
Electrospun Scaffolds from Low Molecular Weight Poly(ester amide)s Based on Glycolic Acid, Adipic Acid and Odd or Even Diamines


Abstract
:
1. Introduction
2. Experimental Section
- (1)
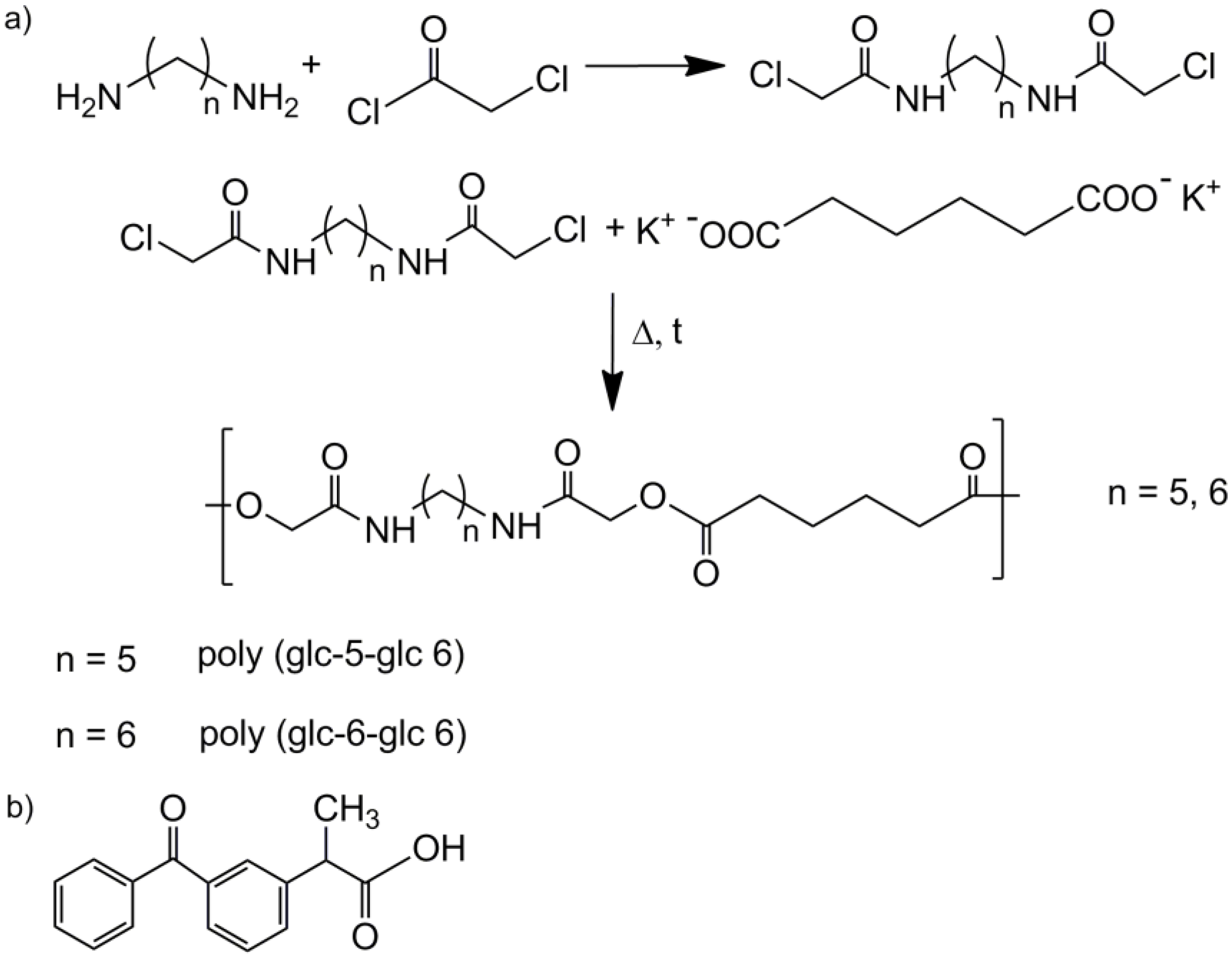
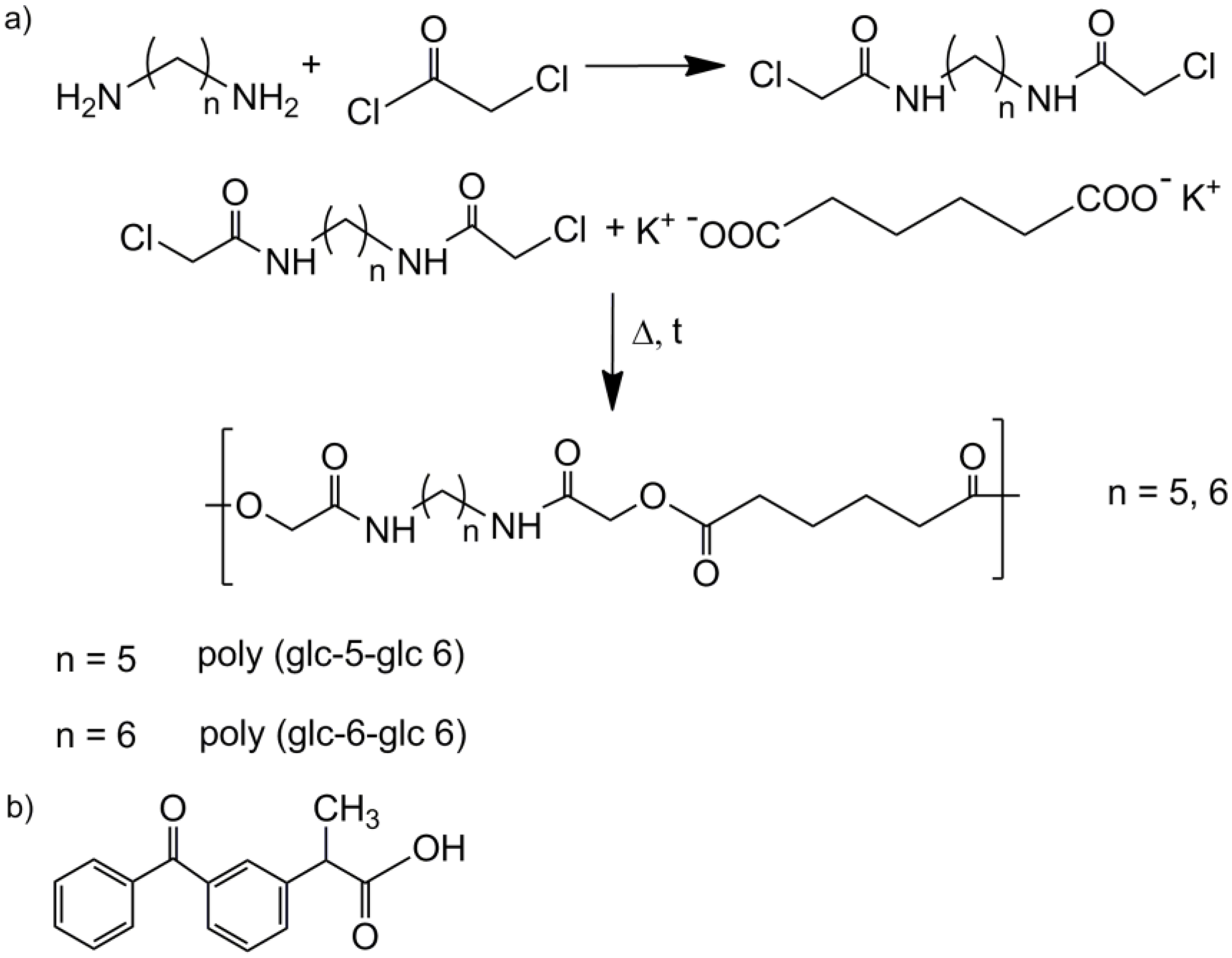
- N,N′-bis(chloroacetyl)-1,5-pentanediamine (glc-5-glc) and N,N′-bis(chloroacetyl)-1,6-hexanediamine (glc-6-glc): 8.75 mL (0.11 mol) of chloroacetyl chloride were dissolved in 20 mL of dried diethyl ether under nitrogen atmosphere in a previously dried pressure-equalizing dropping funnel. The solution was added dropwise to 50 mL of NaOH 4M solution containing 0.05 mol of 1,5-pentanediamine (for glc-5-glc) or 1,6-hexanediamine (for glc-6-glc) in a round bottom flask magnetically stirred at 0 °C. pH was maintained around 11 with additional NaOH 4M solution to neutralize the HCl formed during the reaction. After 3 h, the reaction was stopped and the solution filtrated. The recovered precipitate was exhaustively washed with distilled water and diethyl ether. The white powder was vacuum dried at 30 °C.
- (2)
- Potassium adipate: A certain amount of adipic acid was neutralized by an ethanolic solution of KOH 4M. The solution was rotoevaporated and vacuum dried.
