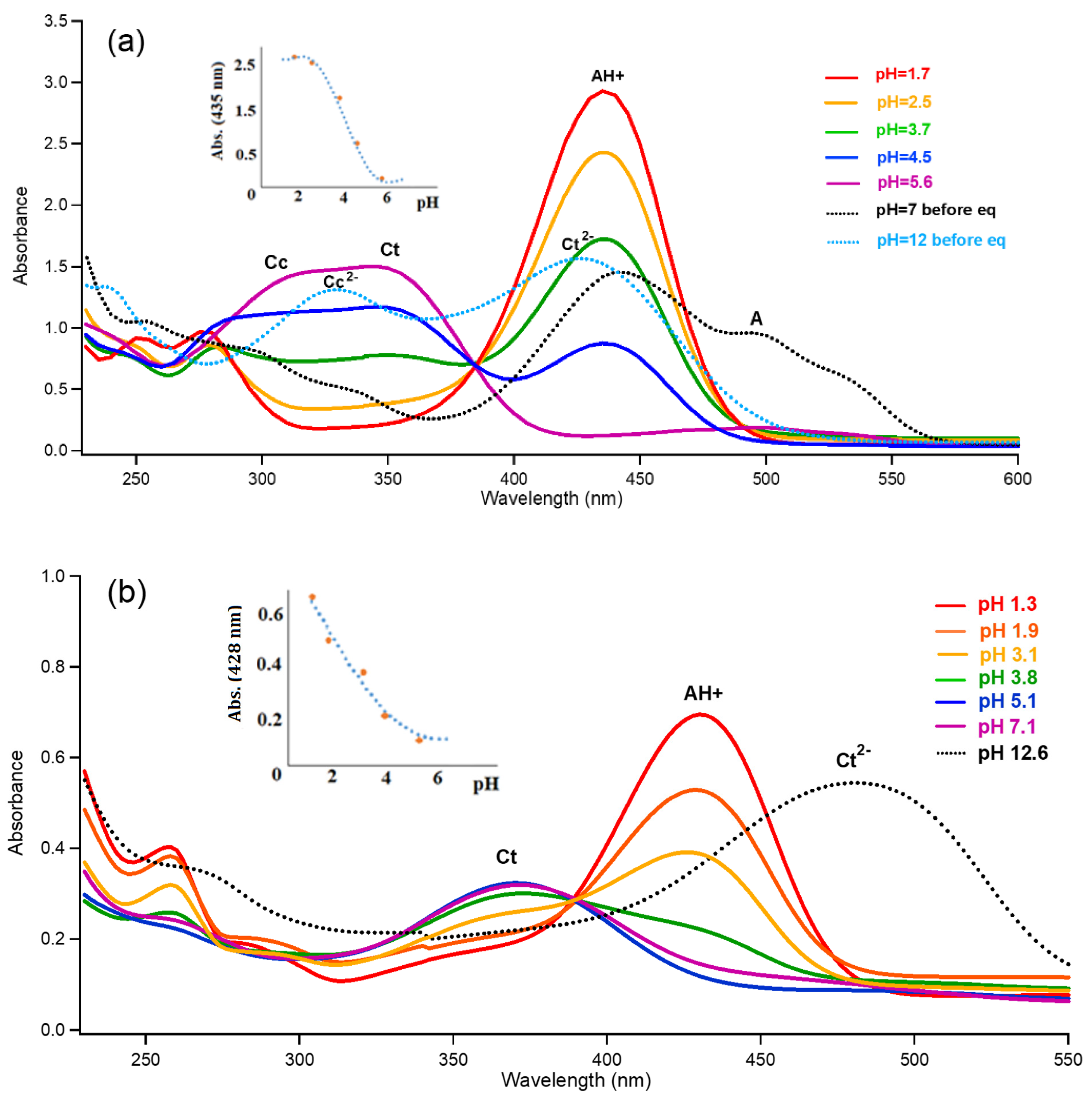
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthetic Procedure [46]
2.2. General Photoreaction Procedure
2.3. Gaussian Calculation
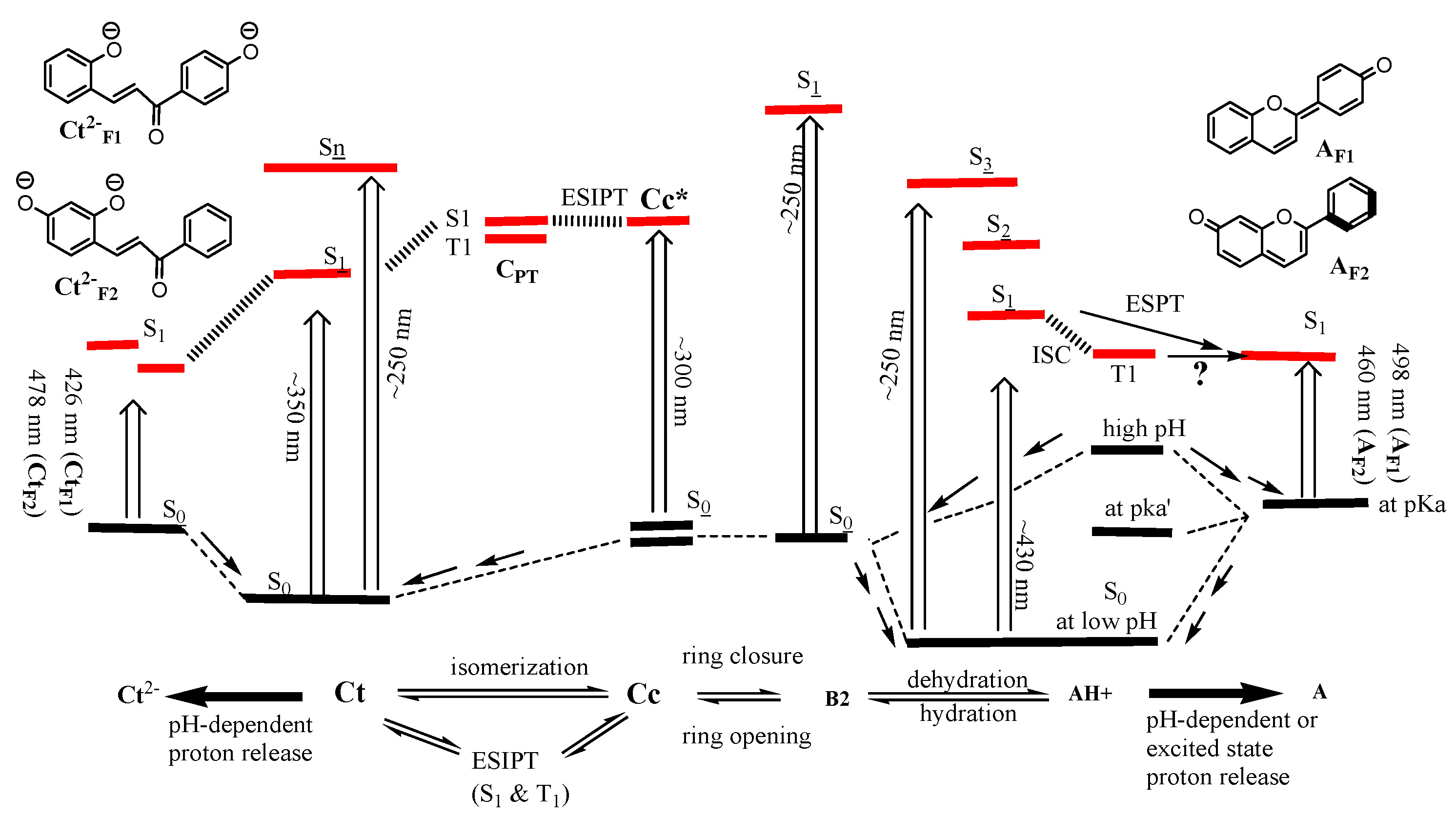
3. Results and Discussion
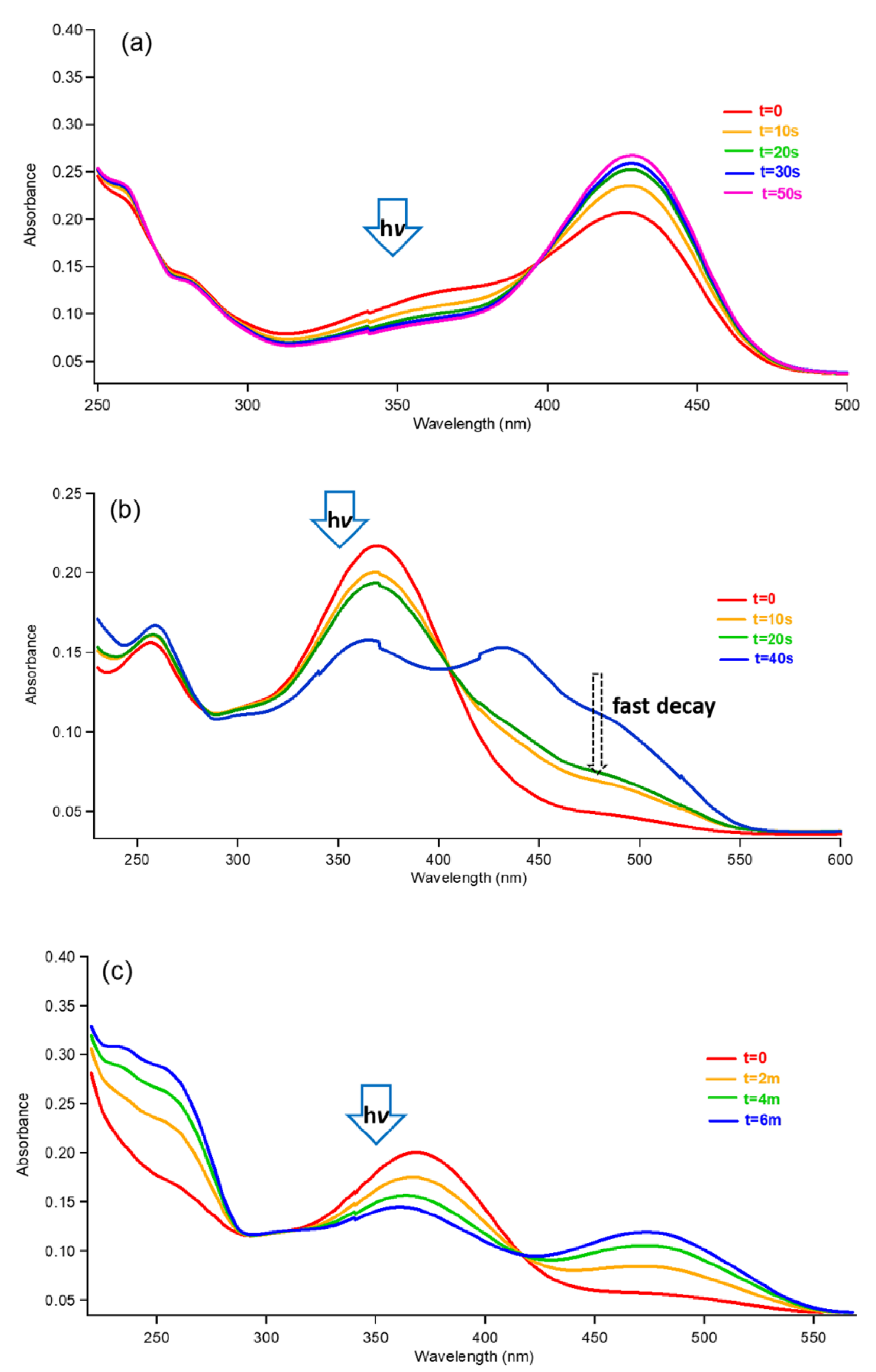
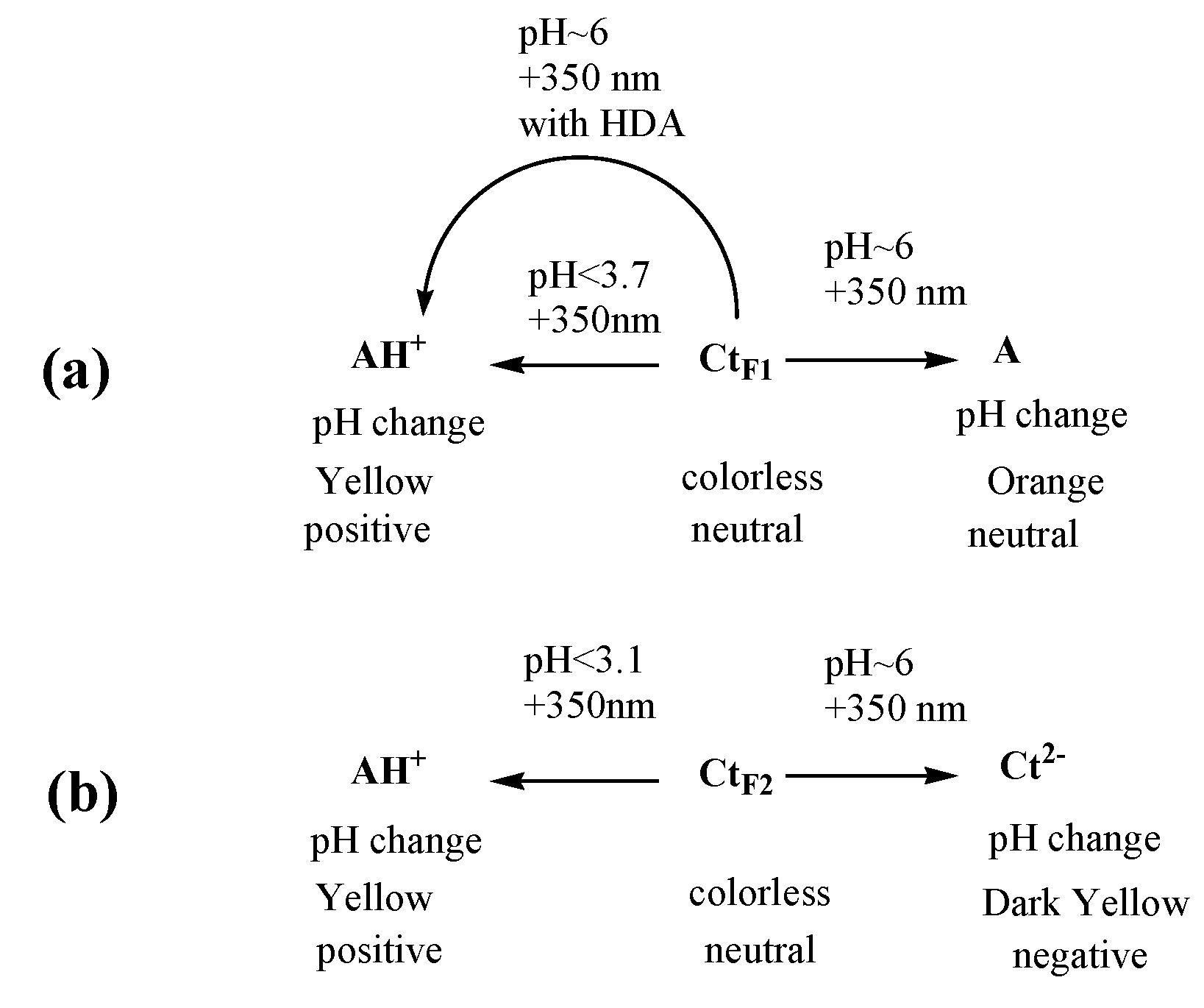
3.1. trans-Chalcone in Equilibrium at Different pHs
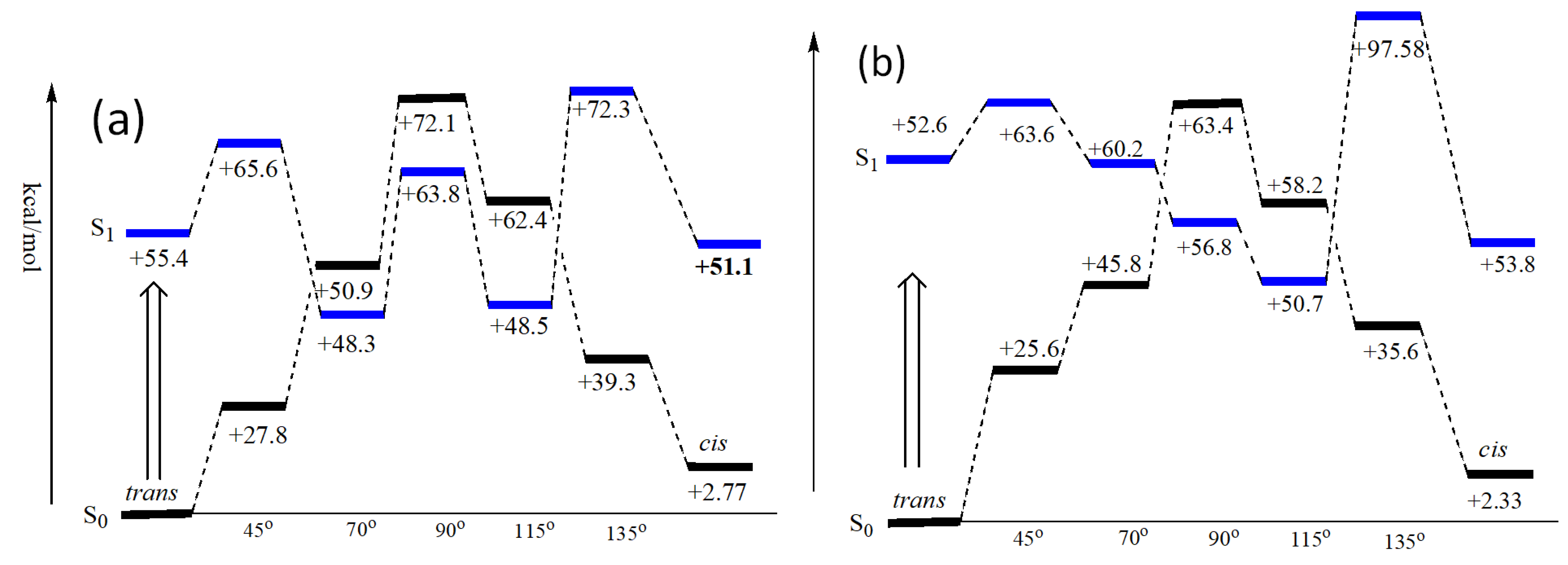
3.2. Photoisomerization of trans-Chalcone
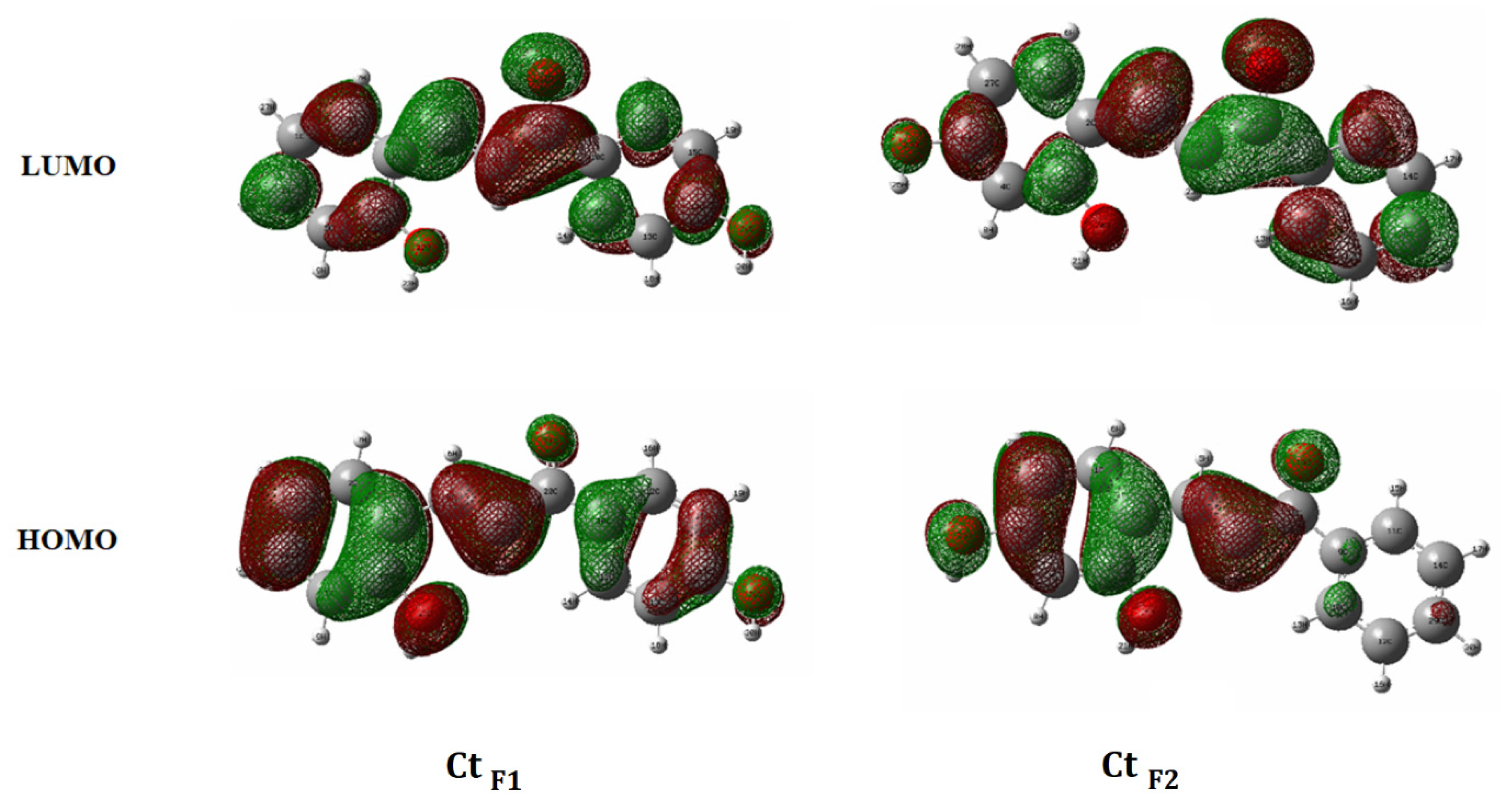
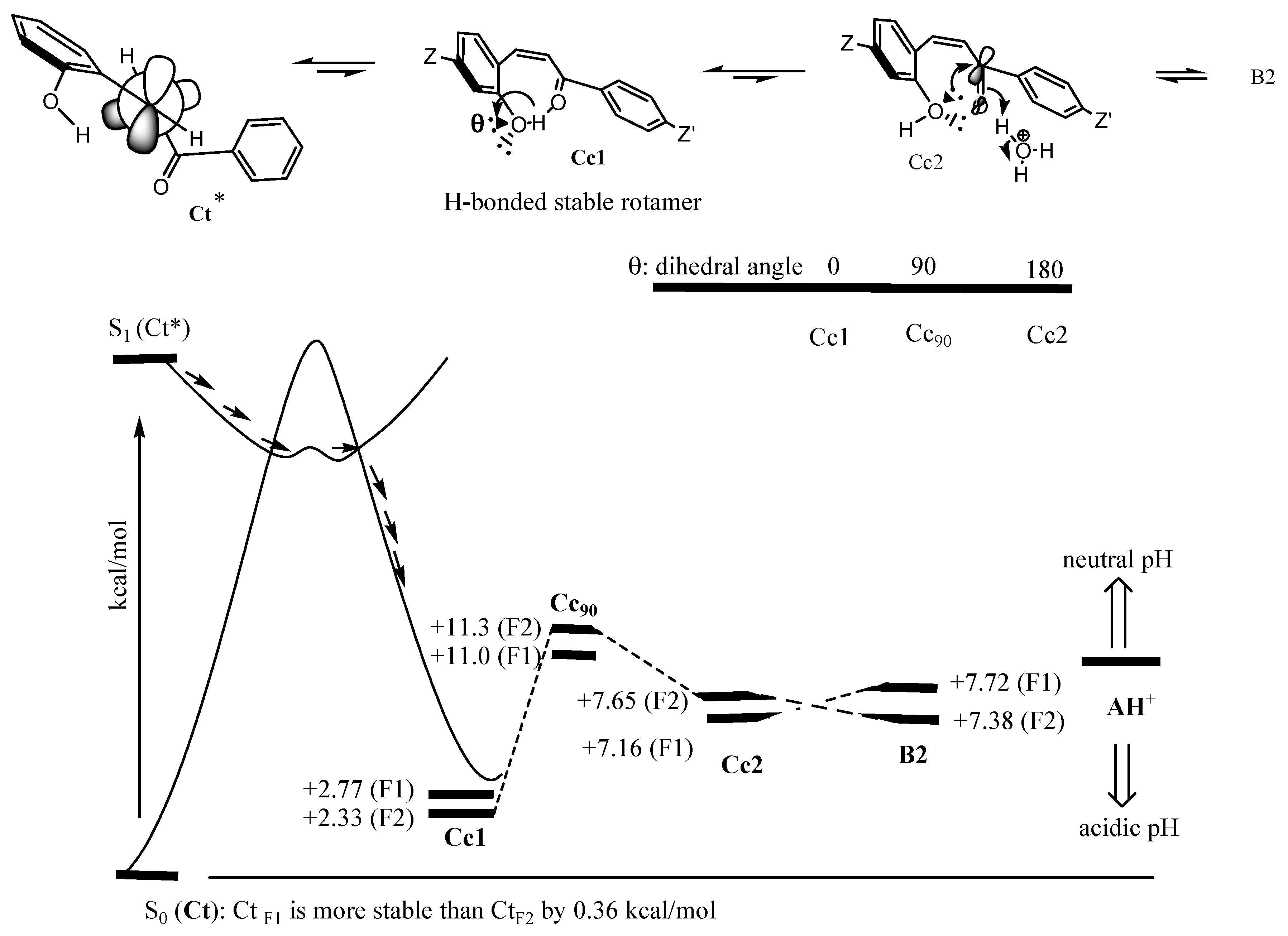
3.3. Insights from DFT Calculation
3.4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Givens, R.S.; Heger, D.; Hellrung, B.; Kamdzhilov, Y.; Mac, M.; Conrad, P.G., II; Cope, E.; Lee, J.I.; Mata-Segreda, J.F.; Showen, R.; et al. The photo-Favorskii reaction of p-hydroxyphenacyl compounds is initiated by water-Assisted, adiabatic extrusion of a triplet biradical. J. Am. Chem Soc. 2008, 130, 3307–3309. [Google Scholar] [CrossRef] [PubMed]
- Barrella, A.; Rene, N.; Ali, K.; Kang, J.; Lee, J.I. Synthesis of first generation dendron, GABA-caged bis-1,2-(4-acetylphenylethynyl)-4,5-dimethoxybenzene for photorelease study. Lett. Org. Chem. 2019, 16, 718–722. [Google Scholar] [CrossRef]
- Cembran, A.; Bernardi, F.; Garavelli, M.; Gagliardi, L.; Orlandi, G. On the mechanism of the cis-trans isomerization in the lowest electronic states of azobenzene: S0, S1, and T1. J. Am. Chem. Soc. 2004, 126, 3234–3243. [Google Scholar] [CrossRef] [PubMed]
