
Kinetics of Thermally Activated Physical Processes in Disordered Media



Abstract
:1. Problem: Dynamic Evolution of Physical Quantities in Disordered Media
1.1. Under an External Action
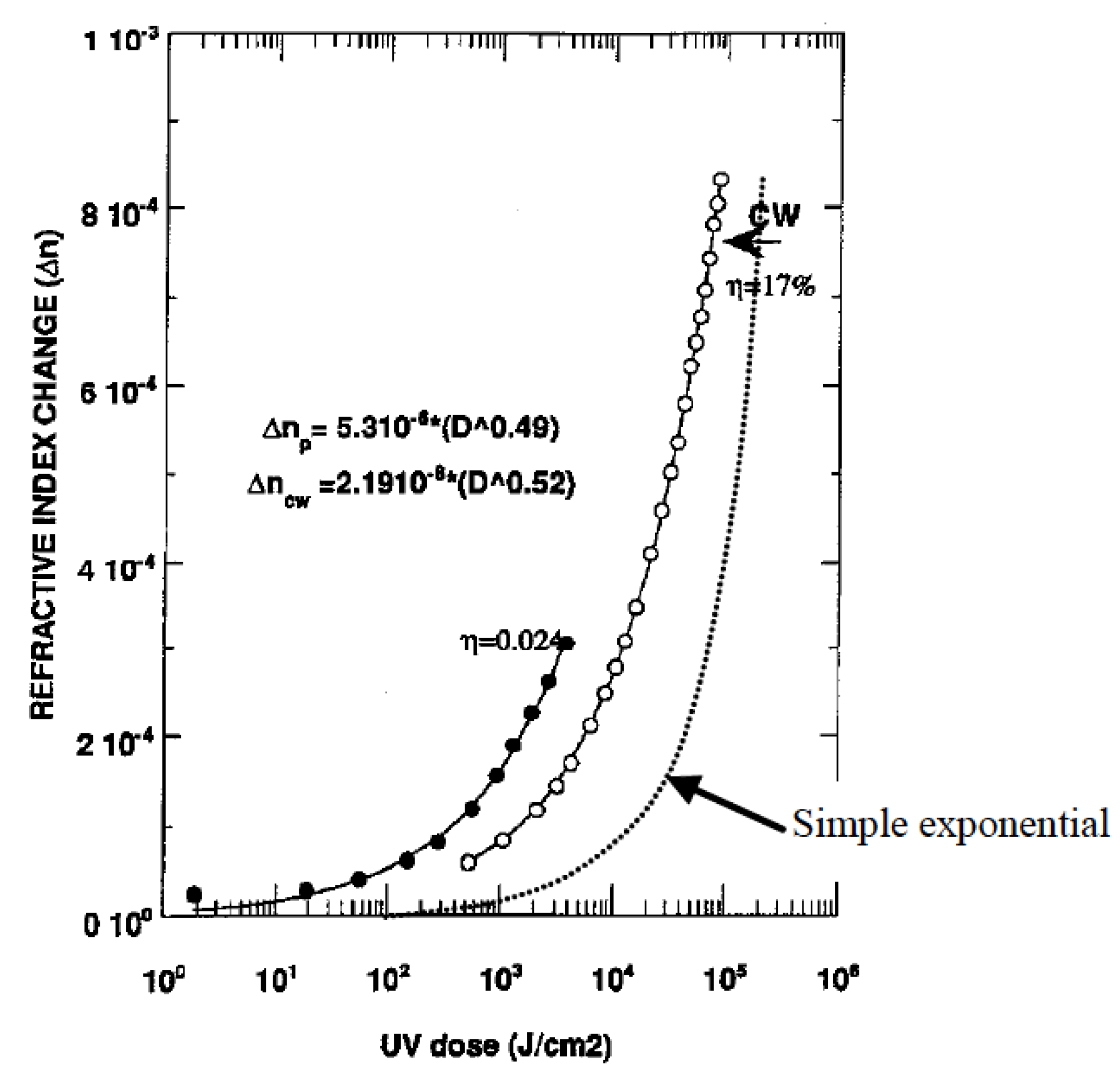
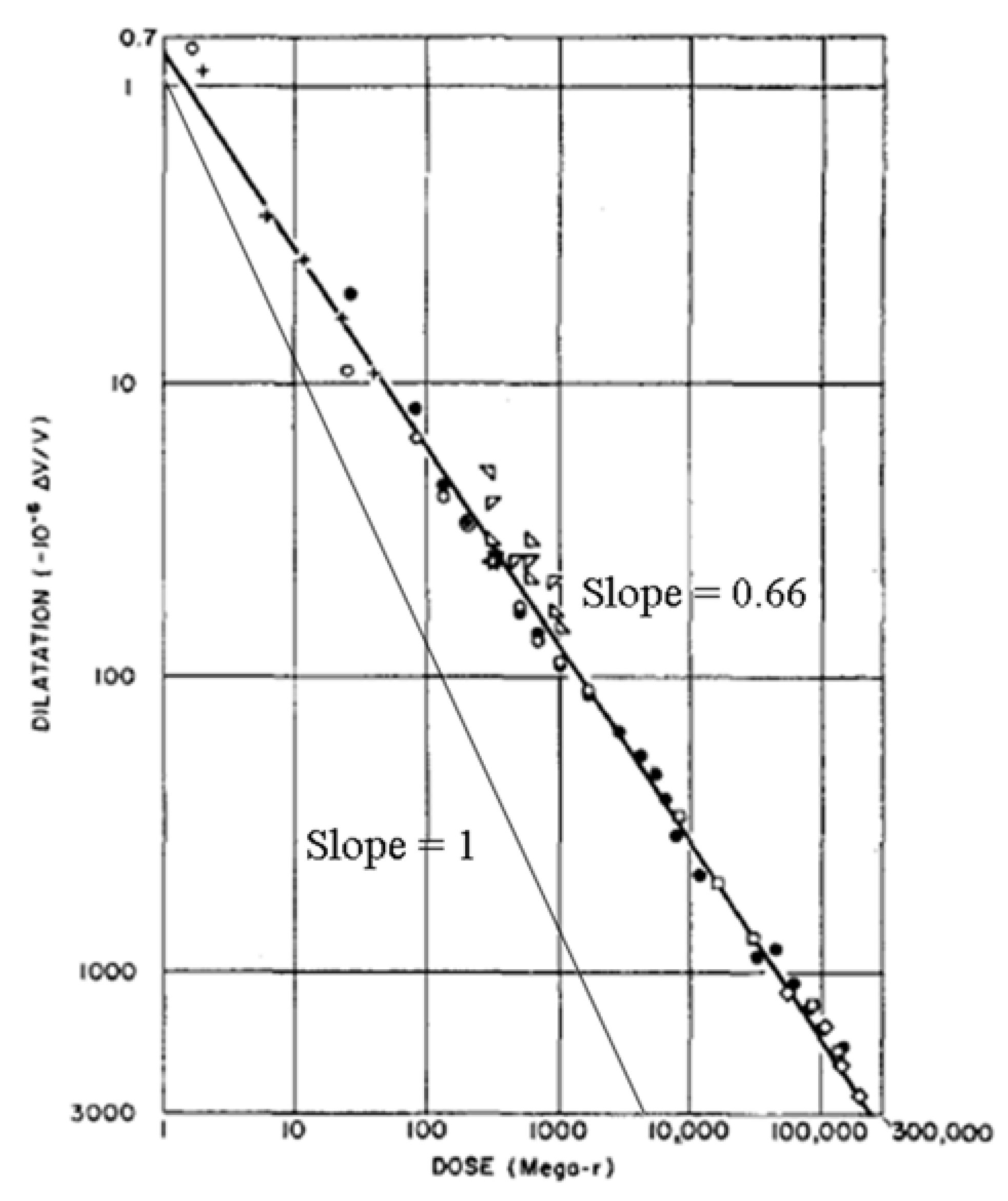
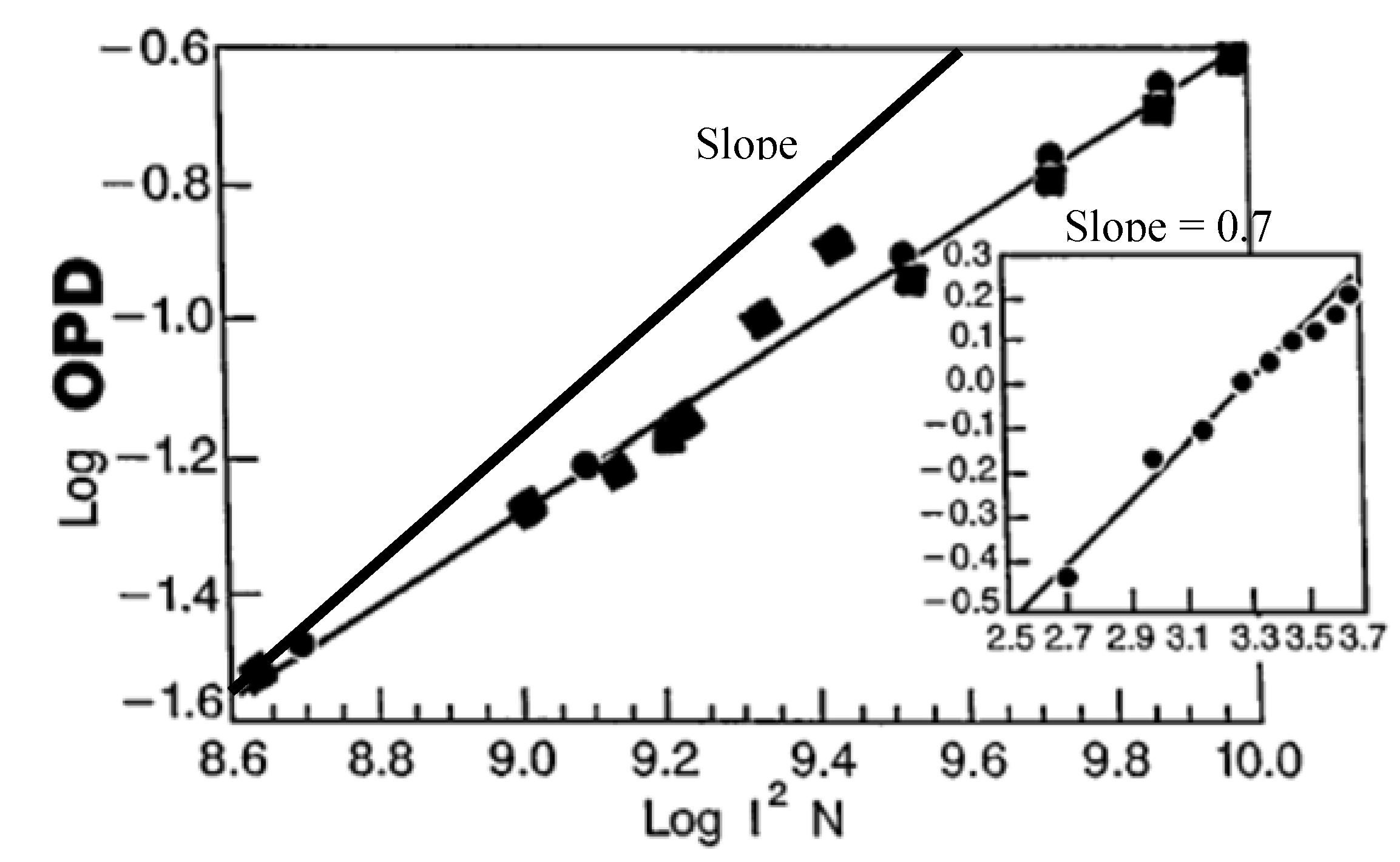
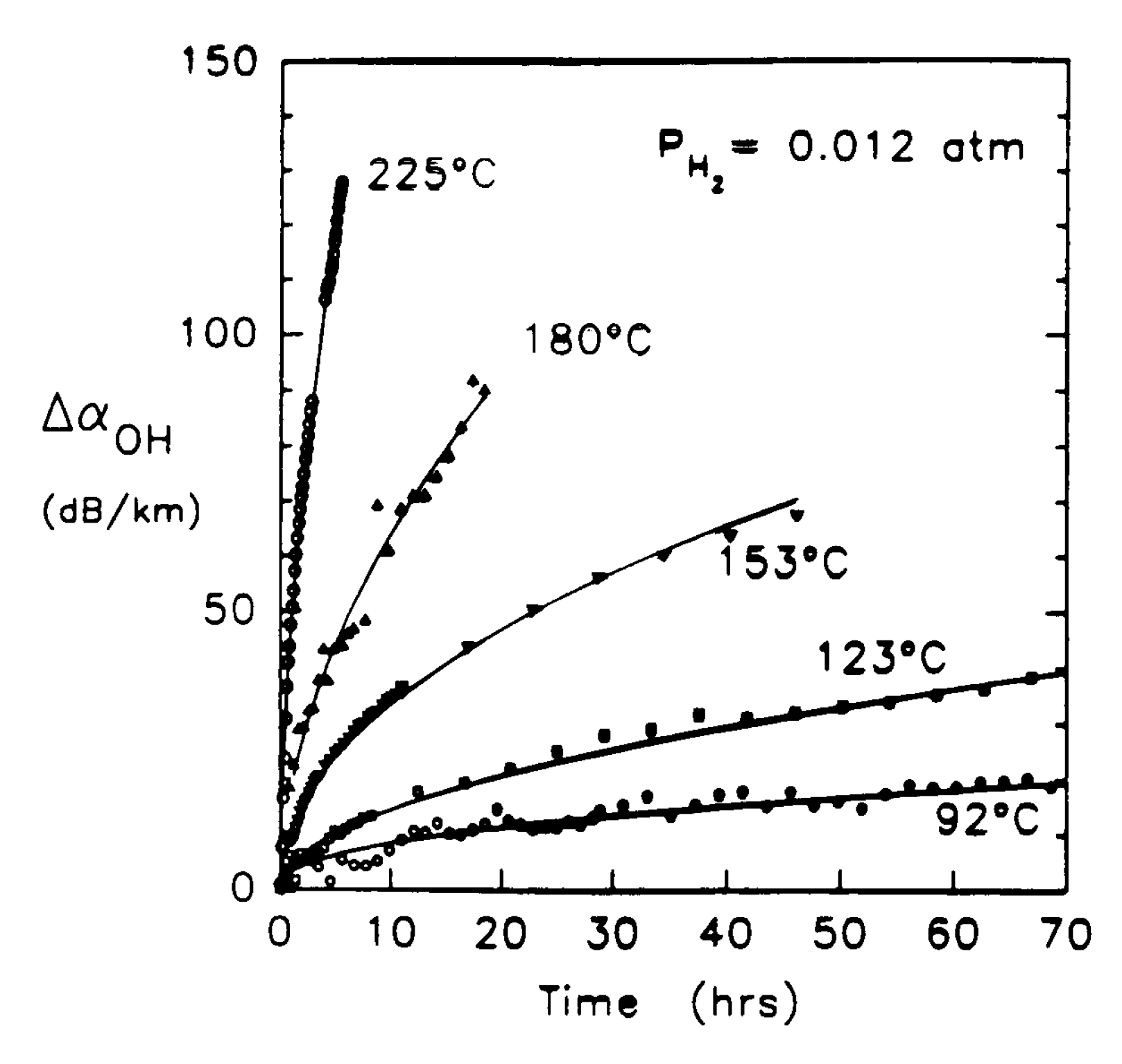
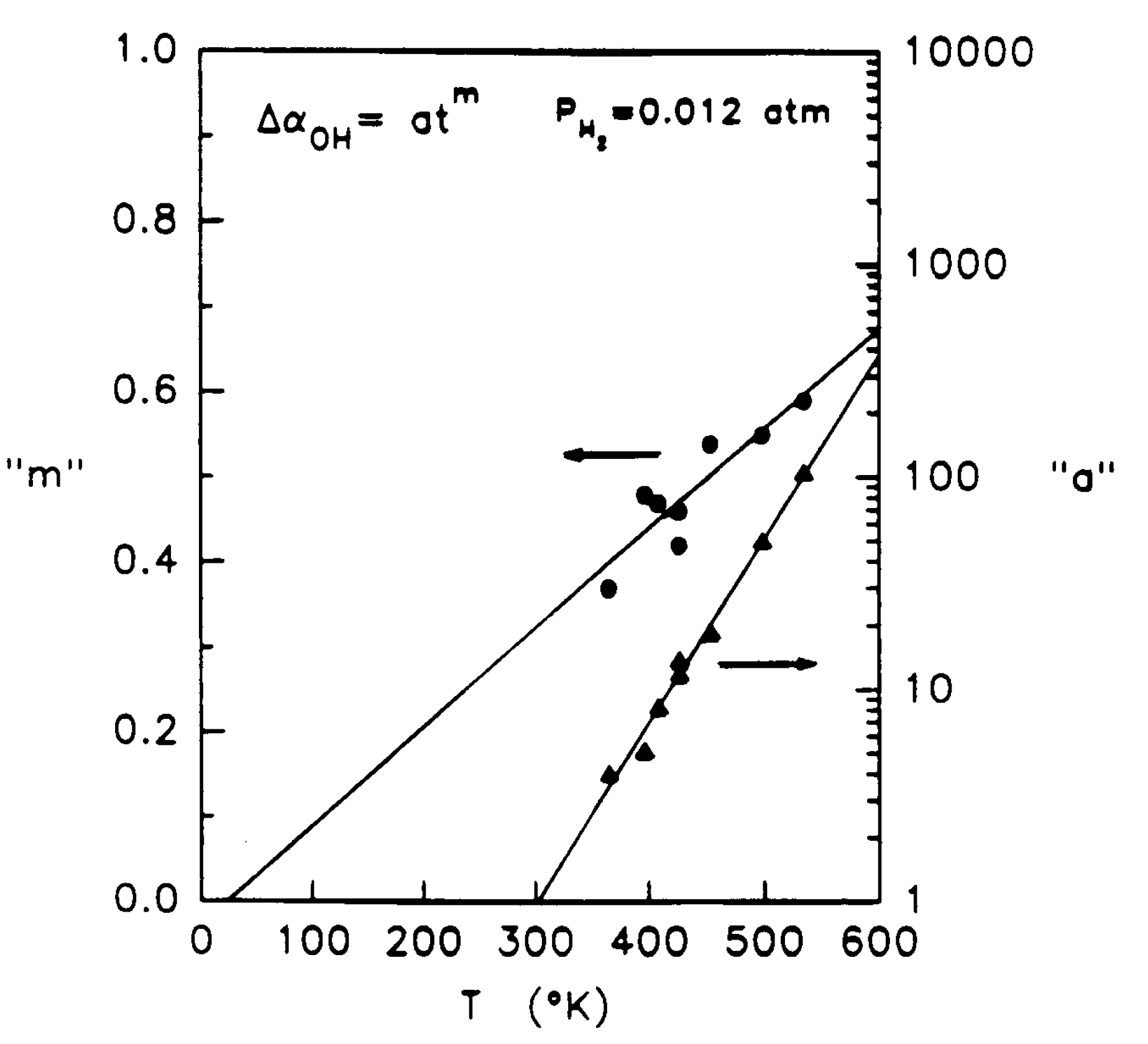
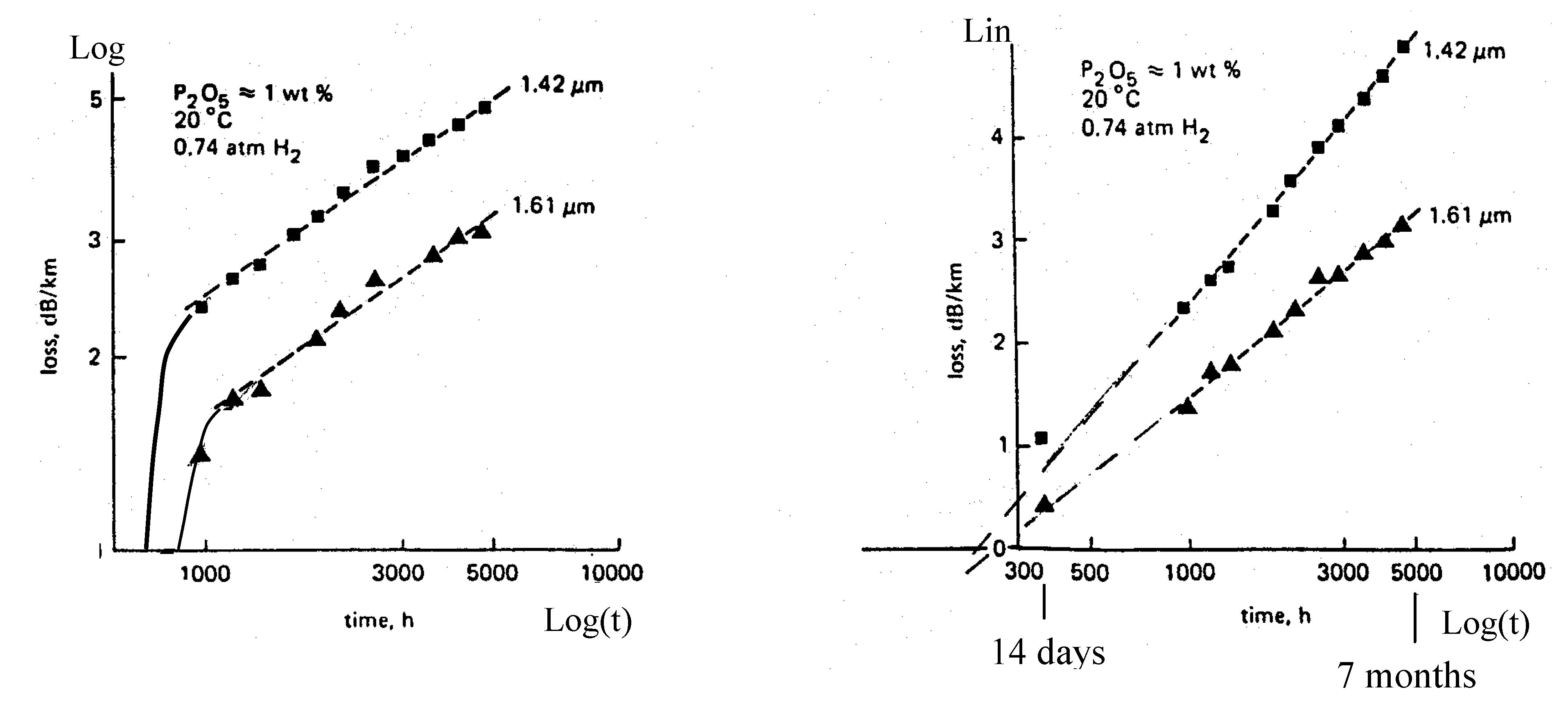
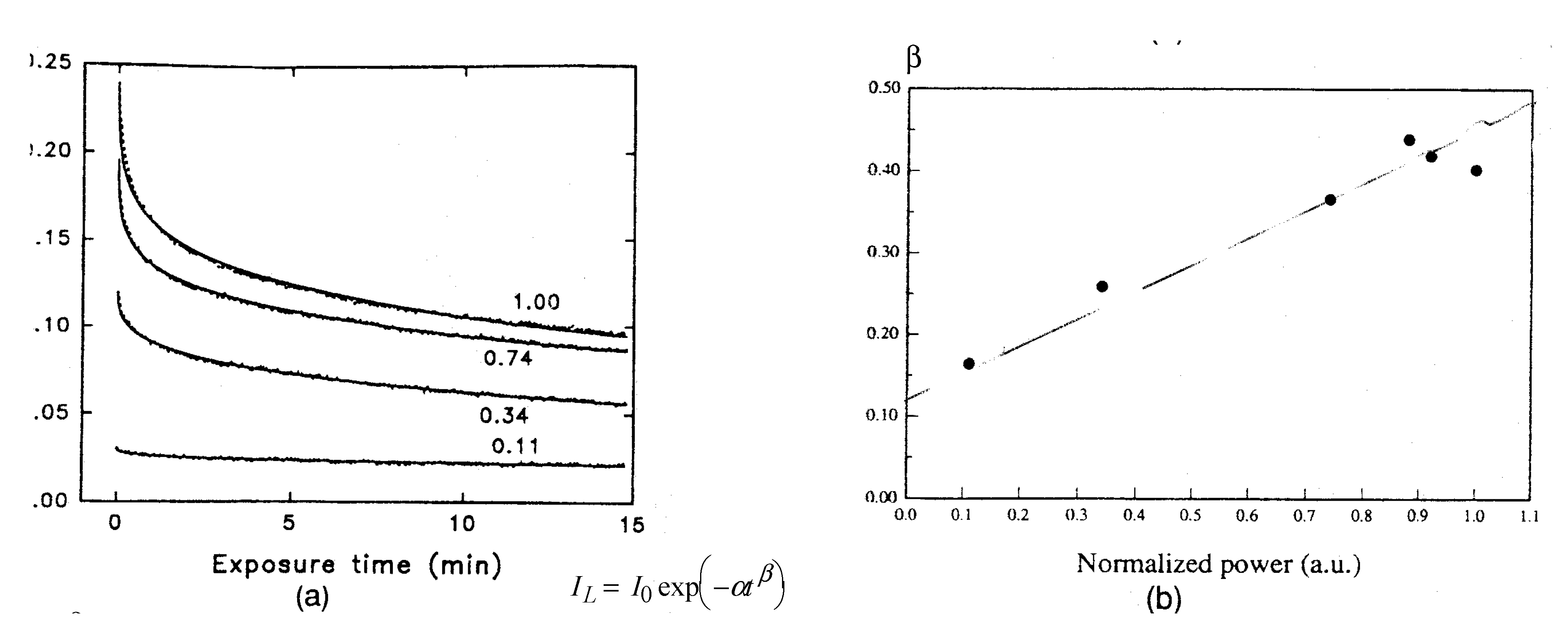
1.2. Thermal Relaxation
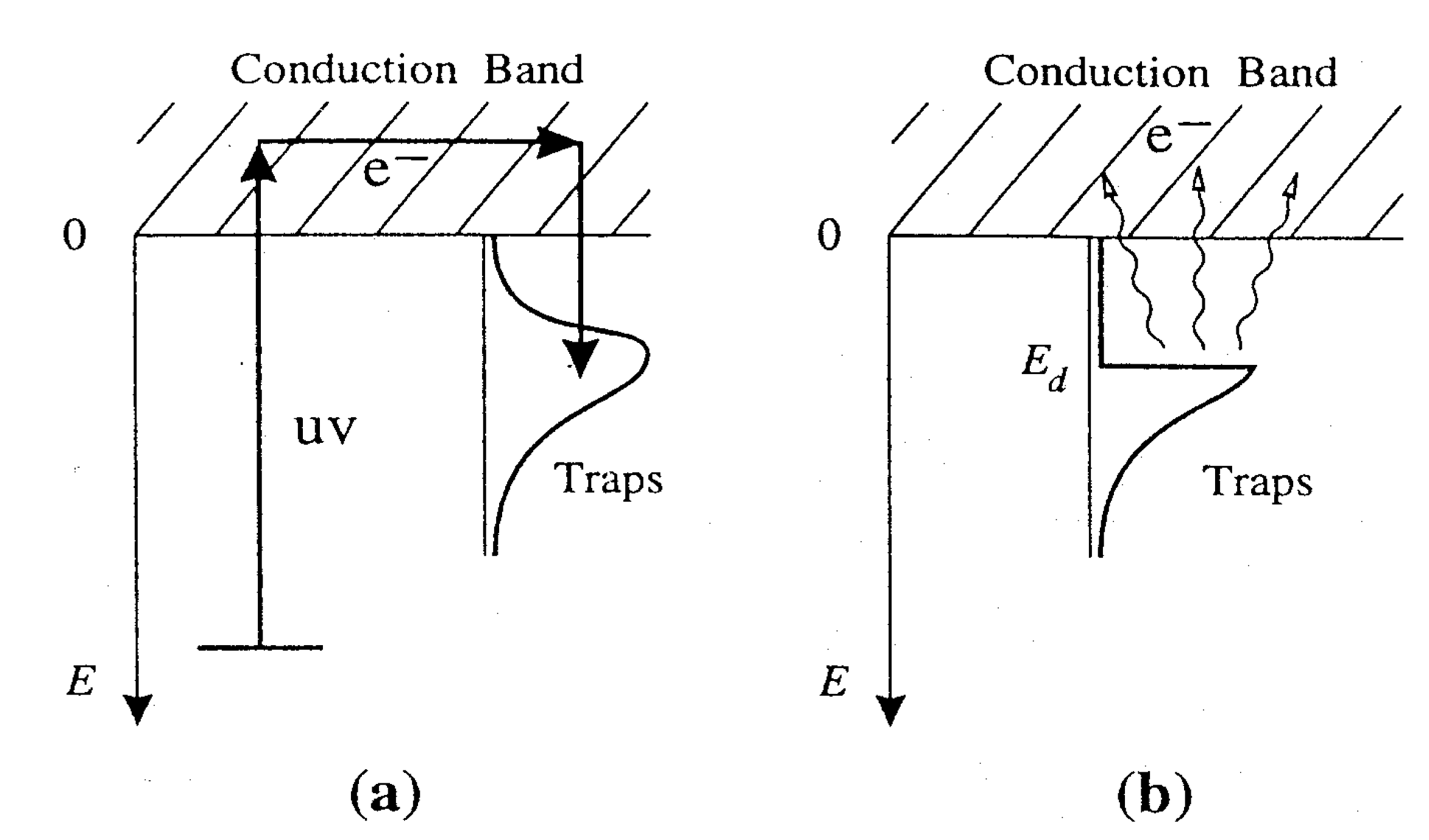
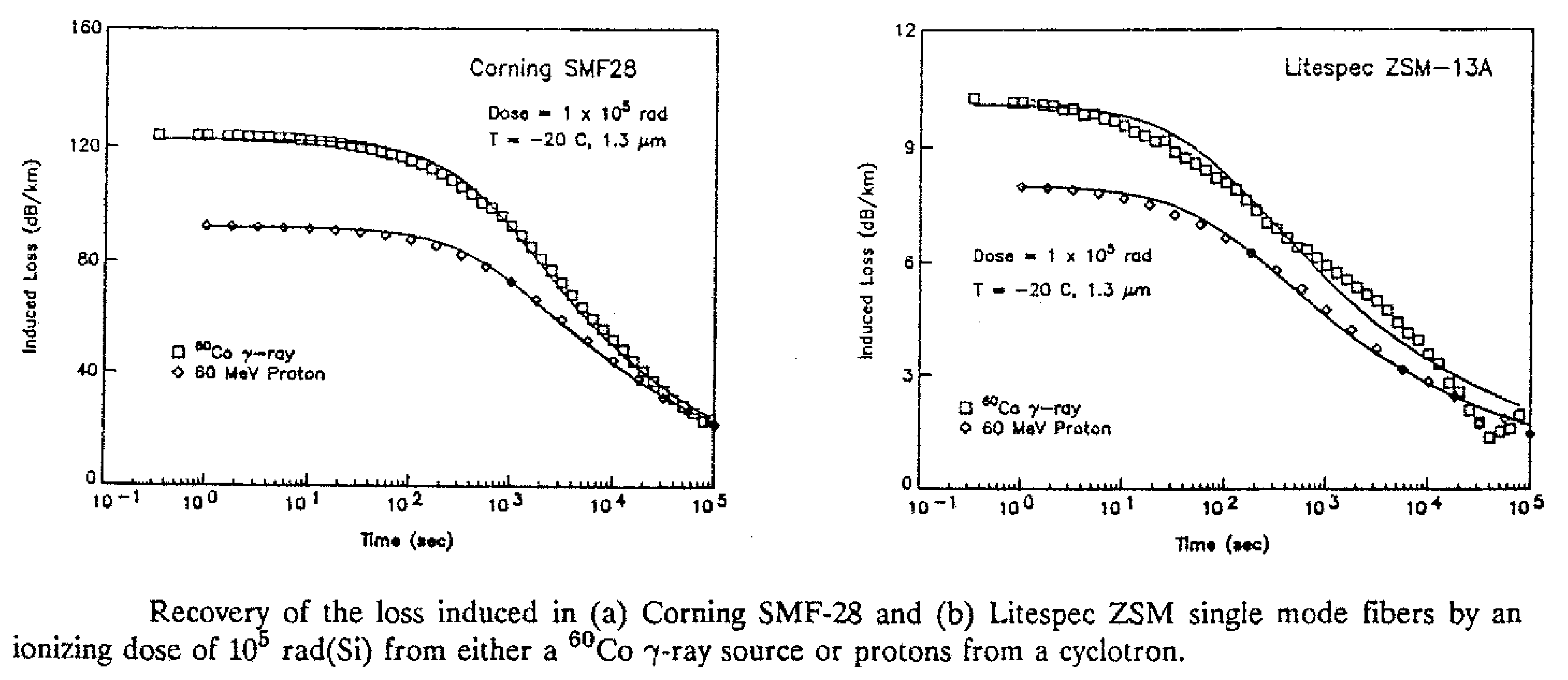
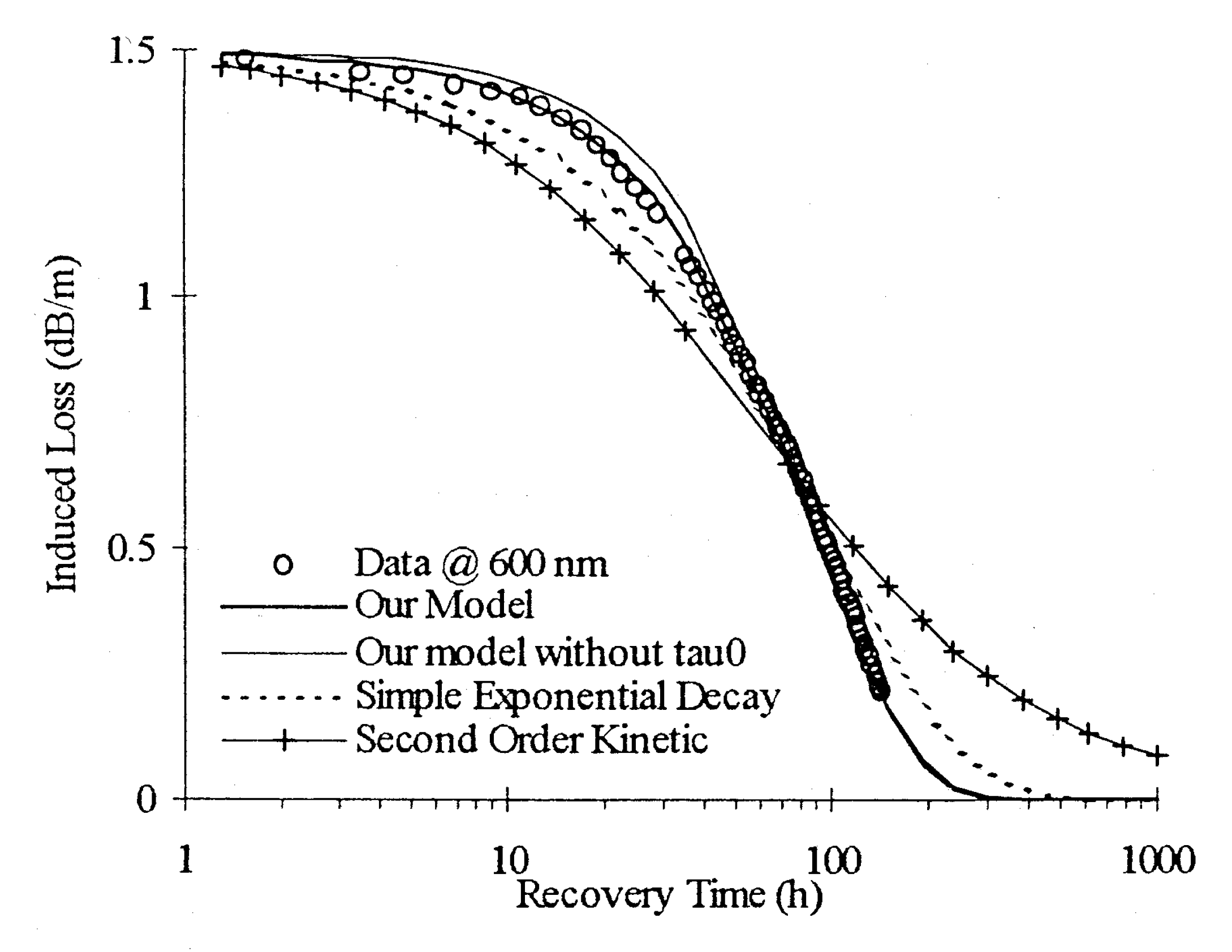
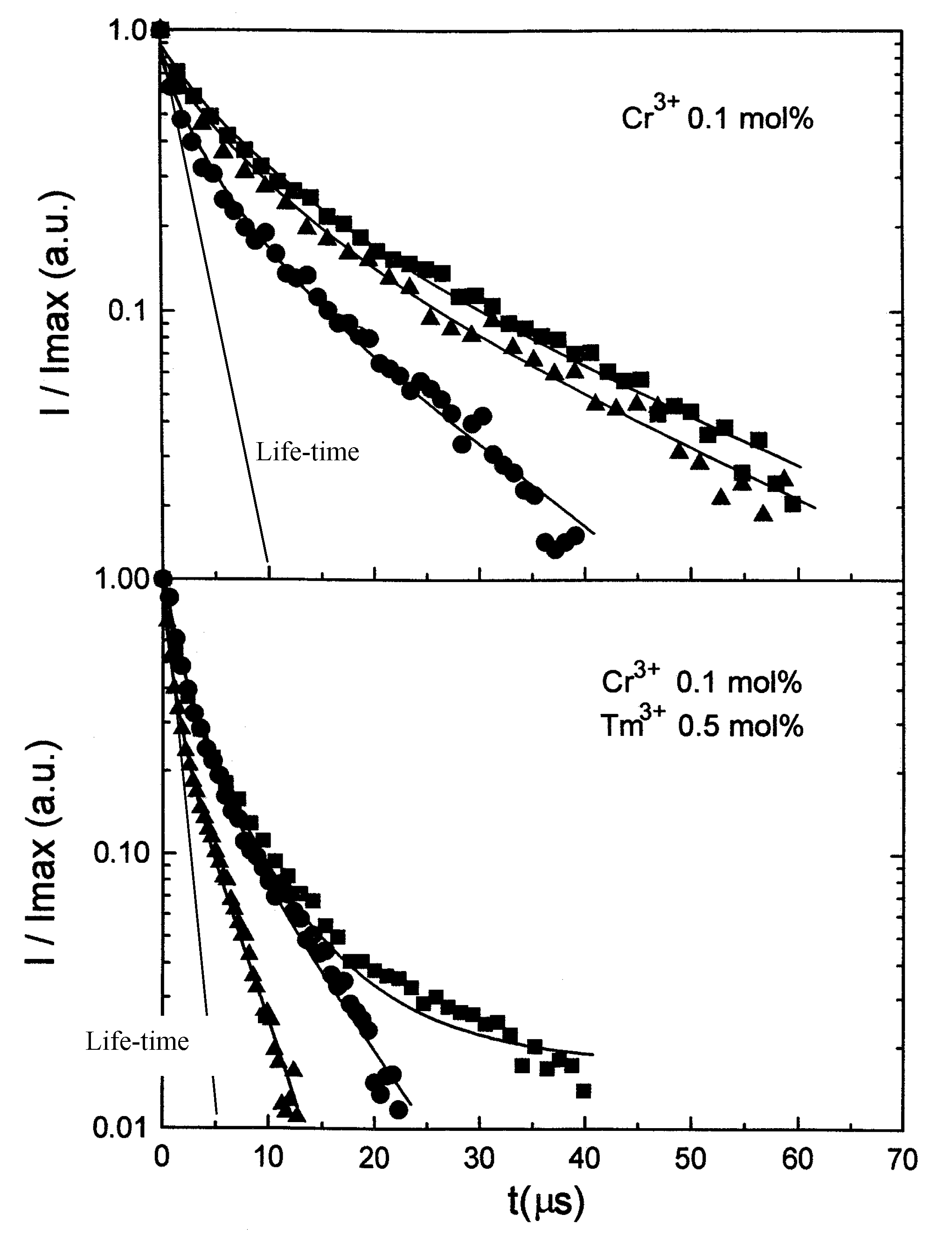


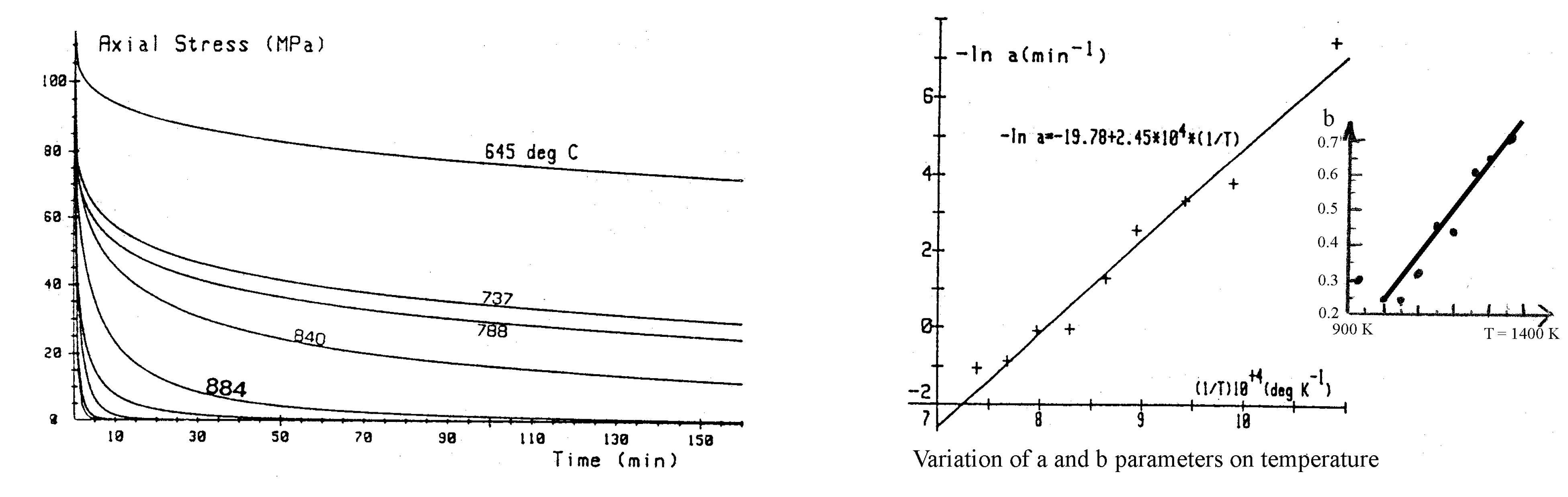
2. Modeling
2.1. Exponential versus Non Exponential Kinetics (Introduction to the Theory)
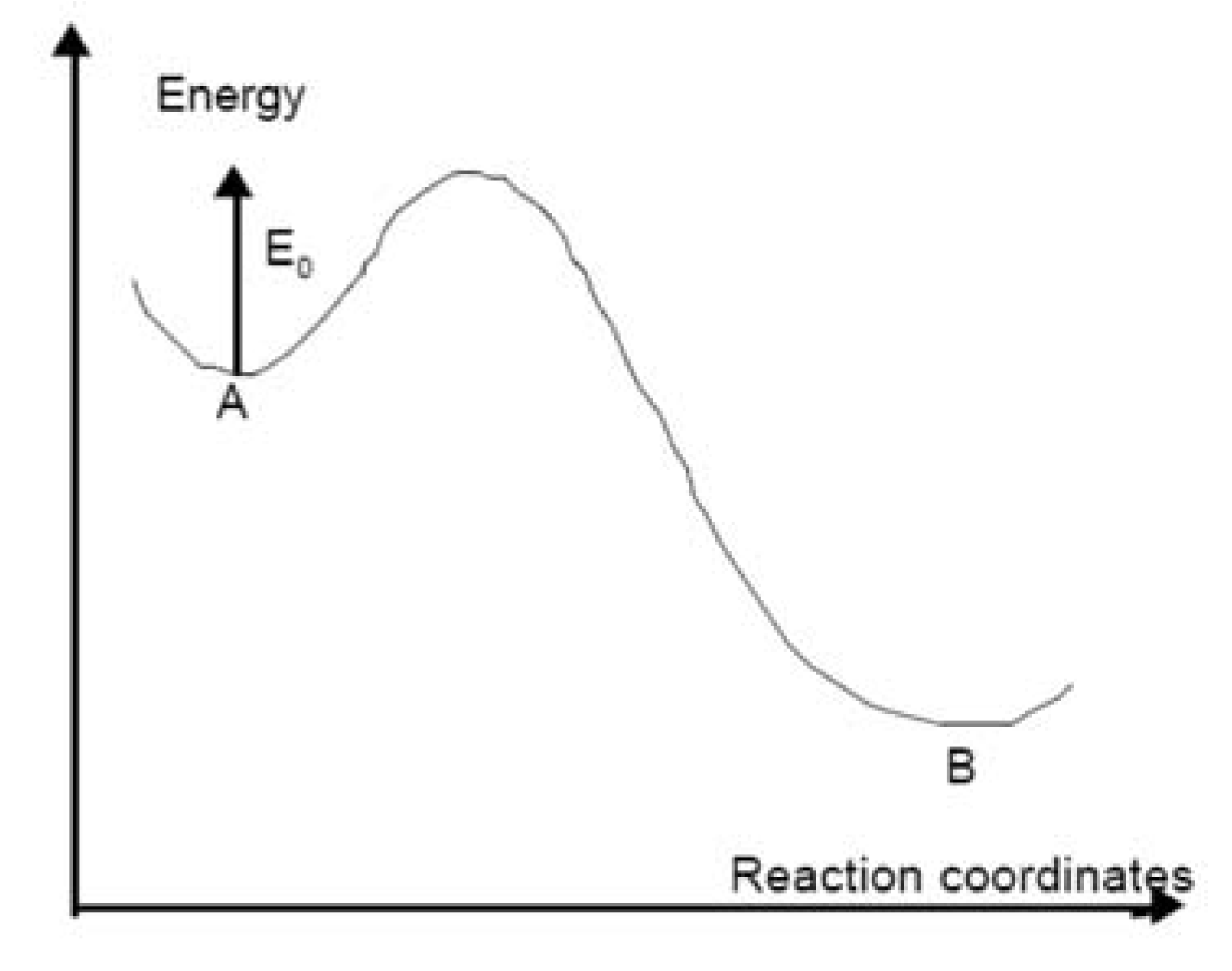
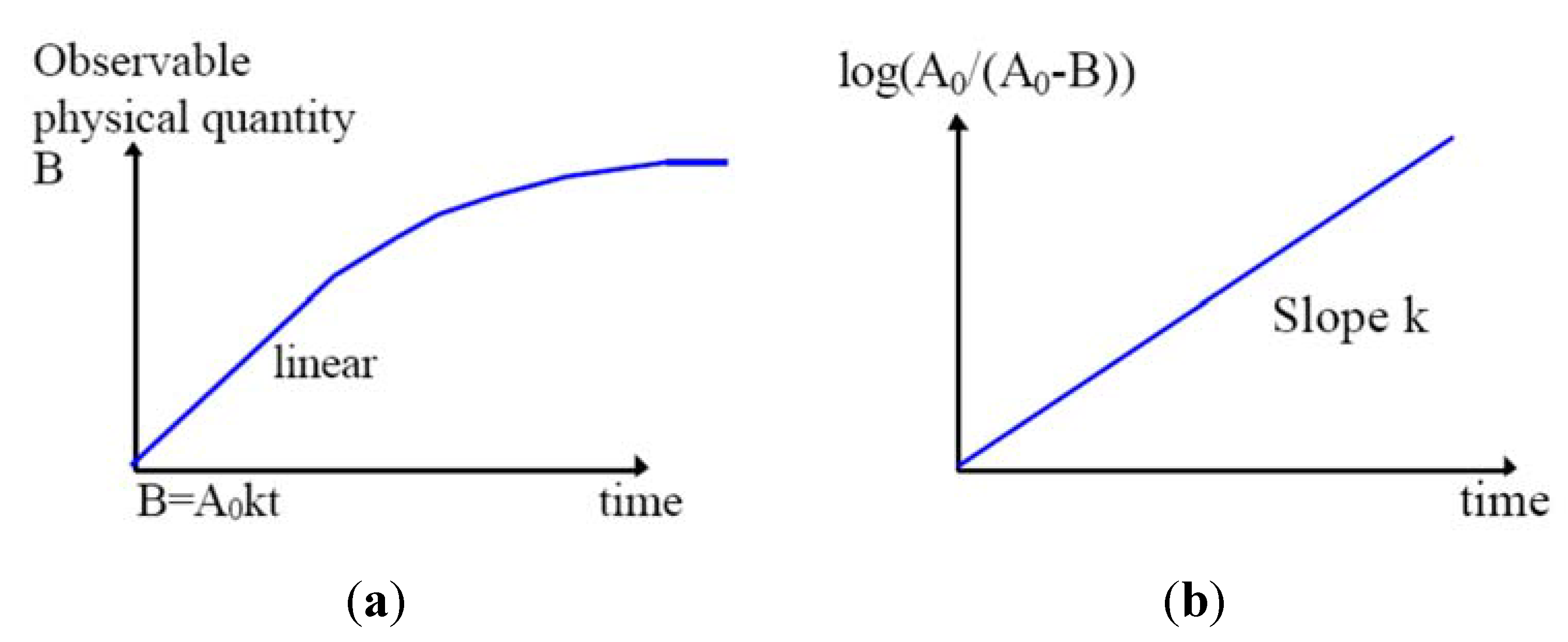
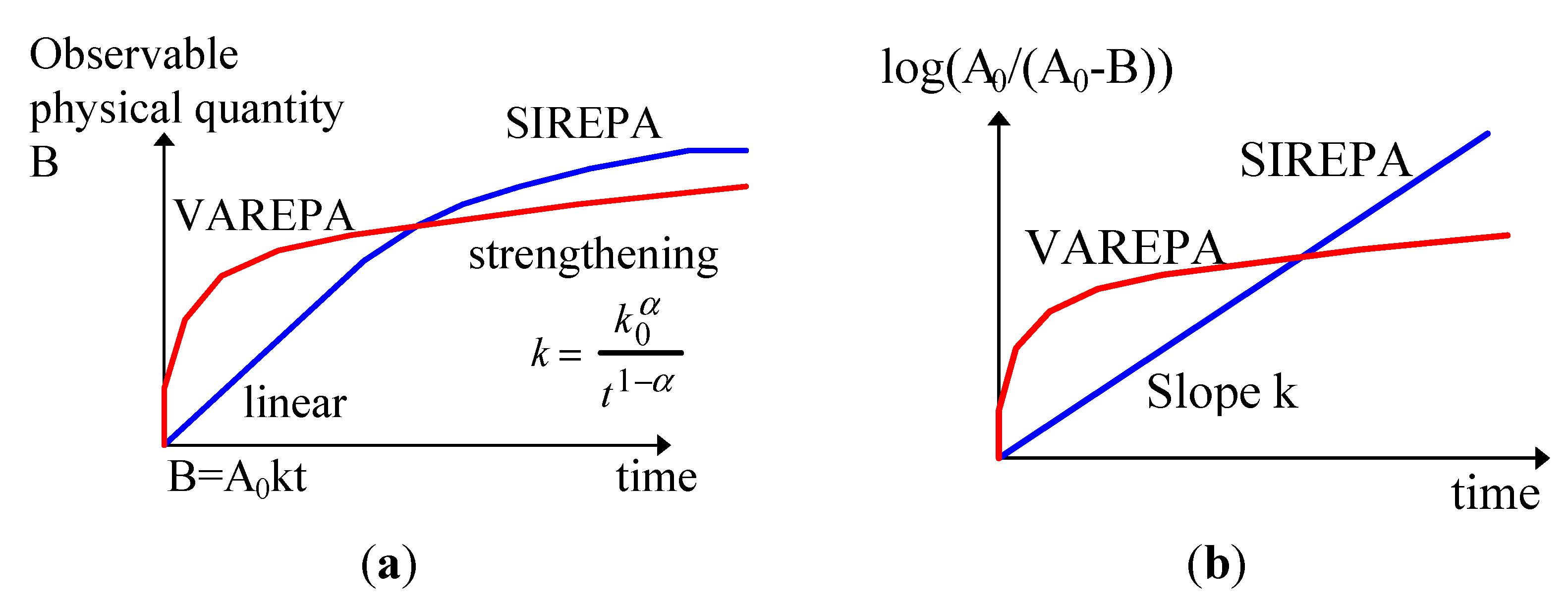

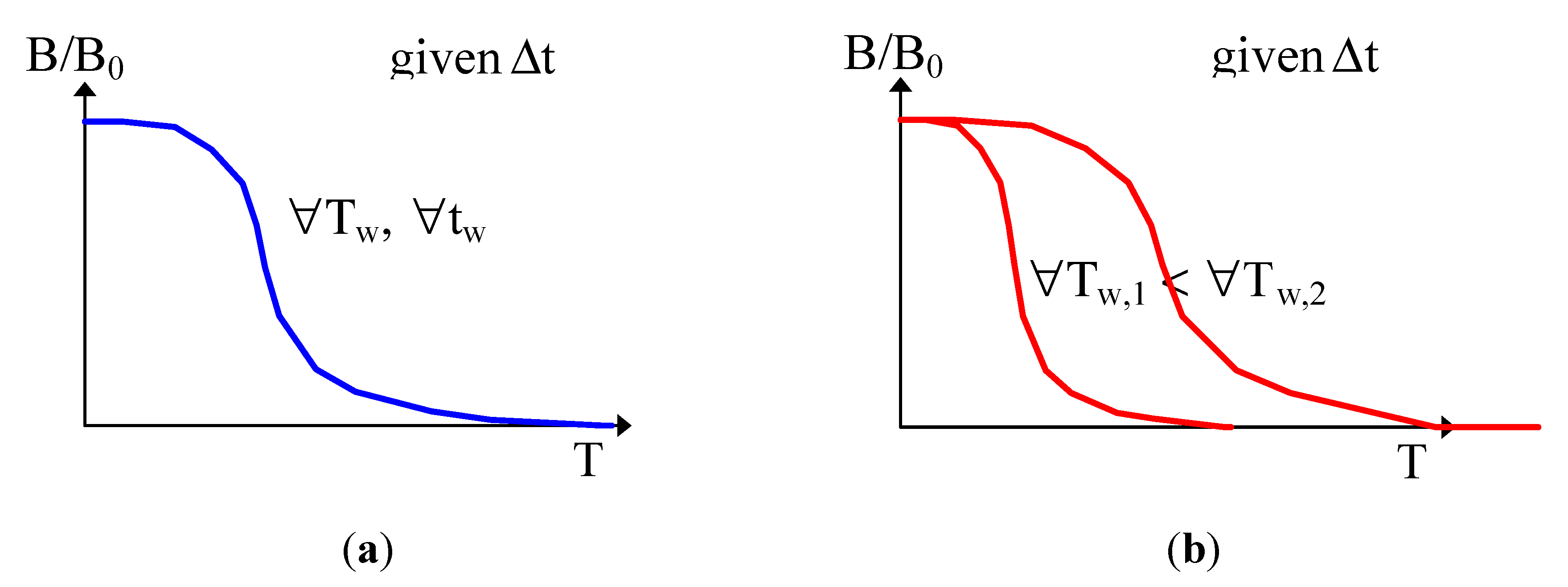