- (3)
- Polymerization of the homopolymers: An equimolar mixture of potassium adipate and N,N′-bis(chloroacetyl)-1,5-pentanediamine or N,N′-bis(chloroacetyl)-1,6-hexanediamine was introduced in a reaction tube provided with a magnetic stirrer and pump-filled with nitrogen atmosphere. The reaction tube was heated in an oil bath at 140 °C for 5 h or 160 °C for 3 h for each mixture, respectively, and stopped introducing the tube in an ice-bath. The polymer was purified by precipitation with methanol of a diluted formic acid solution. The solid was filtrated and repeatedly washed with distilled water, methanol and diethyl ether. Finally, the obtained white powder was dried under vacuum at 30 °C for 2 days.
- (4)
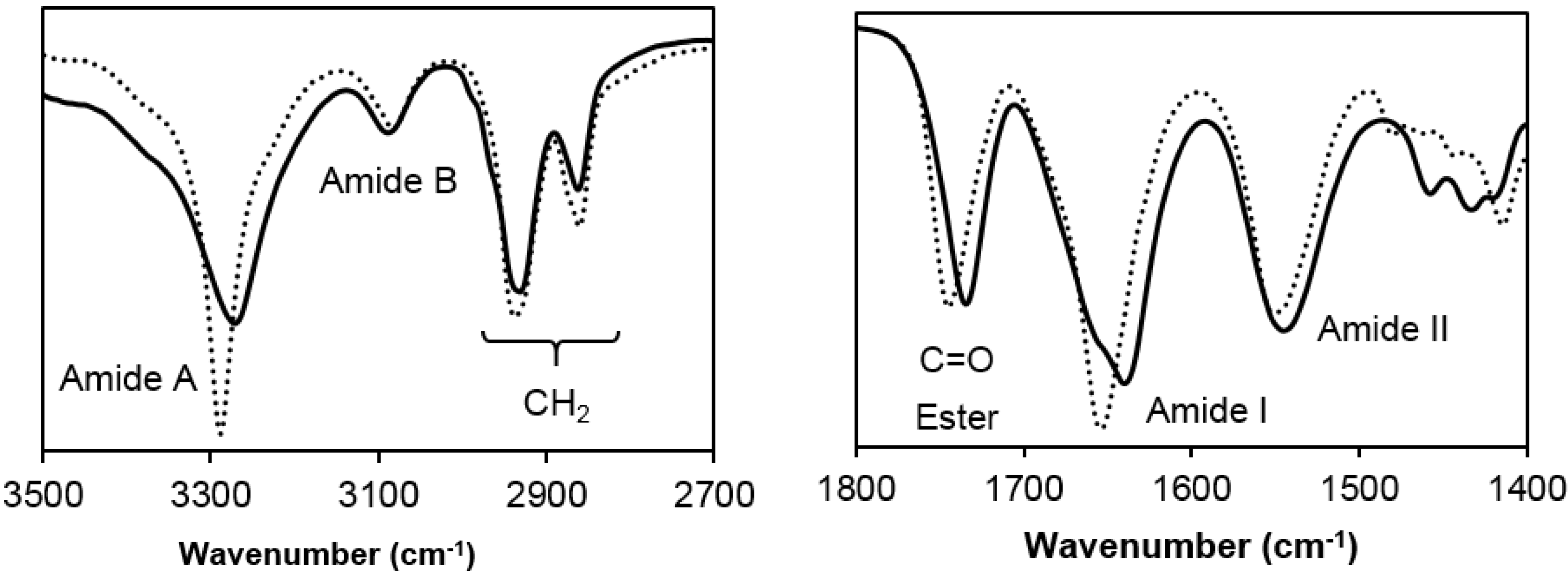
- Poly(glc-5-glc 6): Yield: 70%. m.p. 118 °C. Mw: 18,000 g/mol. Polydispersity: 2.4. FTIR (ATR) cm−1: 3270 (Amide A), 3086 (Amide B), 2927 and 2861 (CH2), 1738 (C=O), 1638 (Amide I), 1540 (Amide II) and 1145 (C-O). 1H-NMR (CDCl3/TFA, TMS as int. ref., ppm): 7.54 (s, 1H, CONH), 4.60 (s, 4H, OCH2CO), 3.49 (m, 4H, CONHCH2), 2.64 (m, 4H, OOCCH2), 1.80 (m, 4H, OOCCH2CH2), 1.71 (m, 4H, NHCH2CH2) and 1.47 (m, 2H, NHCH2CH2CH2).