- Rossegger, E.; Strasser, J.; Rita Höller, R.; Fleisch, M.; Berer, M.; Schlögl, S. Wavelength selective multi-material 3D printing of soft active devices using orthogonal photoreactions. Macromol. Rapid Commun. 2022, 44, 2200586. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Lei, M.; Zhang, P.; Leng, J.; Zheng, Z.; Yu, Y. Orthogonal photochemistry-assisted printing of 3D tough and stretchable conductive hydrogels. Nat. Commun. 2021, 12, 2082. [Google Scholar] [CrossRef]
- Yang, J.; Li, Z.; Gu, X.; Zhan, T.; Cui, J.; Zhang, K. Photogated photoswitchable [2]rotaxane based on orthogonal photoreactions. Tetrahedron 2021, 92, 132284. [Google Scholar] [CrossRef]
- Yamano, Y.; Murayama, K.; Asanuma, H. Dual crosslinking photo-switches for orthogonal photo-control of hybridization between serinol nucleic acid and RNA. Chem. A Eur. J. 2021, 27, 4475. [Google Scholar] [CrossRef]
- Wang, D.; Wagner, M.; Saydjari, A.; Mueller, J.; Winzen, S.; Butt, H.; Wu, S. A photoresponsive orthogonal supramolecular complex based on Host-Guest interactions. Chem. A Eur. J. 2017, 23, 2628–2634. [Google Scholar] [CrossRef]
- Manna, D.; Udayabhaskararao, T.; Zhao, H.; Klajn, R. Orthogonal light-induced self-assembly of nanoparticles using differently substituted azobenzenes. Angew. Chem. Int. Ed. 2015, 54, 12394–12397. [Google Scholar] [CrossRef]
- Olson, J.; Banghart, M.; Sabatini, B.; Ellis-Davies, G. Spectral evolution of a photochemical protecting group for orthogonal two-color uncaging with visible light. J. Am. Chem. Soc. 2013, 135, 15948–15954. [Google Scholar] [CrossRef]