2.2. Kinetics of Writing
2.2.1. First Group of Assumptions
- (1)
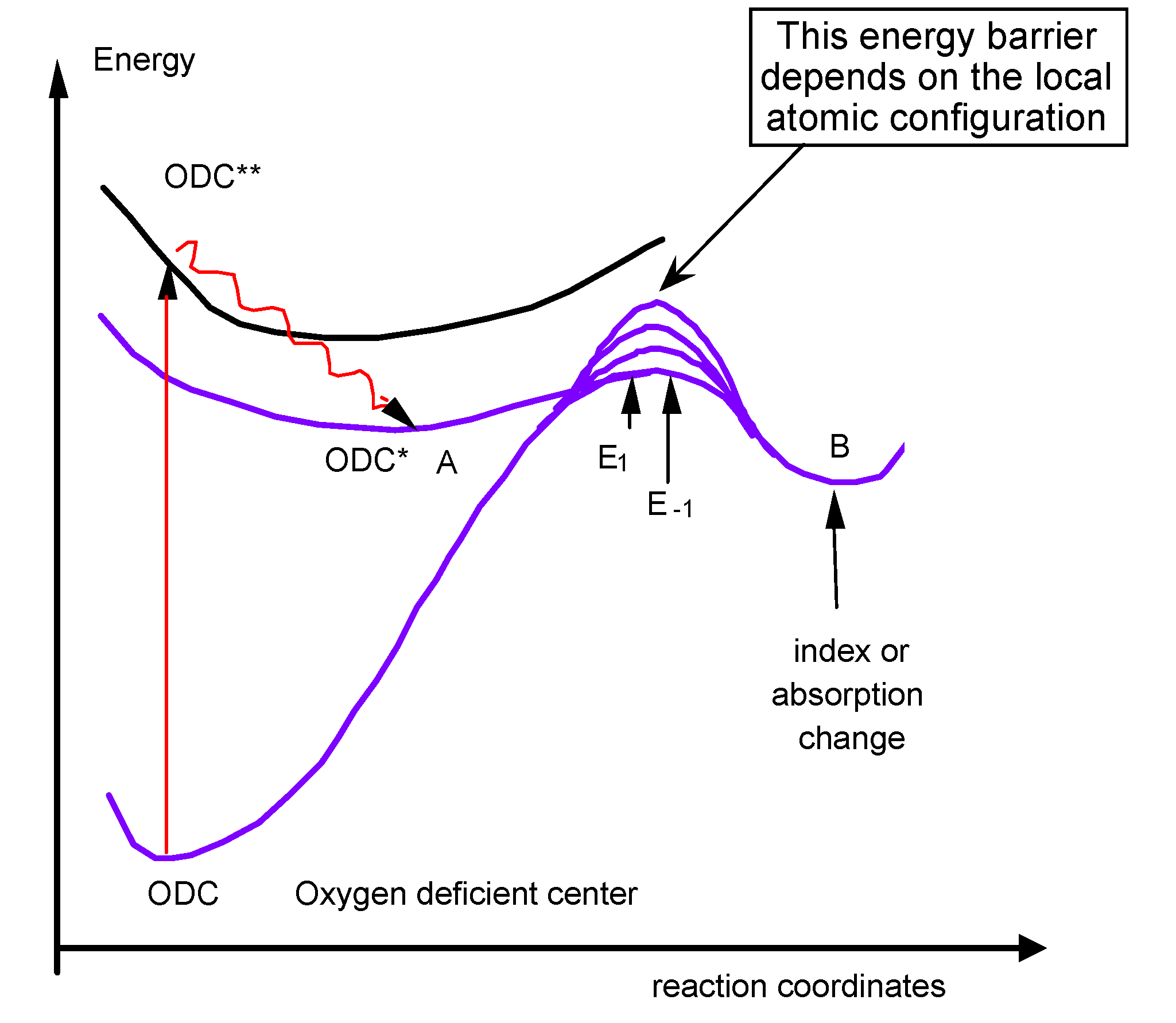
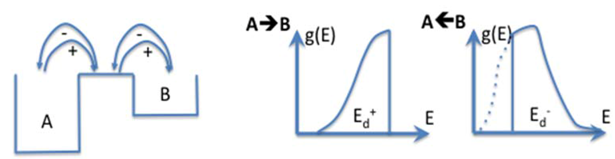
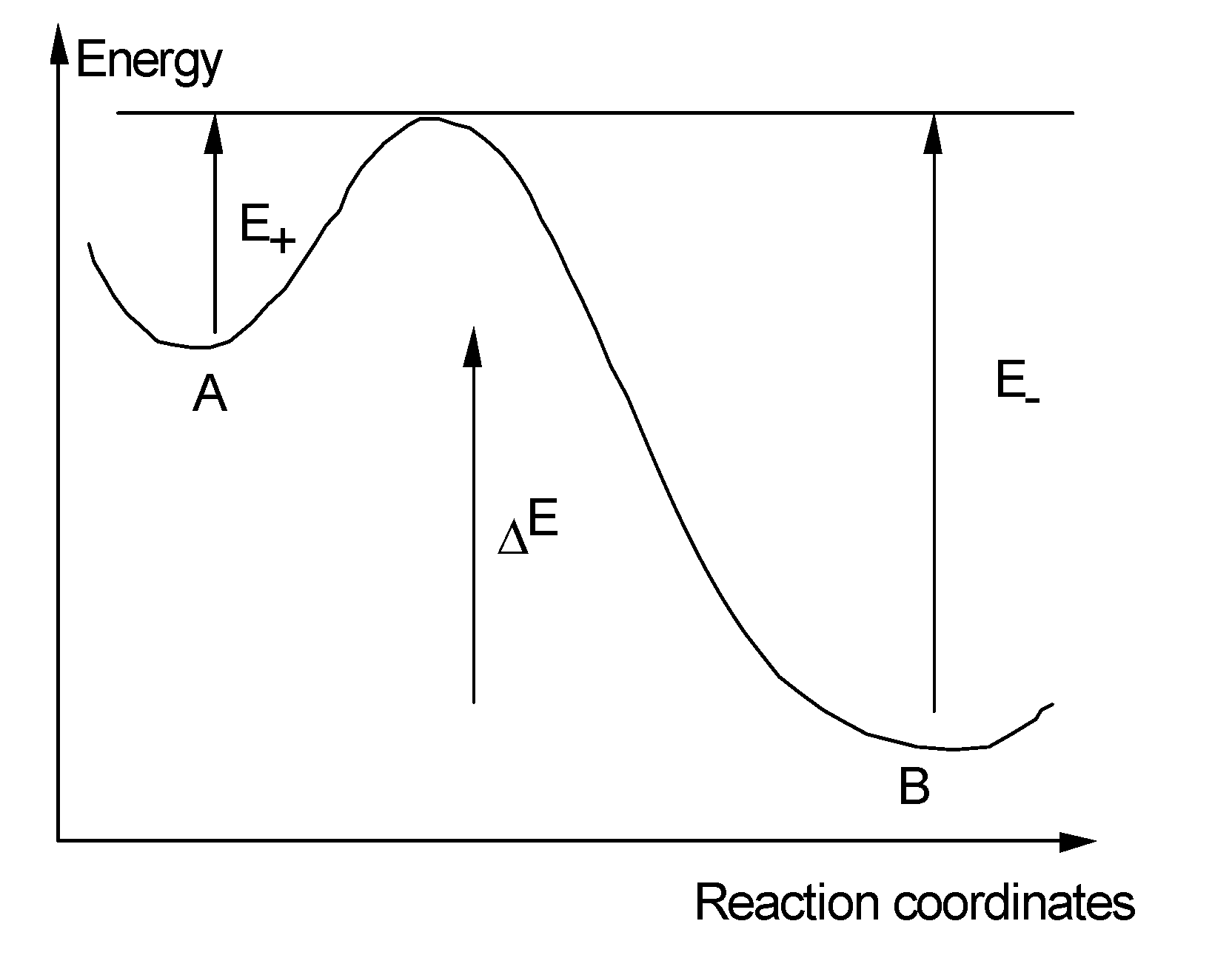
- We can easily imagine that the reaction leading to refractive index change contains an absorption step on a species (for instance Oxygen Deficient Center), followed sometimes by a relaxation to another excited level (Figure 20). Then, several pathways are possible (one to luminescence, another one to bond breaking, migration, and structural rearrangement). All of them finally contribute to refractive index change. The first assumption we make here is that only one elementary reaction is involved: the limiting reaction of the process. We can write the reaction A→B.
- (2)
- The second assumption is that it is thermally activated (with an activation energy E). The Arrhenius law can be used, the d[B]/dt rate is proportional to a reaction constant k0 like k(E, T) = k0exp(−E/kBT), where kB is the Boltzman constant and [B] is the concentration of the B species.
- (3)
- The third assumption is that activation energy is distributed. As a matter of fact, in glasses, E is not unique. The creation energy of B can vary with its atomic environment. The transition state energy (the bottleneck on the reaction pathway) is also sensitive to the disordered environment in such a way that the activation energy, which is the energy difference between B and the transition state, is distributed. This means that chemical pathways are variable depending on the configuration distribution of the transition state and/or the initial stable states throughout the glass. This is at the origin of ergodicity lost because space-time points are no more equivalent. The reaction is faster in some places than in others. This is also heterogeneity, but not at a macroscopic level, just at a microscopic one.