- Corrigan, N.; Boyer, C. 100th anniversary of macromolecular science viewpoint: Photochemical reaction orthogonality in modern macromolecular science. ACS Macro Lett. 2019, 8, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Loffe, I.N.; Granovsky, A.A. Photoisomerization of Stilbene: The detailed XMCQDPT2 treatment. J. Chem. Theory Comput. 2013, 9, 4973–4990. [Google Scholar] [CrossRef]
- Pina, F.; Melo, M.J.; Laia, C.A.; Parola, J.; Lima, J.C. Chemistry and applications of flavylium compounds: A handful of colours. Chem. Soc. Rev. 2012, 41, 869–908. [Google Scholar] [CrossRef] [PubMed]
- Brouillard, R.; Iacobucci, G.A.; Sweeny, J.G. Chemistry of anthocyanin pigments. 9. UV-visible spectrophotometric determination of the acidity constants of apigeninidin and three related 3- deoxyflavylium salts. J. Am. Chem. Soc. 1982, 104, 7585–7590. [Google Scholar] [CrossRef]
- McClelland, R.A.; McGall, G.H. Hydration of the Flavylium Ion. 2. The 4′-Hydroxyflavylium Ion. J. Org. Chem. 1982, 47, 3730–3736. [Google Scholar] [CrossRef]
- Takeda, K.; Osakabe, A.; Saito, S.; Furuyama, D.; Tomita, A.; Kojima, Y.; Yamadera, M.; Sakuta, M. Components of protocyanin, a blue pigment from the blue flowers of Centaurea cyanus. Phytochemistry 2005, 66, 1607–1613. [Google Scholar] [CrossRef]
- Takeda, K. Blue metal complex pigments involved in blue flower color. Proc. Jpn. Acad. Ser. B Phys. Biol Sci. 2006, 82, 142–154. [Google Scholar] [CrossRef]
- Gould, K.S. Nature’s Swiss Army Knife: The Diverse Protective Roles of Anthocyanins in Leaves. J. Biomed. Biotechnol. 2004, 5, 314–320. [Google Scholar] [CrossRef]
- Lima, A.A.; Sussuchi, E.M.; Giovani, W.F. Electrochemical and Antioxidant Properties of Anthocyanins and Anthocyanidins. Croat. Chem. Acta 2007, 80, 29–34. [Google Scholar]
- Gould, K.S.; McKelvie, J.; Markham, K.R. Do anthocyanins function as antioxidants in leaves? Imaging of H2O2 in red and green leaves after mechanical injury. Plant Cell Environ. 2002, 25, 1261–1269. [Google Scholar] [CrossRef]
- Hoch, W.A.; Singsaas, E.L.; McCown, B.H. Resorption protection. Anthocyanins facilitate nutrient recovery in autumn by shielding leaves from potentially damaging light levels. Plant Physiol. 2003, 133, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, J.; Bergquist, G.; Gardeström, P.; Jansson, S.A. Cellular Timetable of Autumn Senescence. Plant Physiol. 2005, 139, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Close, D.C.; Beadle, C.L. The ecophysiology of foliar anthocyanin. Bot. Rev. 2003, 69, 149–161. [Google Scholar] [CrossRef]
- Rechner, A.R.; Kroner, C. Anthocyanins and colonic metabolites of dietary polyphenols inhibit platelet function. Thromb. Res. 2005, 116, 327–334. [Google Scholar] [CrossRef]
- Bell, D.R.; Gochenaur, K. Direct vasoactive and vasoprotective properties of anthocyanin-rich extracts. J. Appl. Physiol. 2006, 100, 1164–1170. [Google Scholar] [CrossRef]
- Toufektsian, M.C.; De Lorgeril, M.; Nagy, N.; Salen, P.; Donati, M.B.; Giordano, L.; Mock, H.-P.; Peterek, S.; Matros, A.; Petroni, K. Chronic dietary intake of plant-derived anthocyanins protects the rat heart against ischemia-reperfusion injury. J. Nutr. 2008, 138, 747–752. [Google Scholar] [CrossRef]
- Bontempo, P.; de Masi, L.; Carafa, V.; Rigano, D.; Scisciola, L.; Iside, C.; Grassi, R.; Molinari, A.M.; Aversano, R.; Nebbioso, A. Anticancer activities of anthocyanin extract from genotyped Solanum tuberosum L. “Vitelotte”. J. Funct. Foods 2015, 19, 584–593. [Google Scholar] [CrossRef]