2.2.2. The Assumption on the Reaction (Connection with Time and Temperature)
- (1)
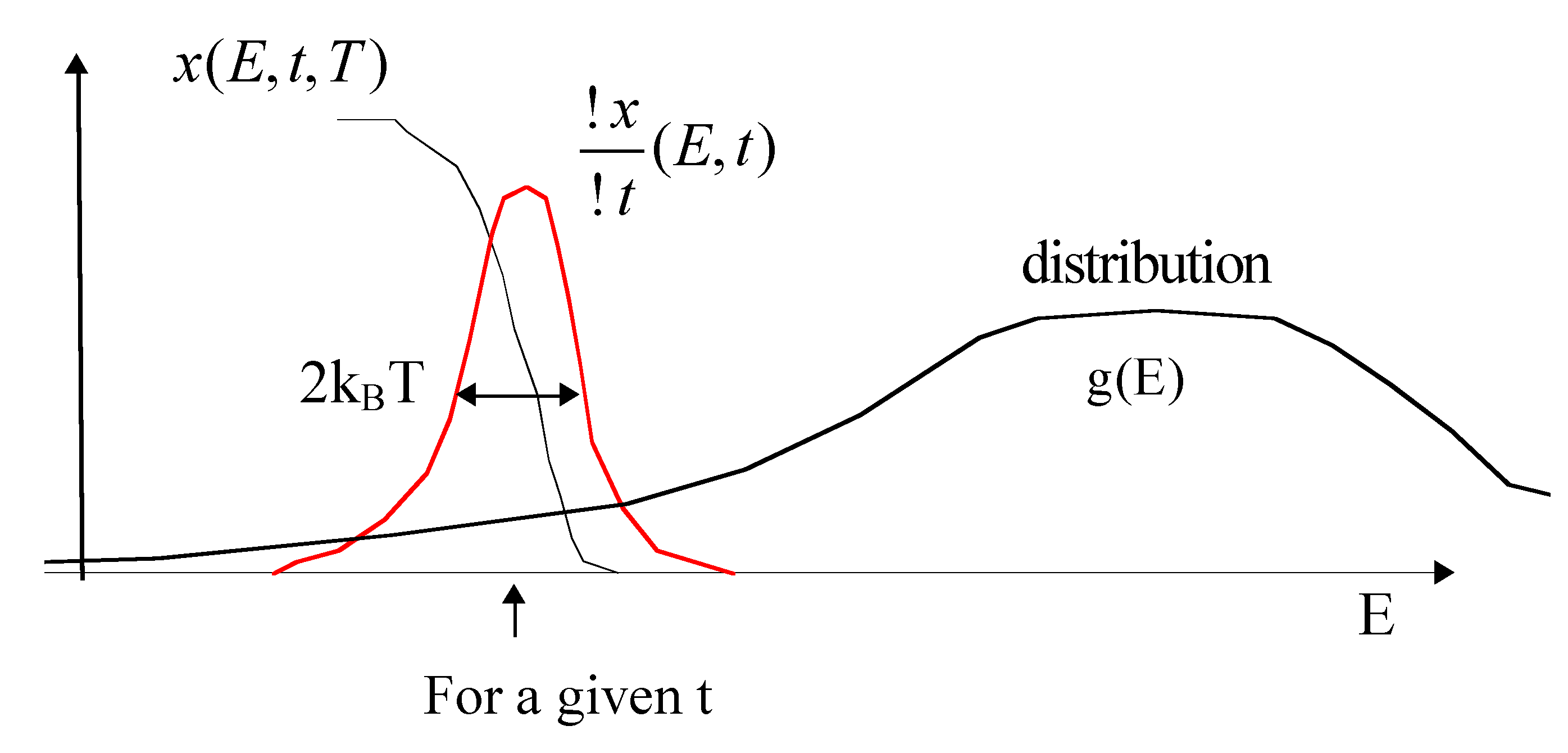
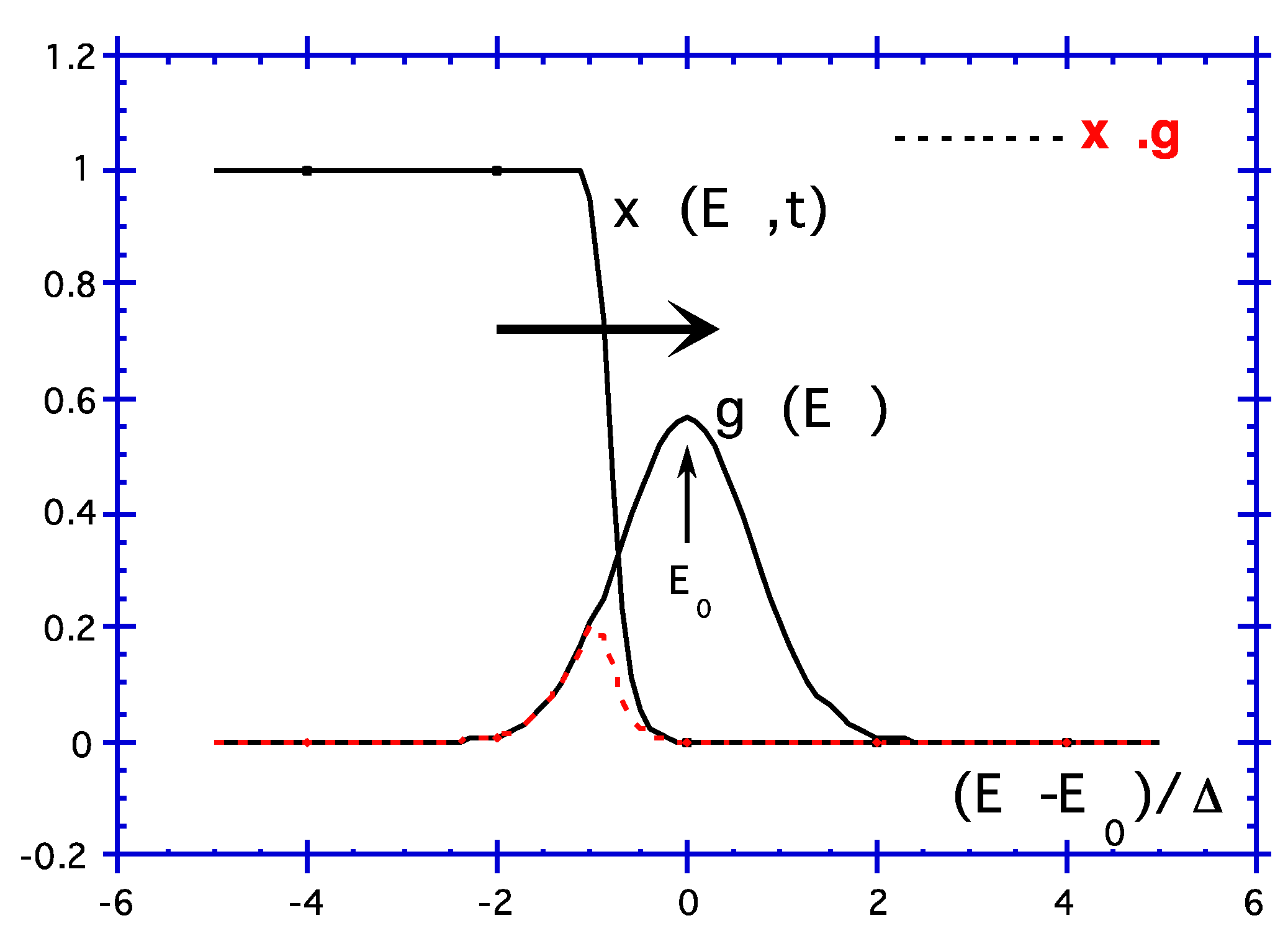
- It is worth noting that the reaction properties appear now only in Ed(t, T), on one hand (the bound of the integral), and, on the other hand, the structural disorder appears in the integrand.
- (2)
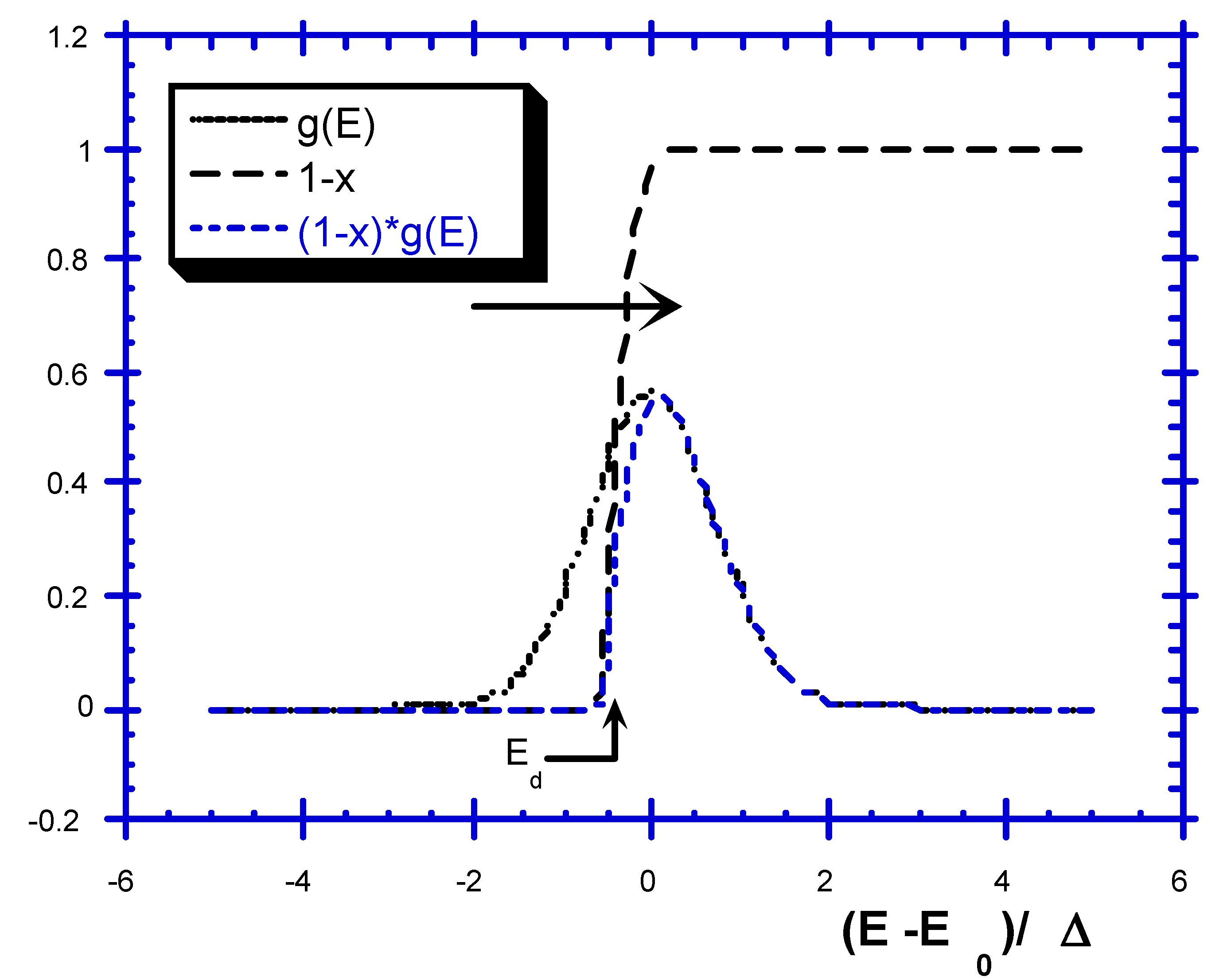
- Note that the demarcation energy approximation is valid only if g(E) varies slower than x(E, t, T). The width of is 2kBT. Otherwise, a correction has to be added to the integral [32]:where ck+1 are constant close to unity (c1 = Euler constant 0.577, c2 = 0.989, c3 = 0.907, c4 = 0.981, c5 also and then equal to 0.999).
- (3)
- Most interesting, the differentiation of B against Ed() yields the shape of the distribution function.
- (4)
- As B is expressed with only one variable B(Ed). It exhibits the same shape as B(T) for given t or B(ln(t)) for given T. Thus, we deduce that if g(E) does not depend on T, isochrons and isotherms are equivalents:
2.2.3. Examples for Time Dependence Computation
- (Step 1)
- Integration using a Gaussian distribution as the following:yields:
- (Step 2)
- The integration of a sigmoid differentiation distribution:yields:
- (Step 3)
- with a Poisson distribution:
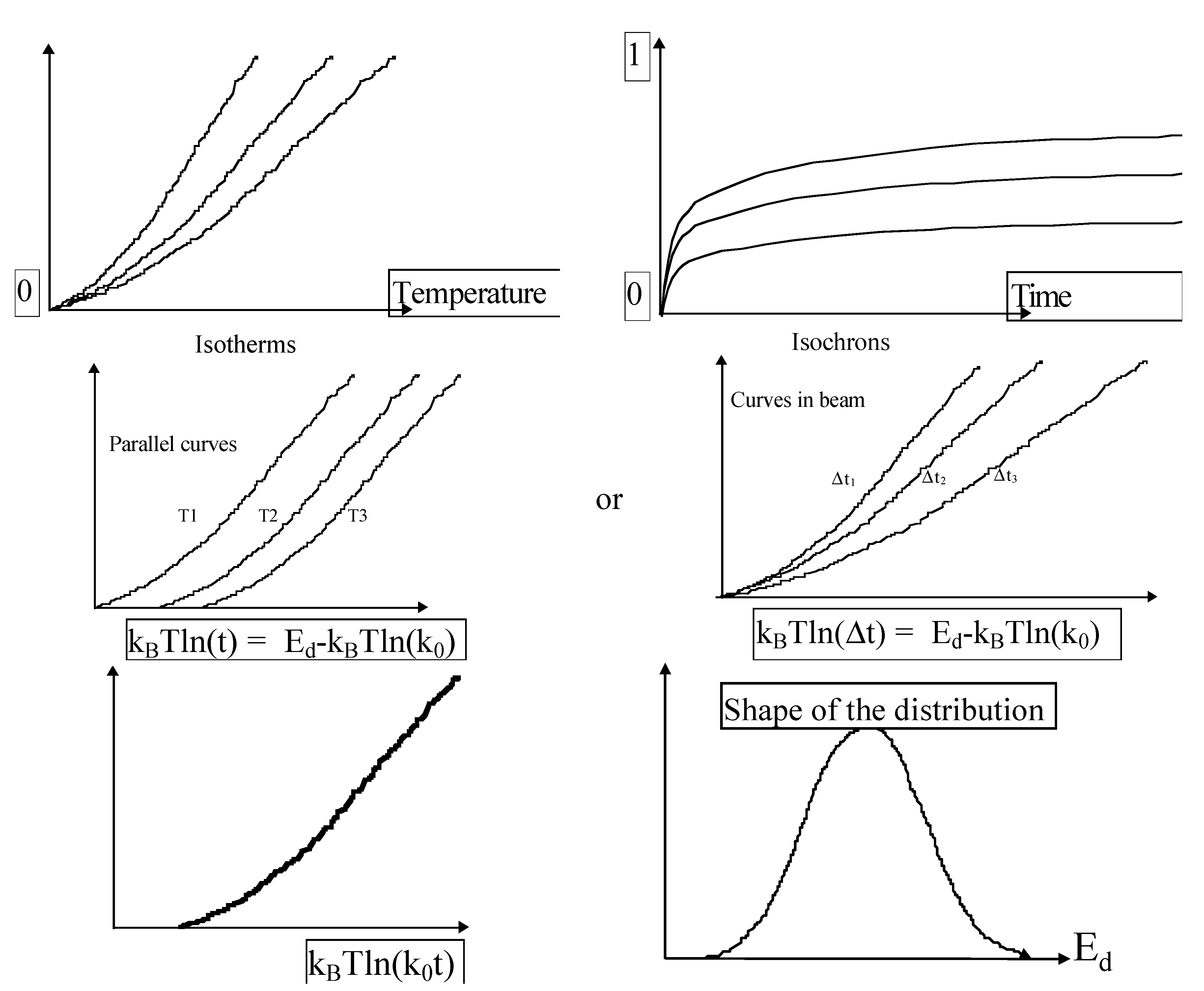
2.2.4. How to Obtain k0 and g(E) (Figure 23)
- (1)
- Measurement of several isochrons or isotherms writing kinetics and replotting against a new abscissa;
- (2)
- k0 fitting for curve collapsing and;
- (3)
- Differentiation .
2.3. Kinetics of Erasing
2.3.1. First Group of Assumptions
- (1)
- One elementary reaction (limiting process), A→B is the writing process;
- (2)
- Thermal activation, E−, ;
- (3)
- E− is distributed, i.e.,
- -
- variable chemical pathways;
- -
- the saddle point or the B state are sensitive to various structural configurations as decrived in Figure 24;
- -
- g(E) is the distribution function for backward reaction, it can be Gaussian for a thermal disorder or Poisson for a diffusional disorder or differentiation of a sigmoid, or also top hat function.

2.3.2. Second Group of Assumptions (towards the Computation of Time Dependence)
- (1)
- Most of the elementary reactions in solids are first order; some are second order, when two species associate, but these are much less likely. Thus, this is not so severe and we can write: , , is the advancement degree of the reaction.
- (2)
- Concept of demarcation energy, Ed
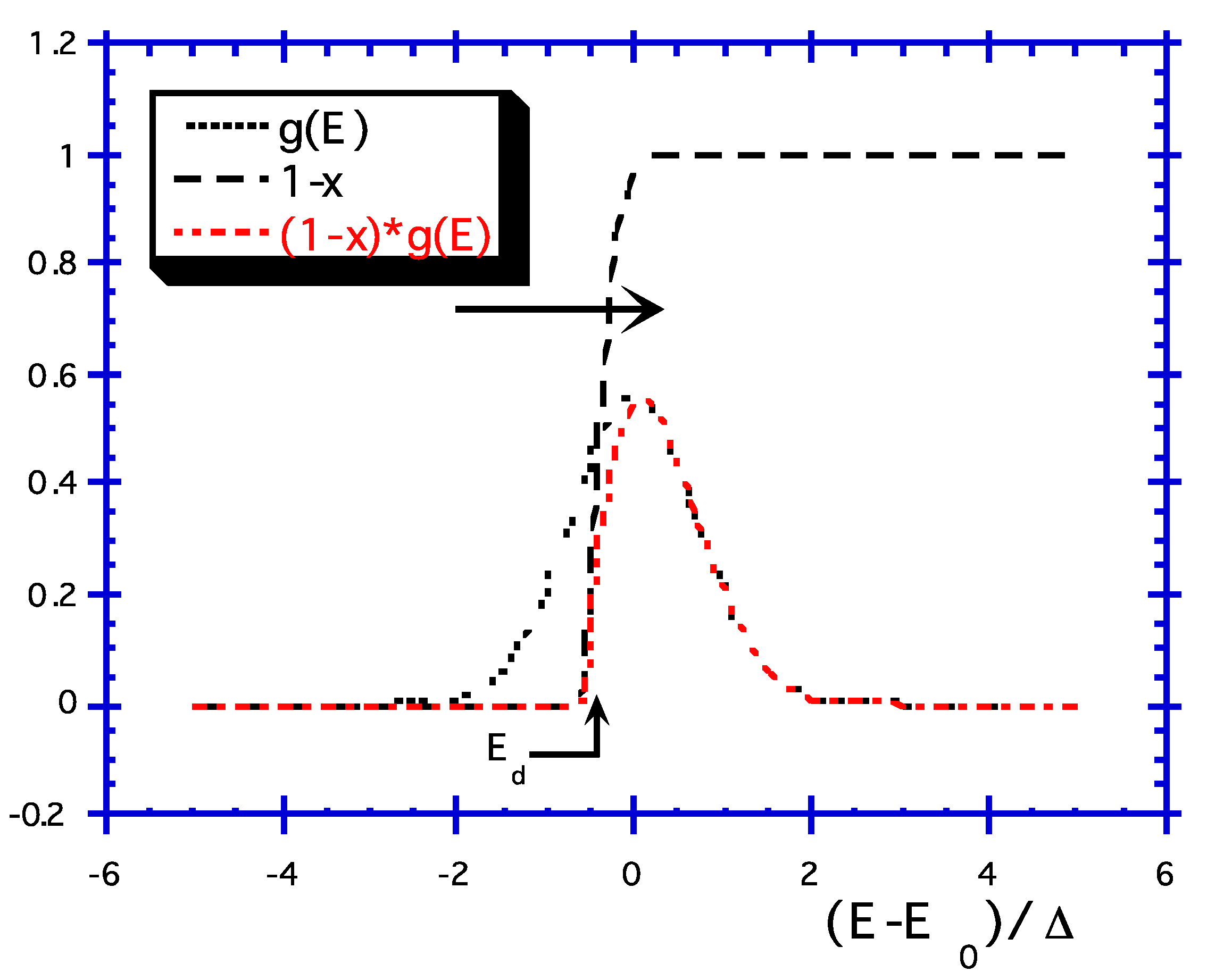
- Some Consequences of the Theory:
- (1)
- [B](t, T) can be expressed as a function of the unique variable called demarcation energy (). The curve is called the master curve (MC), as it is unique, regardless of what the (t, T) couple may be for a given value of Ed. We can note that T is equivalent to lnt in and, thus, isochronal ageing data are equivalent to isothermal ageing data for establishing MC. Note that from a practical point of view, most people consider that NICC() is the master curve. Now, the MC plot allows the user to predict the grating lifetime, providing that the anticipated conditions of BG (i.e., = f(tuse, Tuse) correspond to a point on the MC that has been actually sampled during the annealing experiment.