- Côté, J.; Caillet, S.; Doyon, G.; Dussault, D.; Sylvain, J.-F.; Lacroix, M. Antimicrobial effect of cranberry juice and extracts. Food Control 2011, 22, 1413–1418. [Google Scholar] [CrossRef]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. Methods Enzymol. 1990, 186, 343–355. [Google Scholar] [CrossRef]
- Hardin, B.E.; Hoke, E.T.; Amstrong, P.B.; Yum, J.H.; Comte, P.; Torres, T.; Frechet, J.M.J.; Nazeeruddin, M.K.; Gratzel, M.; McGehee, M.D. Increased light harvesting in dye-sensitized solar cells with energy relay dyes. Nat. Photonics 2009, 3, 406–411. [Google Scholar] [CrossRef]
- Moncada, M.C.; Fernandez, D.; Lima, J.C.; Parola, A.J.; Lodeiro, C.; Folgosa, F.; Melo, M.J.; Pina, F. Multistate properties of 7-(N,N-diethylamino)-40 -hydroxyflavylium. An example of an unidirectional reaction cycle driven by pH. Org. Biomol. Chem. 2004, 2, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Giestas, L.; Folgosa, F.; Lima, J.C.; Parola, A.J.; Pina, F. Bio-Inspired Multistate Networks Responsive to Light, pH and Thermal Inputs–An Example of a Multistate System Operating Through Different Algorithms. Eur. J. Org. Chem. 2005, 19, 4187–4200. [Google Scholar] [CrossRef]
- Furtado, P.; Figueiredo, P.; Chaves das Neves, H.; Pina, F. Photochemical and thermal degradation of anthocyanidins. Photochemical and thermal degradation of anthocyanidins. J. Photochem. Photobiol. A 1993, 75, 113–118. [Google Scholar] [CrossRef]