- (2)
- Notice also that the distribution function is included in the MC differentiation as: .
- (3)
- Thermal stability increases along with the ageing time (hardening). This is due to the fact that the less stable sites are removed along the ageing.
- -
- (1) When the pathways are the same in both senses, the distribution function is the same for forward and backward reactions, as shown in the scheme below. If the writing kinetics are not saturated, the forward distribution function is not saturated and, thus, neither is the backward distribution. The stability is dependent on the writing (time and power density that is included in , see B0(Ed) in definition)
![Fibers 03 00206 i001]()
- -
- (2) When there is no correlation between the distribution functions because the pathways are not the same, there are two independent distribution functions, as shown in the scheme below. The B sites are randomly occupied whatever its stability. The complete distribution function of the backward reaction has to be used. The stability is independent of writing (i.e., on the initial grating strength), see Section 2.3.3.
![Fibers 03 00206 i002]()

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.3.3. A Few Examples of Time Dependence Computations Assuming B0(E) = B0
2.3.4. Incomplete forward Reaction in the Case of Dependent Backward and Forward Pathways (B0(E)) [36]
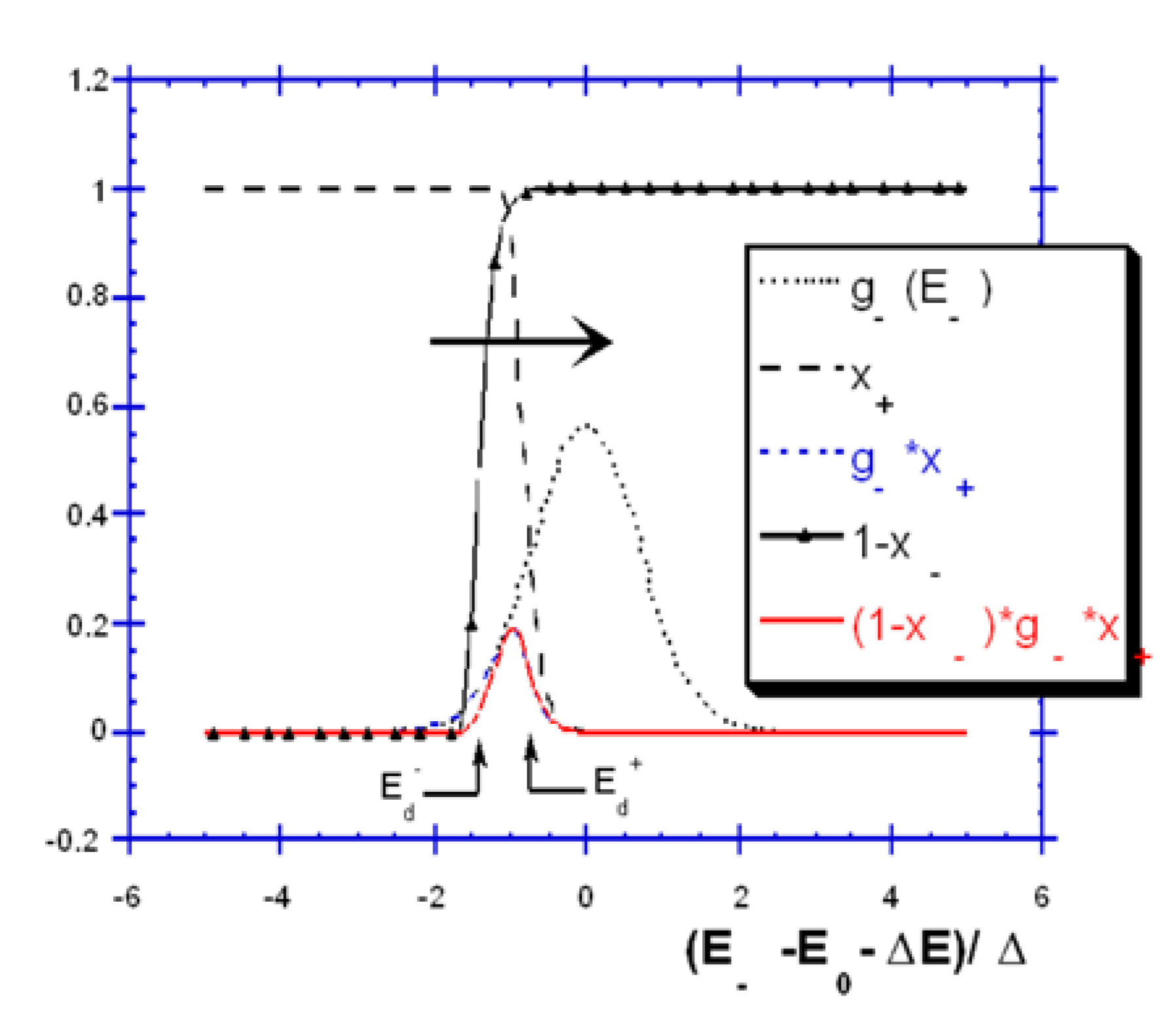
2.3.4.1. Gaussian Distribution
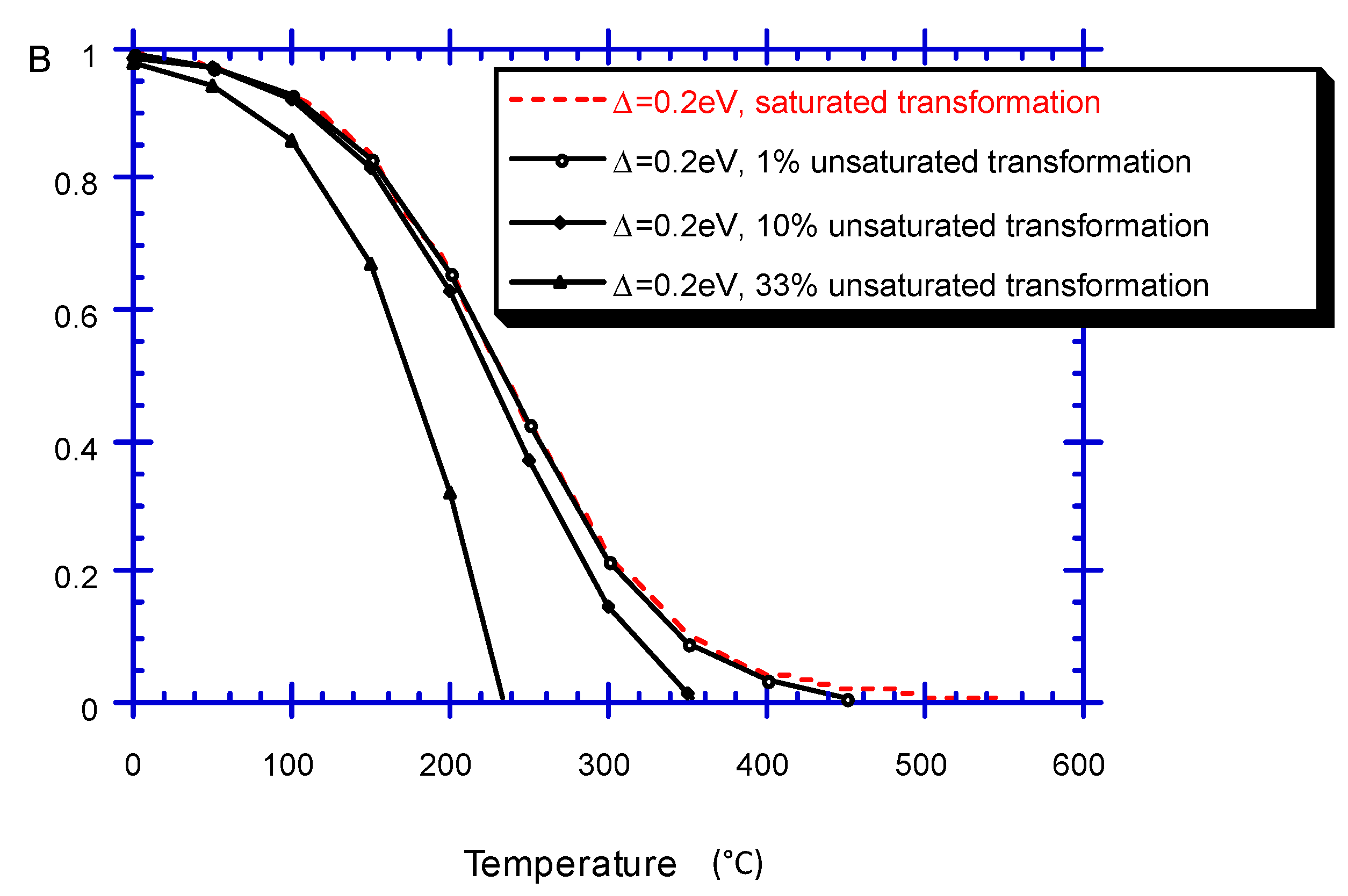
2.3.4.2. Sigmoid Differentiation Distribution
2.3.4.3. Poisson Distribution
3. Basic Experimental Analysis of the Erasure
3.1. Distribution Function Determination for the Reverse Reaction
- (1)
- note:
- (2)
- with the assumptions 2.3.1 and 2.3.2 (and even less, i.e., assumption on first order reaction is not necessary).
- (1)
- Measurement of several isochrons or isotherms:
- (2)
- k0 fitting for curve collapsing. We adjust the constant k0 for obtaining the collapse of the curves like in the Figure 29.
- (3)
- Differentiation of the previous curve. .
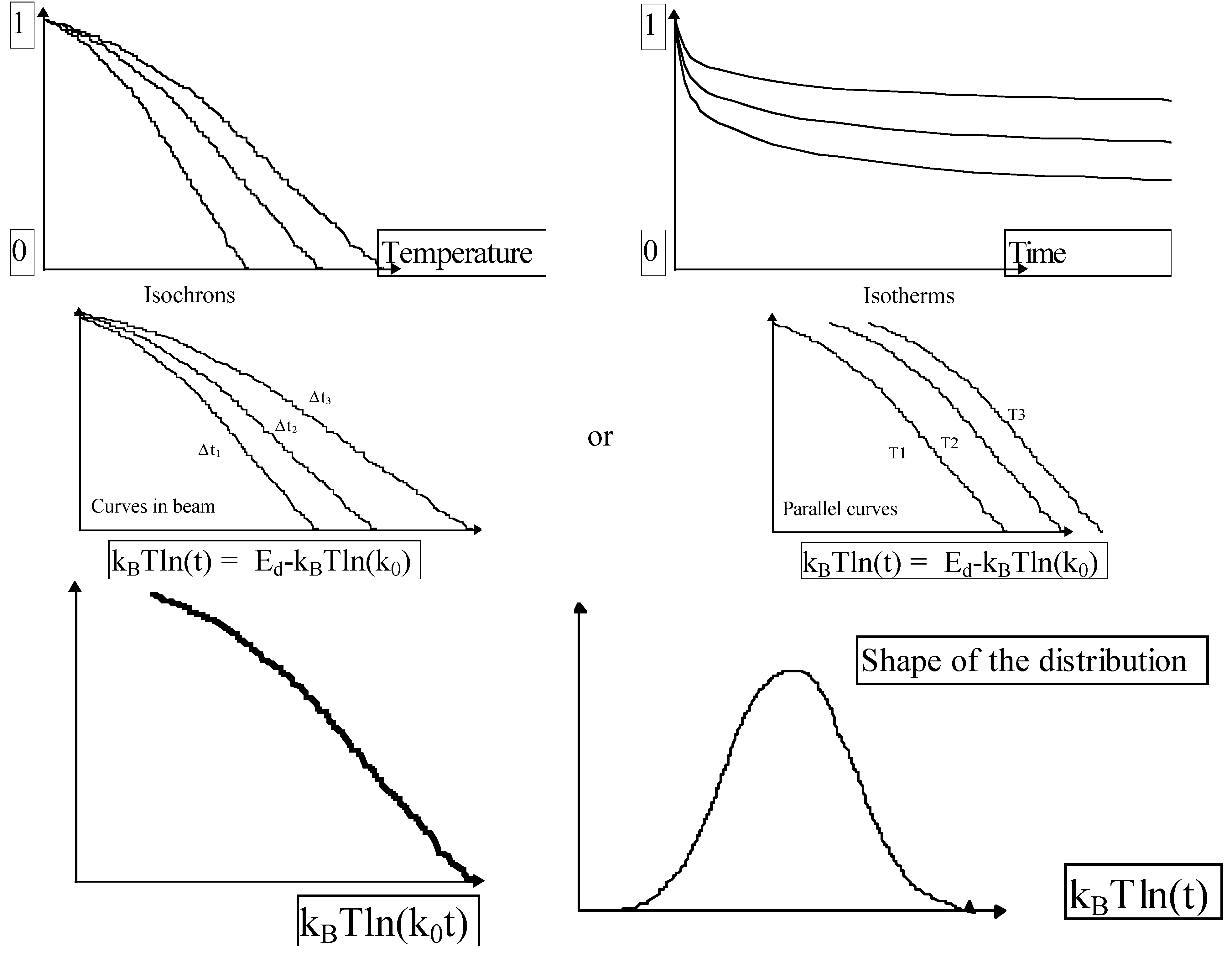
3.2. Practice (or application of the theory)
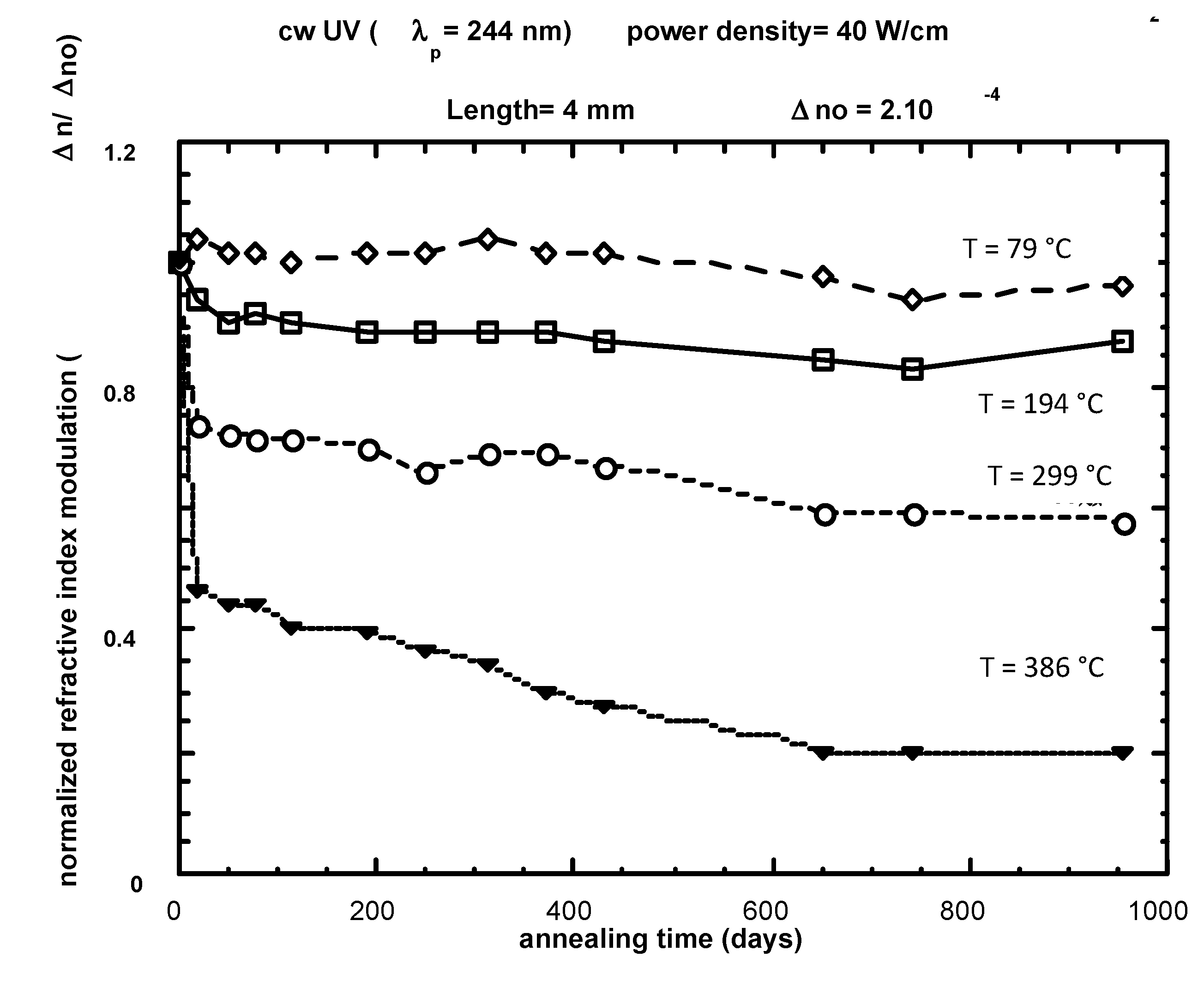
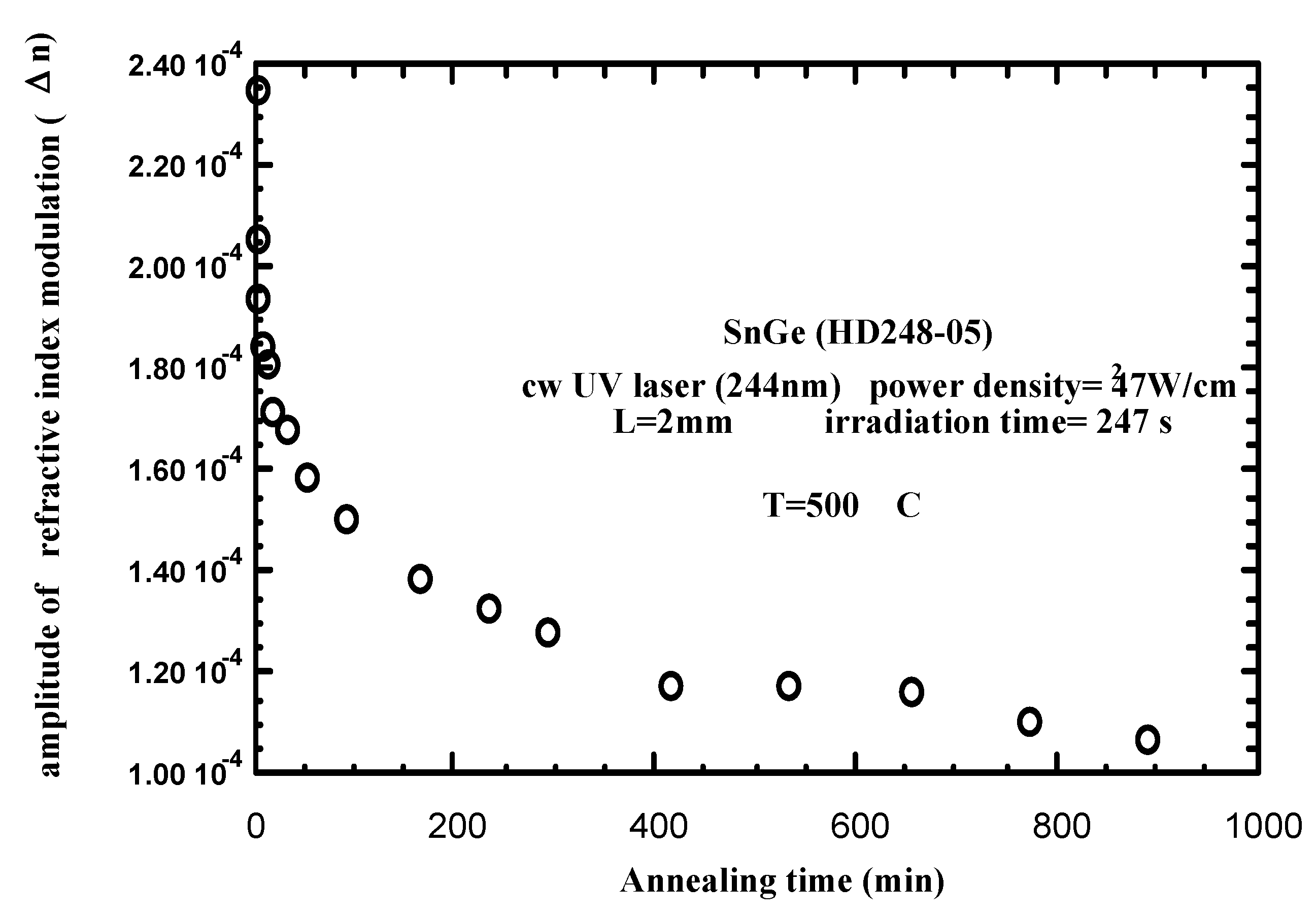
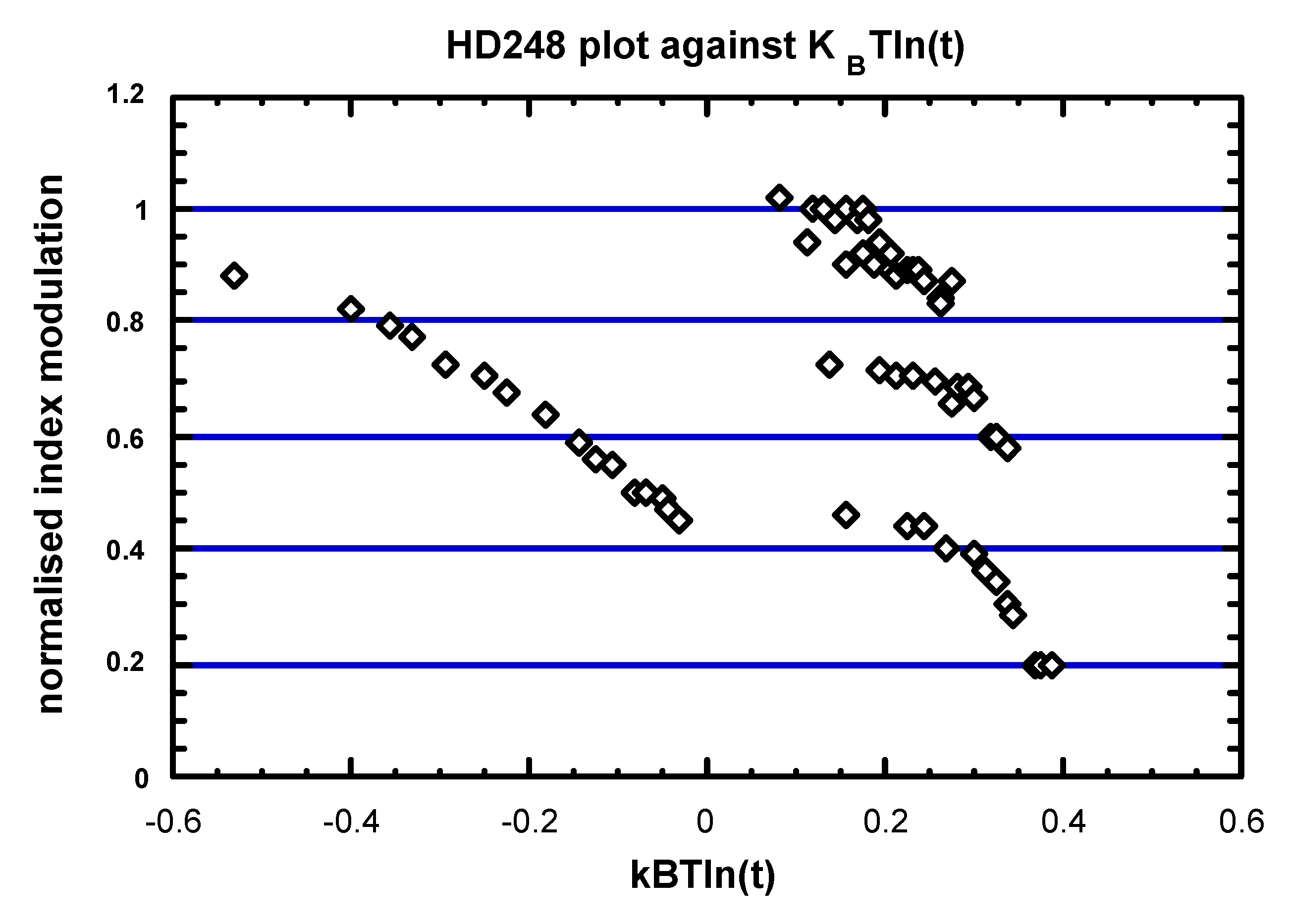
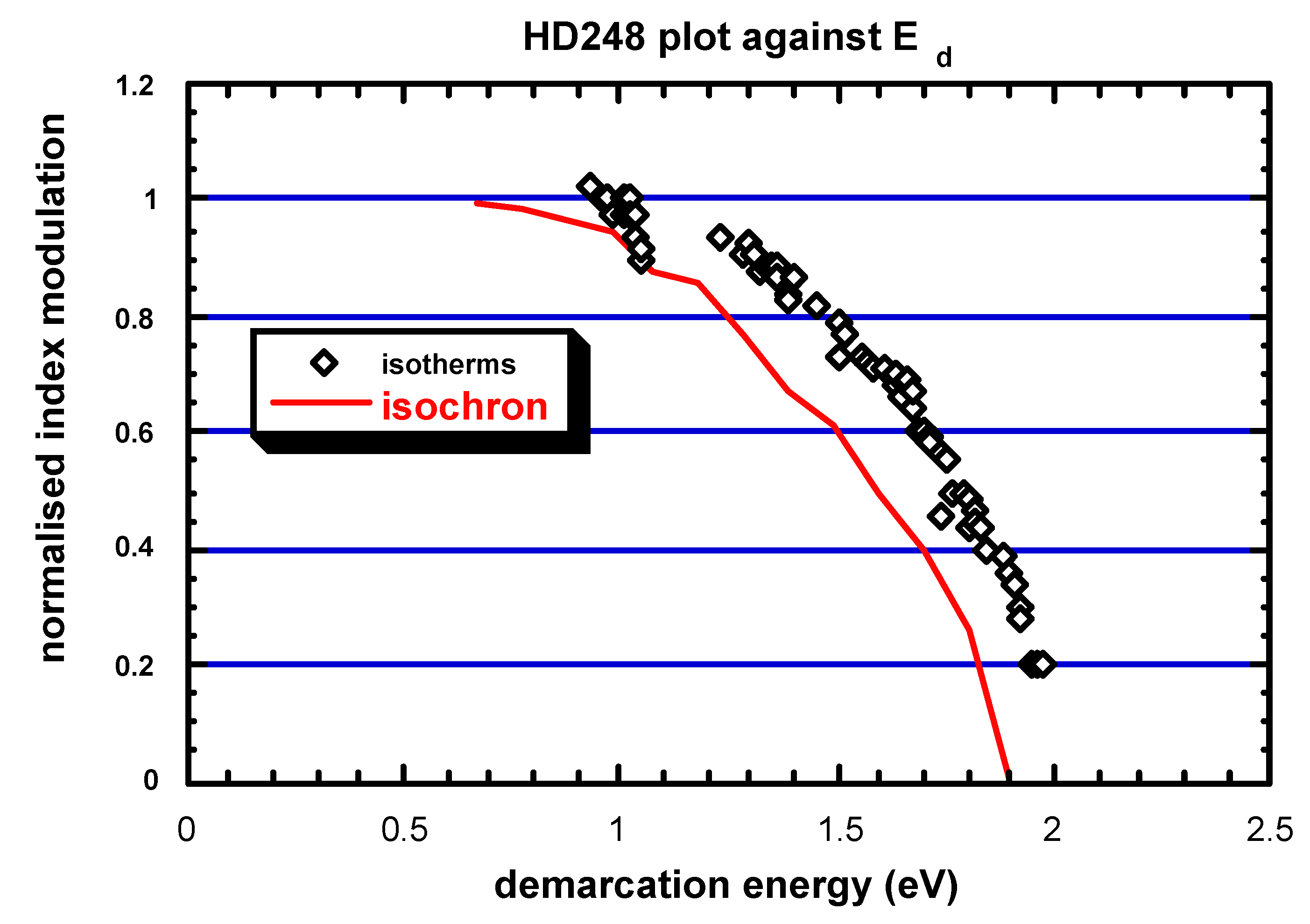
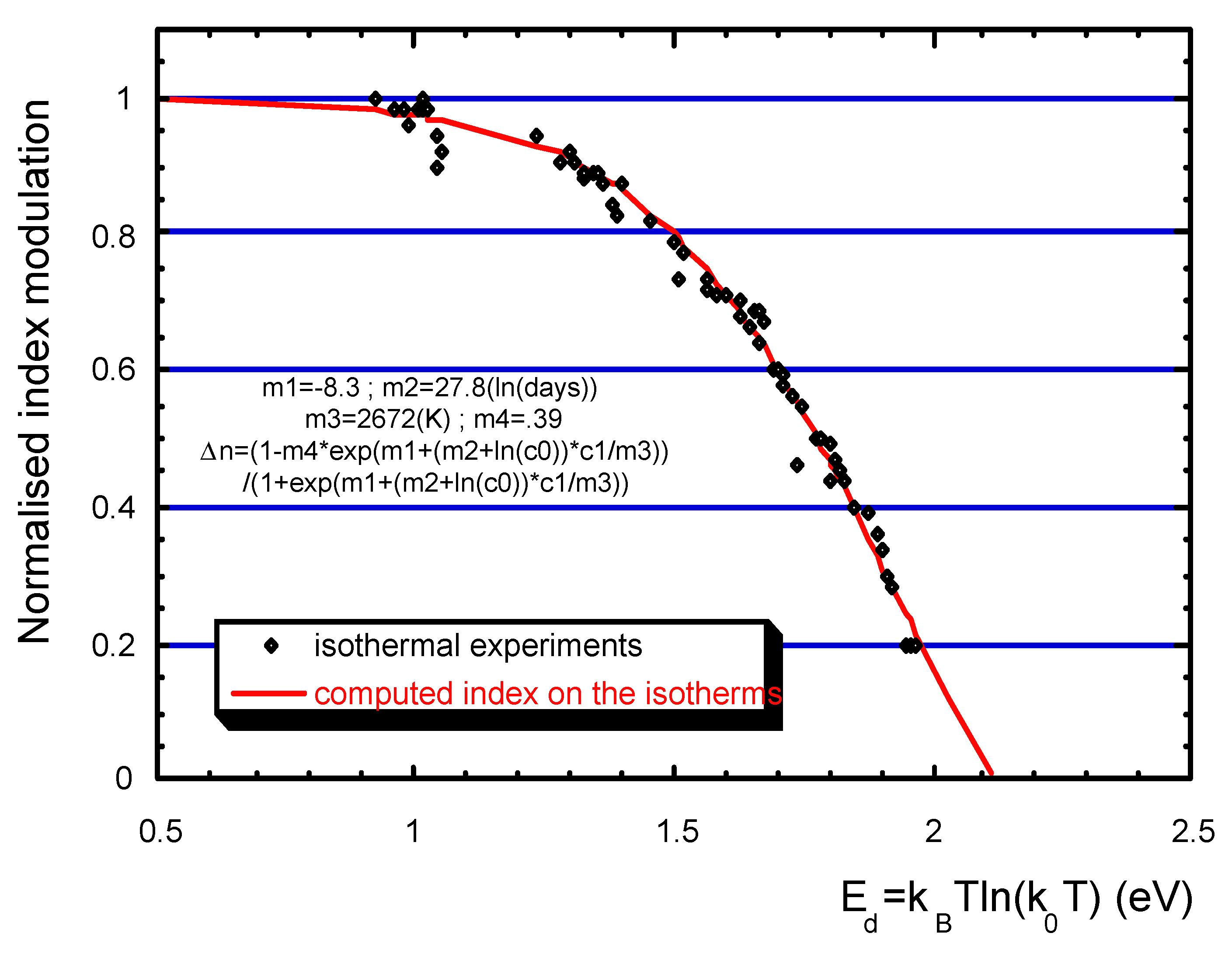
3.3. Stabilization
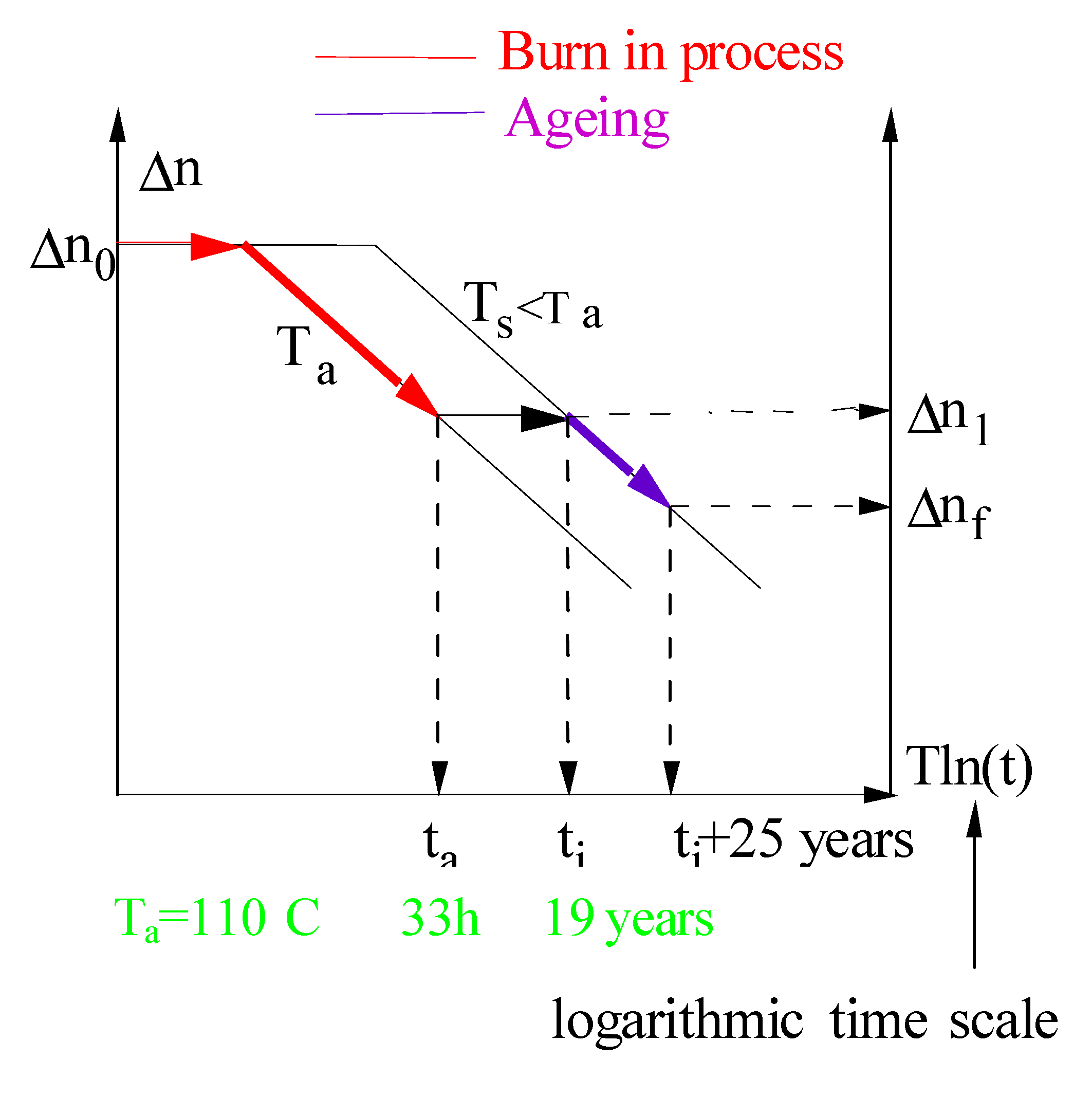
- -
- the non-passivated master curve after annealing is truncated and needs to be renormalized.
- -
- the origin of the time is usually taken from the beginning of the ageing after annealing and, thus, a time shift has to be applied on the previous demarcation energy. This is not negligible in the first 3% of ageing (see page 52 of [38]).
4. Peculiar Properties in the Frame of Assumption Validity (Action of the Reversed Reaction)
4.1. Writing-Erasure Connection (A⇒B, B⇒A)
- (1)
- Case 1 (same pathways): we will show that the distribution function is cut at large Ed at a more elevated temperature. Stability increases because more stable sites are filled [36] (see Section A below).
- (2)
- Case 2 (different pathways, two distribution functions): stability increases because less stable sites are removed in the reverse distribution function during writing (Section B below).
4.1.1. Writing in the Case of Reversible Reaction

4.1.2. Erasure in the Case of Reversible Reaction
4.1.3. Stability When Forward and Backward Reactions Are Independent
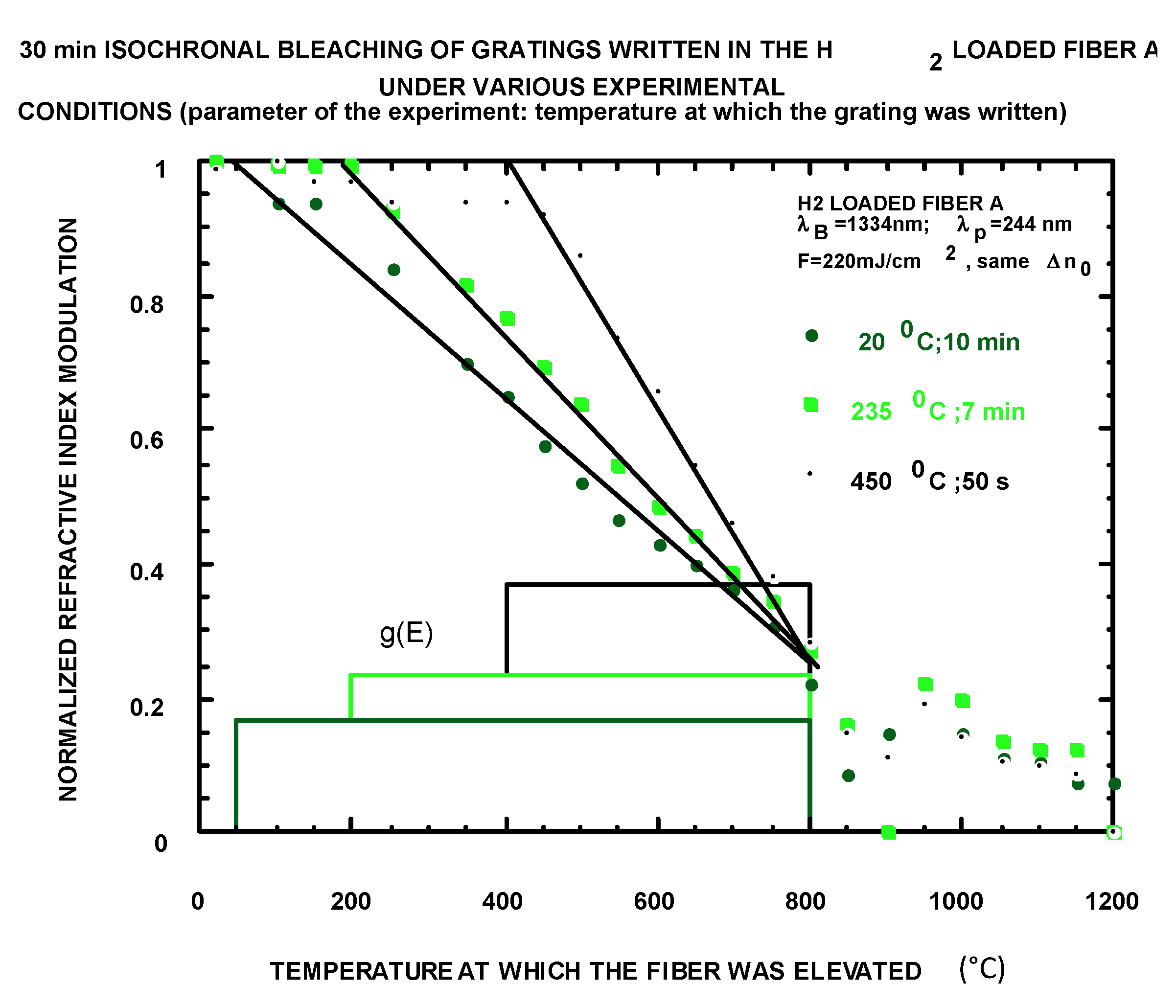
4.1.4. Writing in the Case of Independent Forward and Backward Reactions
4.2. Isotherm-Isochron Equivalence
- -
- the distribution function is not T dependent,
- -
- only one limiting reaction.