- Alejo-Armijo, A.; Parola, A.J.; Pina, F. pH-Dependent Multistate System Generated by a Synthetic Furanoflavylium Compound: An Ancestor of the Anthocyanin Multistate of Chemical Species. ACS Omega 2019, 4, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.J.; Moura, S.; Roque, A.; Maestri, M.; Pina, F. Photochemistry of luteolinidin: “Write-lock-read-unlock-erase” with a natural compound. J. Photochem. Photobiol. A 2000, 135, 33–39. [Google Scholar] [CrossRef]
- Gomes, R.; Diniz, A.M.; Jesus, A.; Parola, A.J.; Pina, F. The synthesis and reaction network of 2-styryl-1-benzopyrylium salts: An unexploited class of potential colorants. Dye. Pigment. 2009, 81, 69–79. [Google Scholar] [CrossRef]
- Sweeny, J.G.; Iacobucci, G.A. Effect of Substitution on the stability of 3-Deoxyanthocyanidins in Aqueous Solutions. J. Agric. Food Chem. 1983, 31, 531–533. [Google Scholar] [CrossRef]
- Petrov, V.; Diniz, A.M.; Cunha-Silva, L.; Parola, A.J.; Pina, F. Kinetic and thermodynamic study of 2′-hydroxy-8-methoxyflavylium. Reaction network interconverting flavylium cation and flavanone. RSC Adv. 2013, 3, 10786–10794. [Google Scholar] [CrossRef]
- Jimenez, A.; Pinheiro, C.; Parola, A.J.; Maestri, M.; Pina, F. The chemistry of 6-hydroxyflavylium: Zwitterionic base and p-quinoidal chalcones. A multiswitchable system operated by proton, electron and photon inputs. Photochem. Photobiol. Sci. 2007, 6, 372–380. [Google Scholar] [CrossRef]
- Gago, S.; Petrov, V.; Parola, A.J.; Pina, F. Synthesis and characterization of 3′-butoxy-flavylium derivatives. J. Photochem. Photobiol. A Chem. 2012, 244, 54–64. [Google Scholar] [CrossRef]
- Fernandez, D.; Parola, A.J.; Branco, L.C.; Afonso, C.A.M.; Pina, F. Thermal and photochemical properties of 4′-hydroxyflavylium in water-ionic liquid biphasic systems. J. Photochem. Photobiol. A Chem. 2004, 168, 185–189. [Google Scholar] [CrossRef]
- Brouillard, R.; Dubois, J. Mechanism of the Structural Transformations of Anthocyanins in Acidic Media. J. Am. Chem. Soc. 1976, 99, 1359–1364. [Google Scholar] [CrossRef]
- Pina, F.; Roque, A.; Melo, M.J.; Maestri, M.; Belladelli, L.; Balzani, V. Multistate/Multifunctional Molecular-Level Systems: Light and pH Switching between the Various Forms of a Synthetic Flavylium Salt. Chem. Eur. J. 1998, 4, 1184. [Google Scholar] [CrossRef]
- Pina, F.; Melo, M.J.; Parola, A.J.; Maestri, M.; Balzani, V. pH-Controlled Photochromism of Hydroxyflavylium Ions. Chem. Eur. J. 1998, 4, 2001. [Google Scholar] [CrossRef]
- Silva, G.T.M.; Silva, C.; Silva, K.M.; Pioli, R.M.; Costa, T.S.; Marto, V.V.; Freitas, A.A.; Rozendo, J.; Martins, L.M.O.S.; Cavalcante, V.F.; et al. Fluorescence and Phosphorescence of Flavylium Cation Analogues of Anthocyanins. Photochem 2022, 2, 423–434. [Google Scholar] [CrossRef]
- Mora-Soumille, N.; Al Bittar, S.; Rosa, M.; Olivier Dangles, O. Analogs of anthocyanins with a 3′,4′-dihydroxy substitution: Synthesis and investigation of their acid–base, hydration, metal binding and hydrogen-donating properties in aqueous solution. Dye. Pigment. 2013, 96, 7–15. [Google Scholar] [CrossRef]
- Moor, K.J.; Schmitt, M.; Erickson, P.R.; McNeill, K. Sorbic acid as a triplet probe: Triplet energy and reactivity with triplet-state dissolved organic matter via 1O2. Environ. Sci. Technol. 2019, 53, 8078–8086. [Google Scholar] [CrossRef]