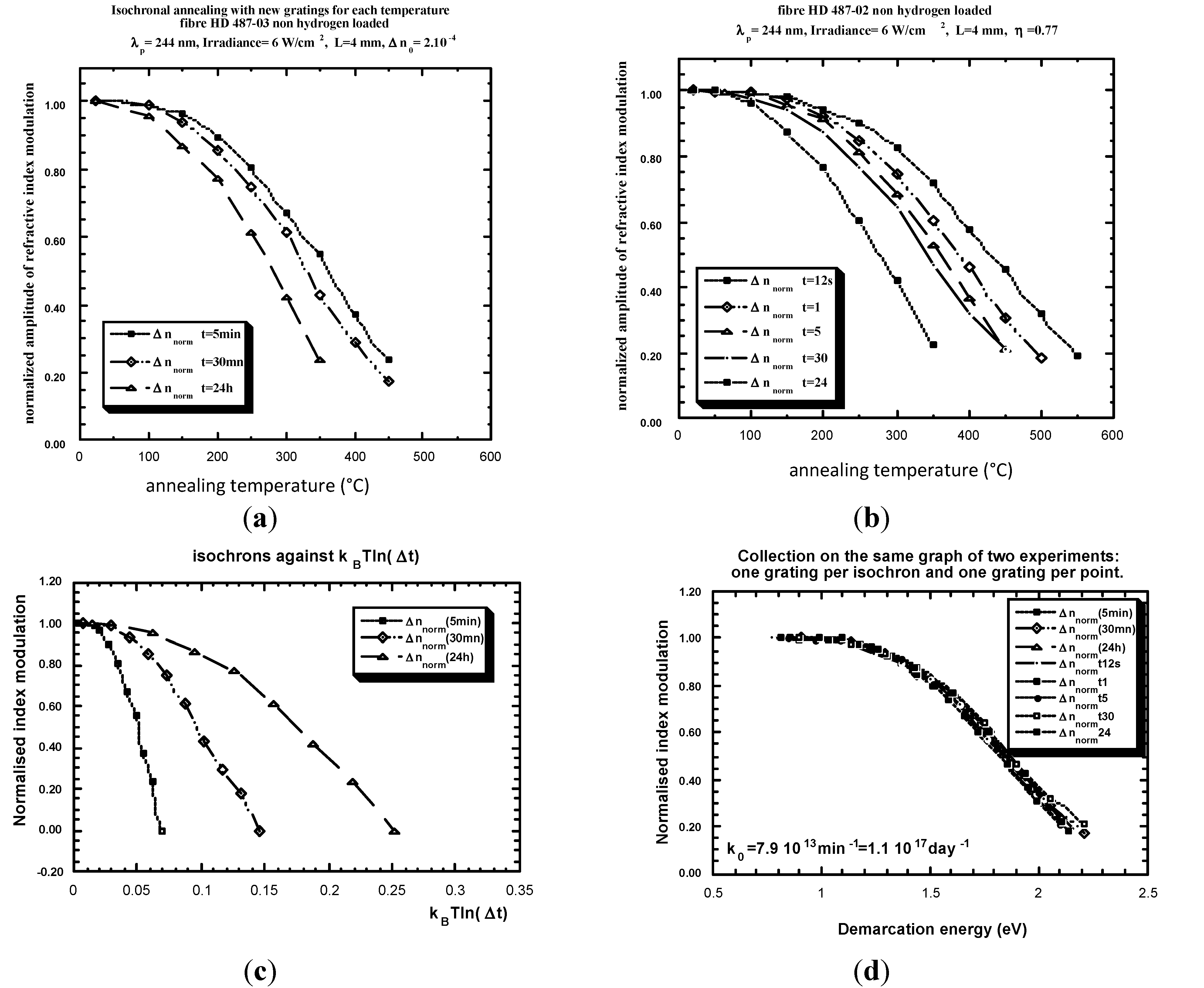
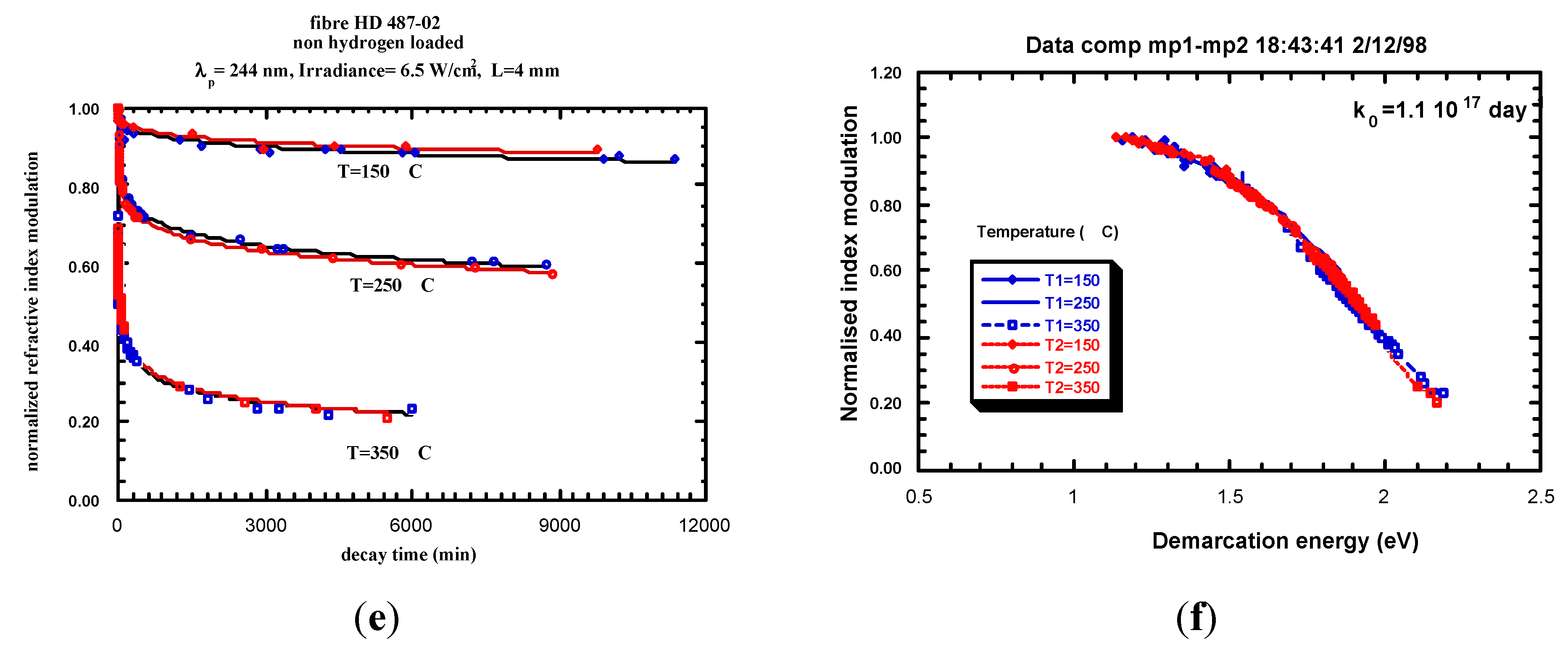
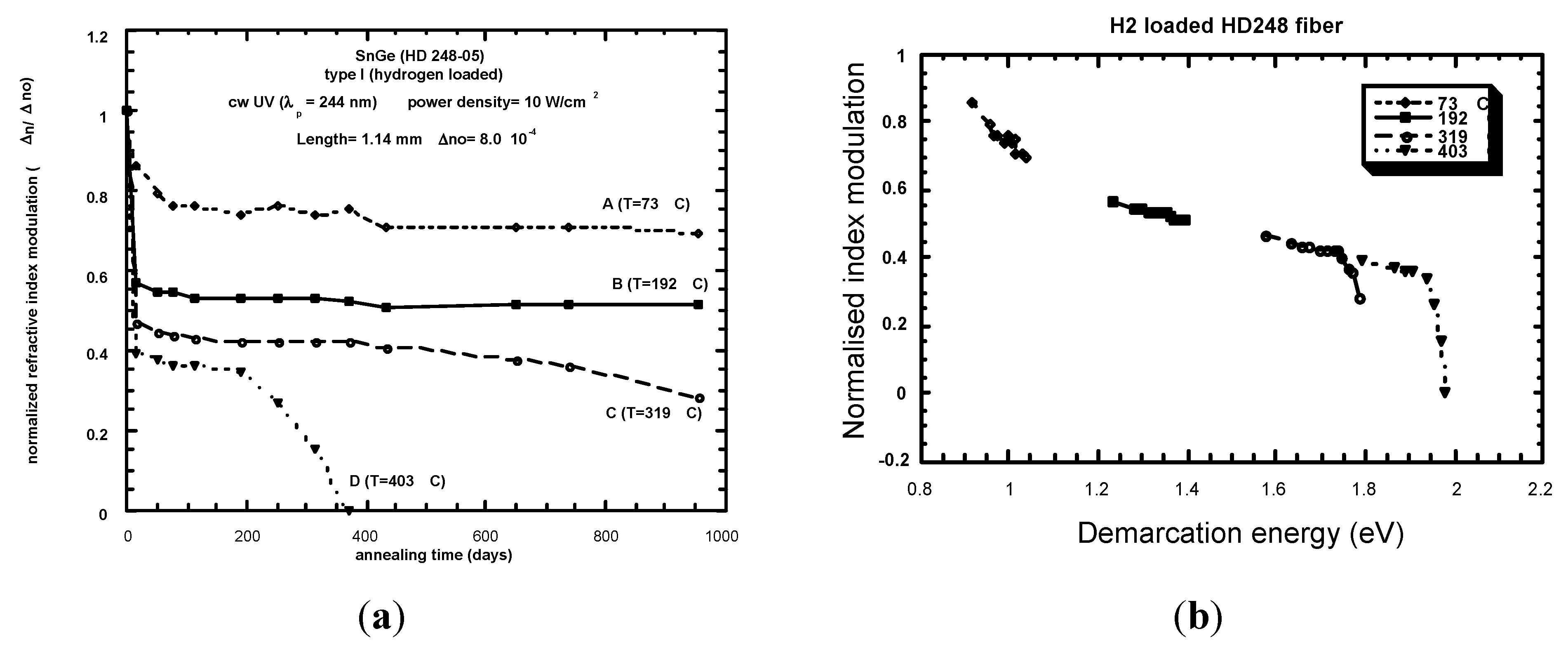
- Example of Equivalence Breaking
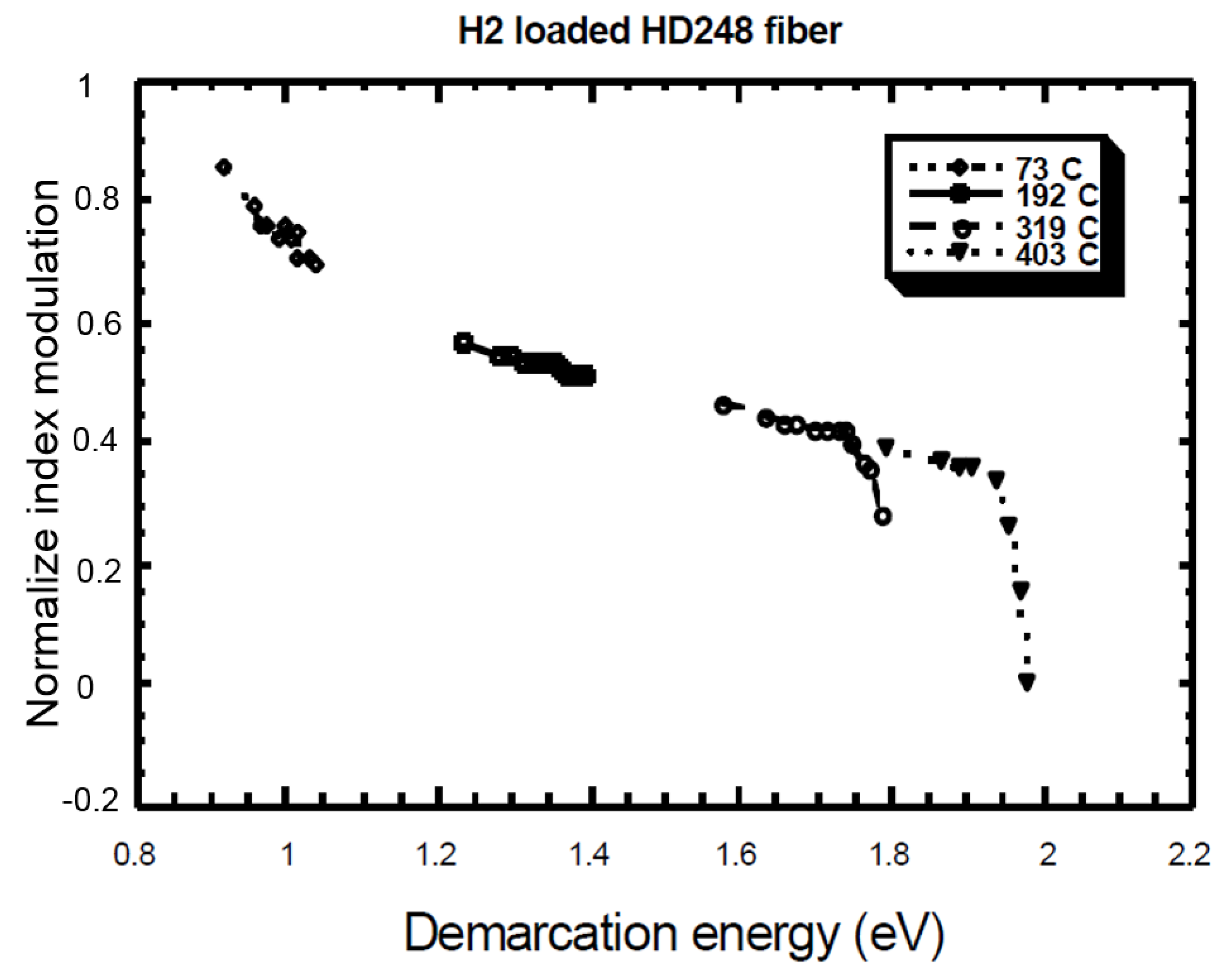
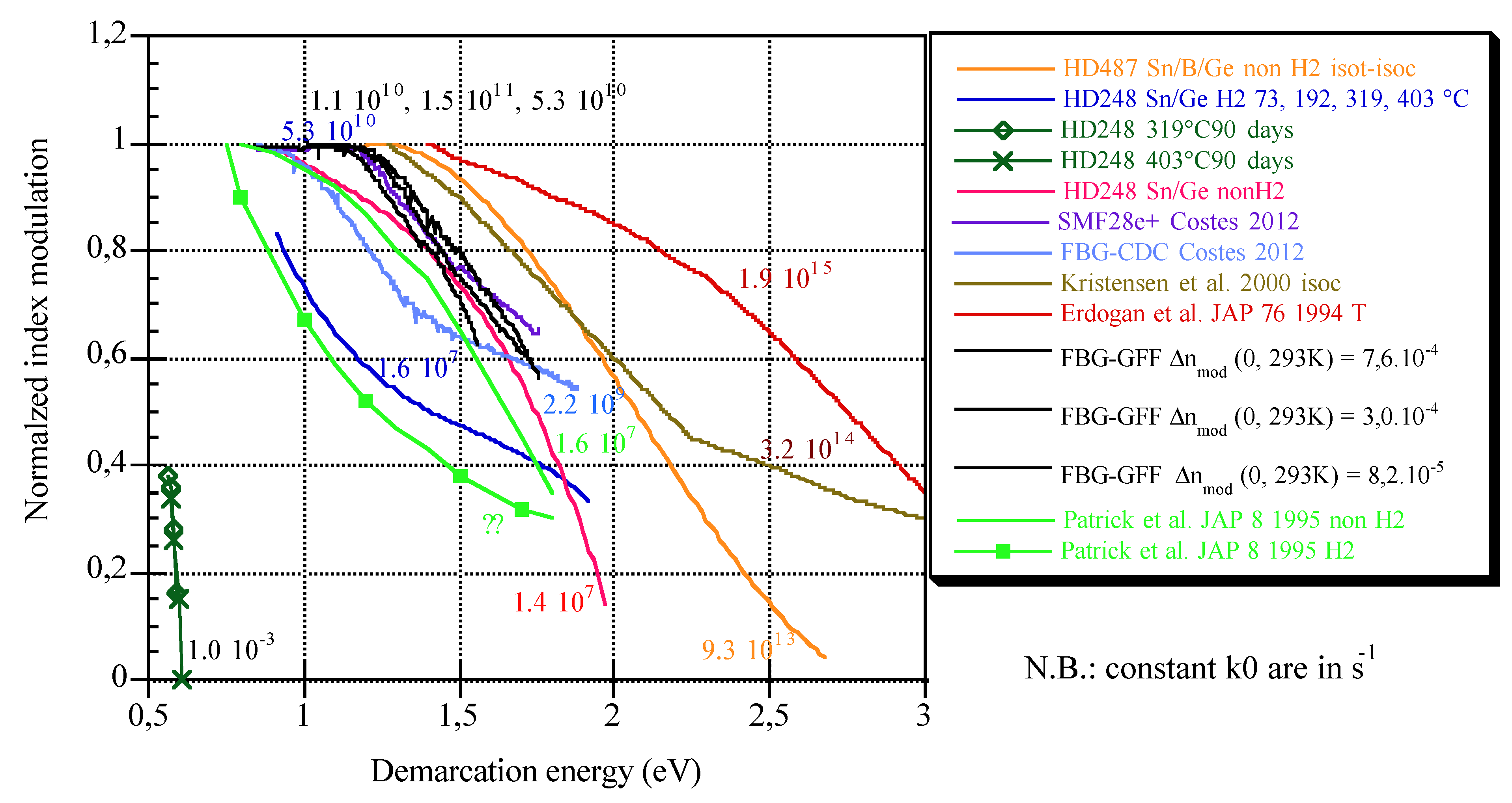
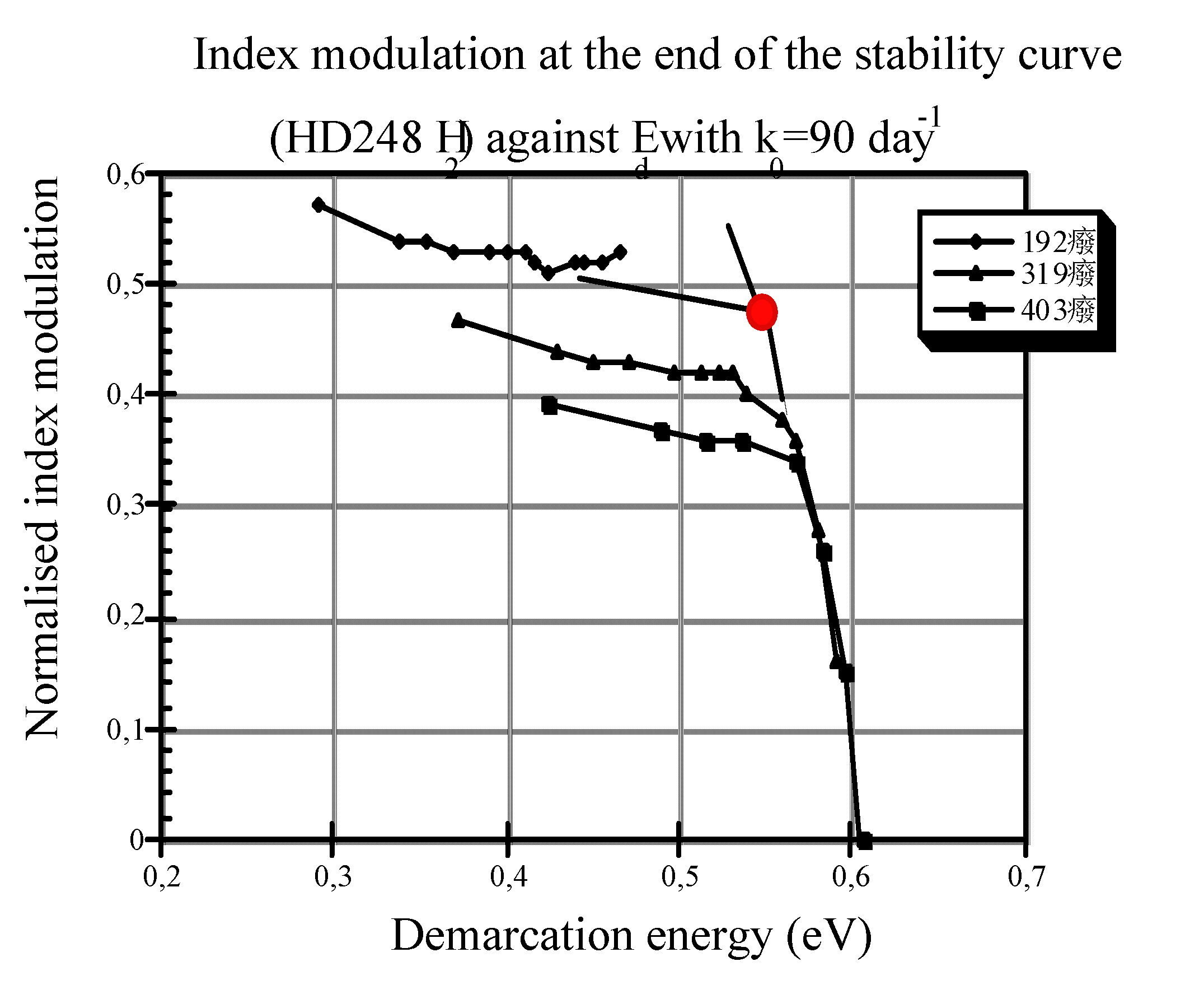
4.3. k0 Comparison
5. Beyond Simple Theory
5.1. Higher Order Reaction
5.2. Two Parallel Reactions
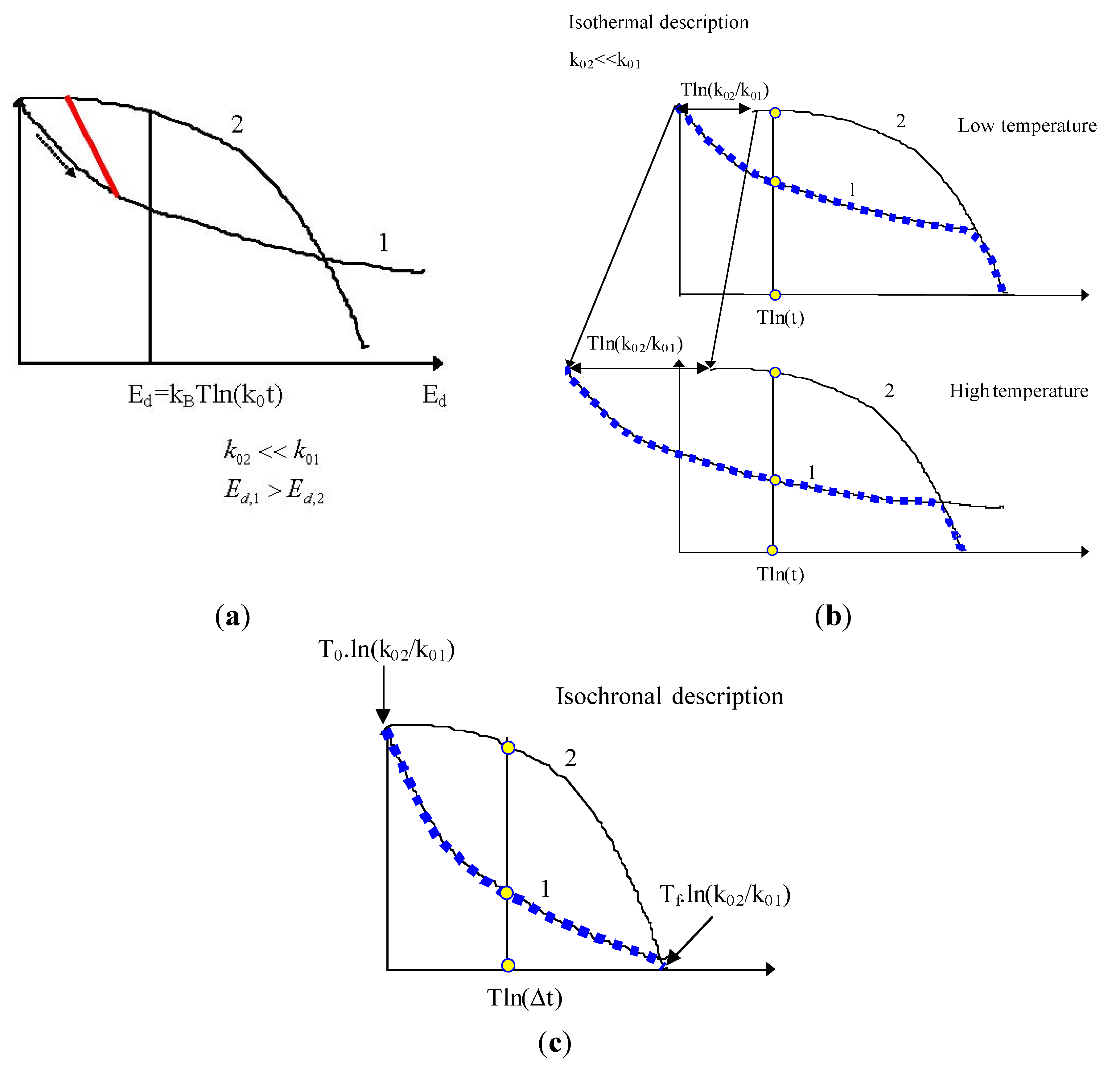
5.3. Serial Reactions
- Theory of Serial Reactions
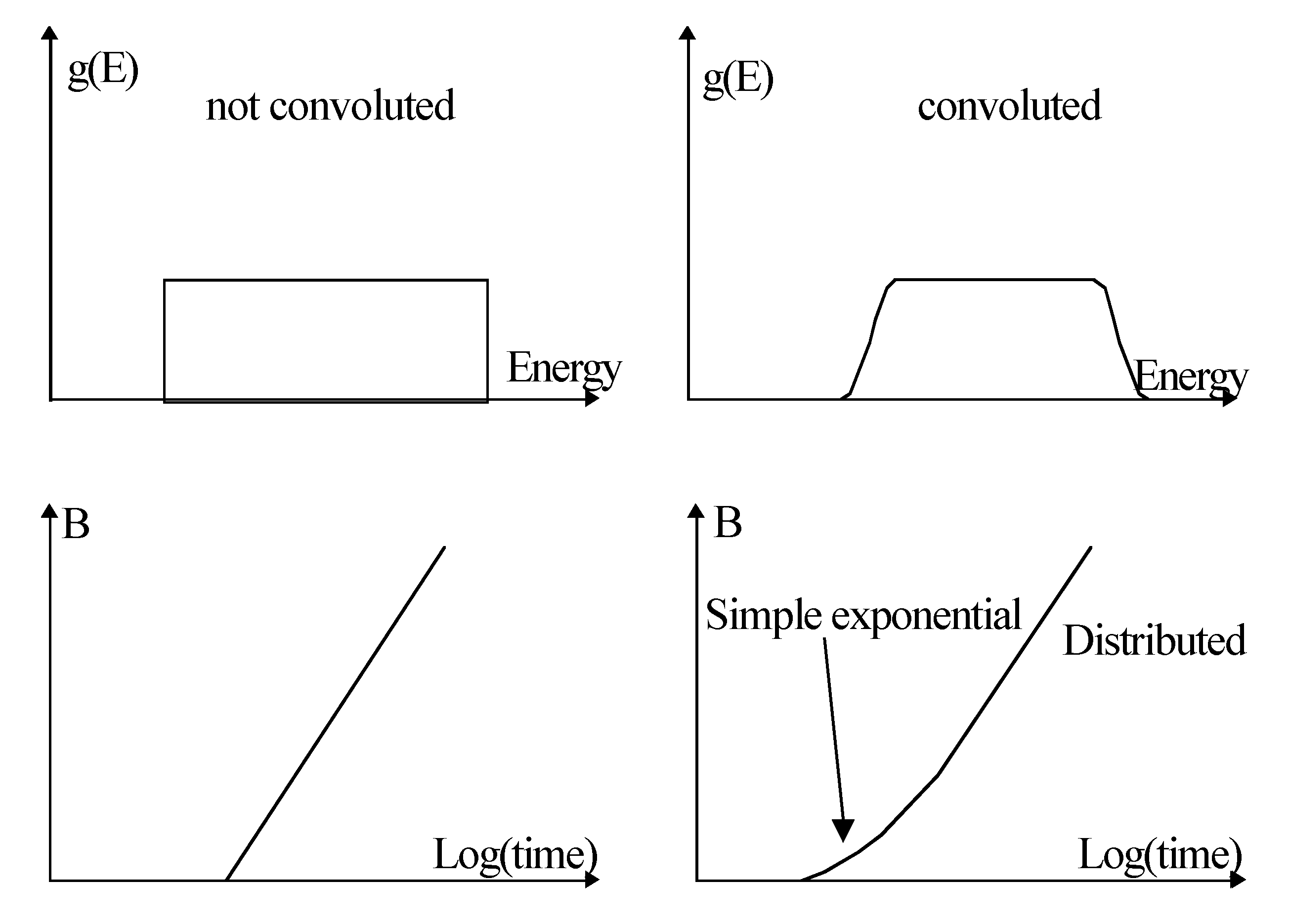
5.4. Narrow and Steep Distribution Function

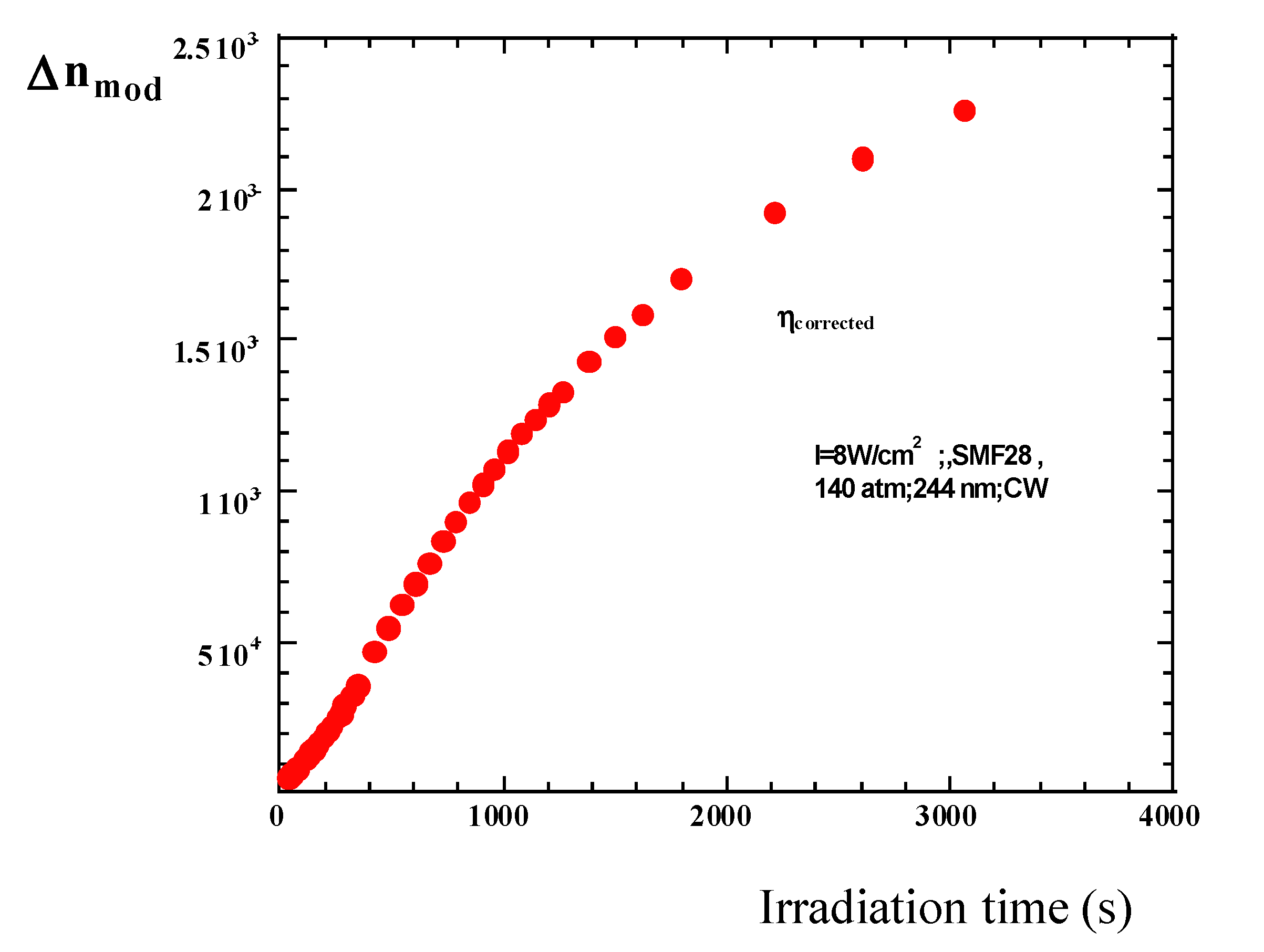
5.5. Distributed Non-Activated Kinetics (Photochromism, Luminescence)
5.5.1. “Writing like”
5.5.2. Erasure
6. Real Analysis of Bragg Grating Writing and Stability
7. Practical Formalism to Describe Generalized Thermally Activated Processes
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Erdogan, T.; Mizrahi, V.; Lemaire, P.J.; Monroe, D. Decay of ultraviolet-induced fiber Bragg gratings. J. Appl. Phys. 1994, 76, 73–80. [Google Scholar] [CrossRef]
- Ishikawa, S.; Inoue, A.I.; Harumoto, M. Adequate aging condition for fiber Bragg grating based on simple power law model. In Optical Fiber Communication Conference and Exhibit, 1998, OFC '98. San Jose, CA, USA, 22–27 February 1998.
- Richert, R.; Blumen, A. Disorder Effects on Relaxational Processes; Springer Verlag: New York, NY, USA, 1994. [Google Scholar]
- Bernage, P.; Taunay, T.; Leconte, B.; Douay, M.; Niay, P.; Bayon, J.F.; Poignant, H.; Herlemont, H.; Legrand, J.; Poumellec, B.; et al. Inscription Kinetics and Thermal Stability of Bragg Gratings Written within Heated Fibers. In Proceedings of the Doped Fiber Devices and systems II, Denver, CO, USA, 20 November 1996.
- Riant, I.; Poumellec, B. Thermal decay of gratings written in hydrogen-loaded germanosilicate fibers. Electron. Lett. 1998, 34, 1603–1604. [Google Scholar] [CrossRef]
- Lancry, M.; Poumellec, B.; Costes, S.; Magné, J. Reliable Lifetime Prediction for Passivated Fiber Bragg Gratings for Telecommunication Applications. Fibers 2014, 2, 92–107. [Google Scholar] [CrossRef]
- Xie, W.X.; Niay, P.; Bernage, P.; Douay, M.; Taunay, T.; Bayon, J.F.; Delevaque, E.; Monnerie, M. Photoinscription of Bragg gratings within preform plates of high NA germanosilicate fibers: Searching for an experimental evidence of type IIA photosensitivity in preform plates. Opt. Commun. 1996, 124, 295–300. [Google Scholar] [CrossRef]
- Poumellec, B.; Niay, P.; Douay, M.; Bayon, J.F. The UV induced refractive index grating in Ge-SiO2 preforms: Additional CW experiments and the macroscopic origin of the change in index. J. Phys. D Appl. Phys. 1996, 29, 1842–1856. [Google Scholar] [CrossRef]
- Fiori, C.; Devine, R.A.B. Ultraviolet irradiation induced compaction and photoetching in amorphous, thermal SiO2. MRS Proc. 1986, 61, 187–195. [Google Scholar] [CrossRef]
- Primak, W.; Kampwirth, R. The radiation compaction of vitreous silica. J. Appl. Phys. 1968, 39, 5651–5658. [Google Scholar] [CrossRef]
- Allan, C.D.; Smith, C.; Borelli, N.F.; Seward, T.P., III. 193-nm excimer laser induced densification of fused silica. Opt. Lett. 1996, 21, 1960–1962. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, P.J. Reliability of optical fibers exposed to hydrogen: Prediction of long-term loss increases. Opt. Eng. 1991, 30, 780–789. [Google Scholar] [CrossRef]
- Marcerou, J.F.; Février, H.; Gabla, P.M.; Augé, J. Sensitivity of erbium-doped fibers to hydrogen: Implications for long-term system performance. In Proceedings of the Optical Fiber Communication’94, San Jose, CA, USA, 20–25 February 1994.
- Beales, K.J.; Cooper, K.J.; Rush, J.D. Increased attenuation in optical fibers caused by diffusion of molecular hydrogen at room temperature. Electron. Lett. 1983, 19, 917–919. [Google Scholar] [CrossRef]
- Smith, C.L. A theory of transient creep in metals. Proc. Phys. Soc. 1948, 61, 201–205. [Google Scholar] [CrossRef]
- Poirier, M.; Thibault, S.; Lauzon, J.; Ouellette, F. Dynamics and orientational behaviour of U.V. induced luminescence bleaching in Ge-doped silica optical fiber. Opt. Lett. 1993, 18, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, T.; Mizrahi, V.; Lemaire, P.J.; Monroe, D. Decay of U.V.-induced fiber Bragg gratings. In Proceedings of the Optical Fiber Communication’94, San Jose, CA, USA, 20–25 February 1994.
- Kyung, J.H.; Lawandy, N.M. Direct observation of the effective χ(2) grating in bulk glasses encoded for second-harmonic generation. Opt. Lett. 1996, 21, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Friebele, E.J.; Gingerich, M.E.; Griscom, D.L. Survivability of optical fibers in space. In Proceedings of the Optical Materials Reliability and Testing, Boston, MA, USA, country, 8 September 1992.
- Deparis, O.; Griscom, D.L.; Mégret, P.; Decréton, M.; Blondel, M. Influence of the cladding thickness on the evolution of the NbOHC band in optical fibers exposed to gamma radiations. J. Non Cryst. Solids 1996, 216, 124–128. [Google Scholar] [CrossRef]
- Rodriguez, V.D.; Rodriguez-Mendoza, U.R.; Martin, I.R.; Lavin, V.; Nunez, P. Site distribution in Cr3+ and Cr3+-Tm3+ doped alkaline silicate glasses. In Proceedings of the SiO2 and Advanced Dielectrics, L’Aquila, Italy, 15–17 June 1997.
- Monroe, D.; Kastner, M.A. Exactly exponential band tail in a glasssy semiconductor. Phys. Rev. B 1986, 33, 8881–8884. [Google Scholar] [CrossRef]
- Kroide, M. Apparent viscosity and relaxation mechanism of glasses below glass transition temperature. Phys. Chem. Glasses 1997, 38, 83–86. [Google Scholar]
- Phillips, J.C. Kohlrausch relaxation and glass transitions in experiment and in molecular dynamics simulations. J. Non Cryst. Solids 1995, 182, 155–161. [Google Scholar] [CrossRef]
- Mohanna, Y.; Saugrain, J.-M.; Rousseau, J.-C.; Ledoux, P. Relaxation of internal stresses in optical fibers. J. Lightwave Technol. 1990, 8, 1799–1802. [Google Scholar] [CrossRef]
- Lemaire, P.J.; Tomita, A. Behavior of single mode MCVD fibers exposed to hydrogen. In Proceedings of the European Conference on Optical Communications, Stuttgart, Germany, 3–6 September 1984.
- Kohlrausch, R. Ueber das Dellmann’sche Elektrometer. Ann. Phys. 1847, 72, 353–405. (In German) [Google Scholar] [CrossRef]
- Vand, V. A theory of the irreversible electrical resistance changes of metallic films evaporated in vacuum. Proc. Phys. Soc. Lond. 1943, A55, 222–246. [Google Scholar] [CrossRef]
- Primak, W. Kinetics of Processes distributed in activation energy. Phys. Rev. B 1955, 100, 1677–1689. [Google Scholar] [CrossRef]
- Primak, W. Large temperature range annealing. J. Appl. Phys. 1960, 31, 1524–1533. [Google Scholar] [CrossRef]
- Orenstein, J.; Kastner, M. Photocurrent transient spectroscopy: Measurement of the density of localized states in a-AS2Se3. Phys. Rev. Lett. 1981, 46, 1421–1424. [Google Scholar] [CrossRef]
- Miller, S.L.; McWhorter, P.J.; Miller, W.M.; Dressendorfer, P.V. A practical predictive formalism to describe generalised activated physical processes. J. Appl. Phys. 1991, 70, 4555–4568. [Google Scholar] [CrossRef]
- Lemaire, P.J.; Monroe, D.P.; Watson, H.A. Hydrogen-induced-loss increases in erbium-doped amplifier fibers: Revised predictions. In Proceedings of the Optical Fiber Communication’94, San Jose, CA, USA, 20–25 February 1994.
- Vandenbrink, J.P. Master stress relaxation function of silica glasses. J. Non Cryst. Solids 1996, 196, 210–215. [Google Scholar] [CrossRef]
- Kannan, S.; Lemaire, P.J.; Guo, J.; LuValle, M.J. Reliability predictions on fiber gratings through alternate methods. In Proceedings of the Bragg Gratings Photosensitivity, and Poling in Glass Fibers and Waveguides: Applications and Fundamentals, Williamsburg, VA, USA, 26–28 October 1997.
- Poumellec, B. Links between writing and erasure (or stability) of Bragg gratings in disordered media. J. Non Cryst. Solids 1998, 239, 108–115. [Google Scholar] [CrossRef]
- Razafimahatratra, D.; Thesis, P. Etude de la Stabilité de la Variation D’indice de Réfraction Photoinduite par Insolation Laser Dans les Guides D’onde Optiques Germanosilicates. PhD Thesis, University of Sciences and Technologies of Lille, Lille, France, 2000. [Google Scholar]
- Costes, S. Extension de L’approche par la Courbe Maîtresse de la Prédiction des Durées de vie de Réseaux D’indice Complexes Inscrits par UV dans les Fibres; NNT: 2013PA112102. Ph.D. Thesis, Université Paris Sud, Orsay, France, 2013. [Google Scholar]
- Patrick, H.; Gilbert, S.L. Growth of Bragg gratings produced by continuous-wave ultraviolet light in optical fiber. Opt. Lett. 1993, 18, 1484–1486. [Google Scholar] [CrossRef] [PubMed]
- Razafimahatratra, D.; Niay, P.; Douay, M.; Poumellec, B.; Riant, I. Comparison of isochronal and isothermal decays of Bragg gratings written through continuous-wave exposure of an unloaded germanosilicate fiber. Appl. Opt. 2000, 39, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.R.; Rourke, H.N.; Baker, V.; Goodchild, D. Thermal decay of fiber Bragg gratings written in Boron and Germanium codoped silica fiber. J. Lightwave Technol. 1997, 15, 1470–1477. [Google Scholar] [CrossRef]
- Chabrerie, C. De L’utilisation des Recuits Isothermes et Isochrones pour la Caractérisation de Structures MOS Irradiées. Ph.D. Thesis, Paris 7 University, Paris, France, 1997. [Google Scholar]
- Ngai, K.L.; Roland, C.M.; Greaves, G.N. An interpretation of quasielastic neutron scattering and molecular dynamics simulation results on the glass transition. J. Non Cryst. Solids 1995, 182, 172–179. [Google Scholar] [CrossRef]
- Mashkov, V.A.; Austin, W.R.; Zhang, L.; Leisure, R.G. Fundamental role of creation and activation in radiation-induced defect production in high-purity amorphous SiO2. Phys. Rev. Lett. 1996, 76, 2926–2929. [Google Scholar] [CrossRef] [PubMed]
- Ramecourt, D.; Niay, P.; Bernage, P.; Riant, I.; Douay, M. Growth of strength in H2 loaded telecommunication fiber during CW UV post-exposure. Electron. Lett. 1999, 35, 329–330. [Google Scholar] [CrossRef]
- Lancry, M.; Poumellec, B. UV laser processing and multiphoton absorption processes in optical telecommunication fiber materials. Phys. Rep. 2013, 523, 207–229. [Google Scholar] [CrossRef]
- Perlmutter, S.H.; Levenson, M.D.; Shelby, R.M.; Weissman, M.B. Polarization properties of quasielastic light scattering in fused-silica optical fiber. Phys. Rev. B 1990, 42, 5294–5305. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poumellec, B.; Lancry, M. Kinetics of Thermally Activated Physical Processes in Disordered Media. Fibers 2015, 3, 206-252. https://doi.org/10.3390/fib3030206
Poumellec B, Lancry M. Kinetics of Thermally Activated Physical Processes in Disordered Media. Fibers. 2015; 3(3):206-252. https://doi.org/10.3390/fib3030206
Chicago/Turabian StylePoumellec, Bertrand, and Matthieu Lancry. 2015. "Kinetics of Thermally Activated Physical Processes in Disordered Media" Fibers 3, no. 3: 206-252. https://doi.org/10.3390/fib3030206
APA StylePoumellec, B., & Lancry, M. (2015). Kinetics of Thermally Activated Physical Processes in Disordered Media. Fibers, 3(3), 206-252. https://doi.org/10.3390/fib3030206