- Pina, F.; Melo, M.J.; Maestri, M.; Passaniti, P.; Camaioni, N.; Balzani, V. Photo- and pH-induced transformations of flavylium cation: “Write-Lock-Read-Unlock-Erase” cycles. Eur. J. Org. Chem. 1999, 1999, 3199–3207. [Google Scholar] [CrossRef]
- Johns, V.K.; Wang, Z.; Li, X.; Liao, Y. Physicochemical study of a metastable-state photoacid. J. Phys. Chem. A 2013, 117, 13101–13104. [Google Scholar] [CrossRef]
- Shi, Z.; Peng, P.; Strohecker, D.; Liao, Y. Long-lived photoacid based upon a photochromic reaction. J. Am. Chem. Soc. 2011, 133, 14699–14703. [Google Scholar] [CrossRef]
- Halbritter, T.; Kaiser, C.; Wachtveitl, J.; Heckel, A. Pyridine-spiropyran derivative as a persistent, reversible photoacid in water. J. Org. Chem. 2017, 82, 8040–8047. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.; Basilashvili, K.; Yoo, B.; Lee, J.I. pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria. Colorants 2023, 2, 58-72. https://doi.org/10.3390/colorants2010005
Kang J, Basilashvili K, Yoo B, Lee JI. pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria. Colorants. 2023; 2(1):58-72. https://doi.org/10.3390/colorants2010005
Chicago/Turabian StyleKang, Jeonghee, Ketevan Basilashvili, Barney Yoo, and Jong I. Lee. 2023. "pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria" Colorants 2, no. 1: 58-72. https://doi.org/10.3390/colorants2010005
APA StyleKang, J., Basilashvili, K., Yoo, B., & Lee, J. I. (2023). pH-Induced Orthogonal Photoresponse of trans-Chalcone Isomers and Related Compounds in Equilibria. Colorants, 2(1), 58-72. https://doi.org/10.3390/colorants2010005