- (5)
- Poly(glc-6-glc 6): Yield: 75%. m.p. 118 °C. Mw: 7,900 g/mol, Polydispersity: 2.8. FTIR (ATR) cm−1: 3288 (Amide A), 3083 (Amide B), 2937 and 2861 (CH2), 1744 (C=O), 1654 (Amide I), 1551 (Amide II) and 1142 (C-O). 1H-NMR (CDCl3/TFA, TMS as int. ref., ppm): 7.25 (s, 1H, CONH), 4.82 (s, 4H, OCH2CO), 3.41 (m, 4H, CONHCH2), 2.59 (s, 4H, OOCCH2), 1.75 (s, 4H, OOCCH2CH2), 1.62 (s, 4H, NHCH2CH2) and 1.39 (s, 2H, NHCH2CH2CH2).
3. Results and Discussion
3.1. Preparation of Electrospun Nanofibers of poly(glc-5-glc 6) and poly(glc-6-glc 6)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Polymer Concentration (%) | Flow Rate (mL/min) | Voltage (kV) | Diameter b (nm) |
|---|---|---|---|---|
| Poly(glc-5-glc 6) | 40 | 1 | 20 | 370, 630 |
| Poly(glc-5-glc 6) + 1% KTP | 40 | 1 | 20 | 410, 600 |
| Poly(glc-5-glc 6) + 5% KTP | 40 | 1 | 20 | 430, 590 |
| Poly(glc-6-glc 6) | 50 | 0.5 | 15 | 500, 2300 |
| Poly(glc-6-glc 6) + 1% KTP | 50 | 0.5 | 15 | 510, 1800 |
| Poly(glc-6-glc 6) + 5% KTP | 50 | 0.5 | 15 | 520, 1500 |

3.2. Spectroscopic and Calorimetric Data of Poly(glc-5-glc 6) and poly(glc-6-glc 6) Samples
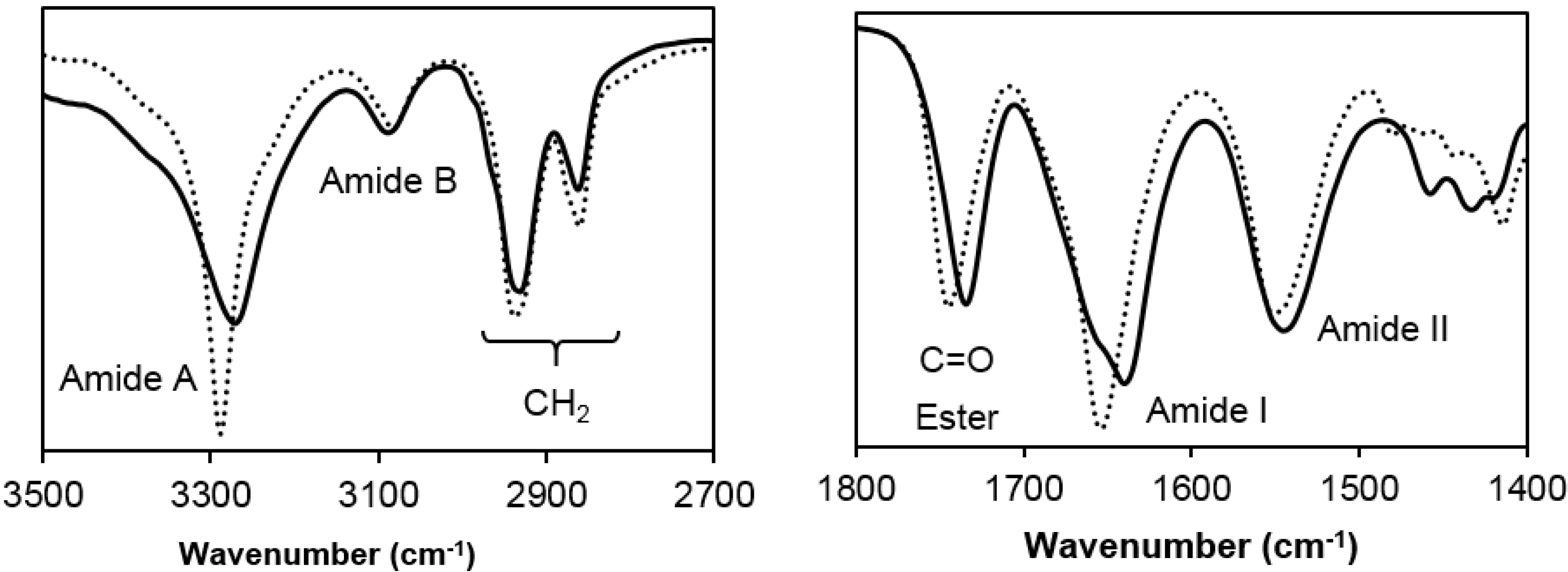
- (1)
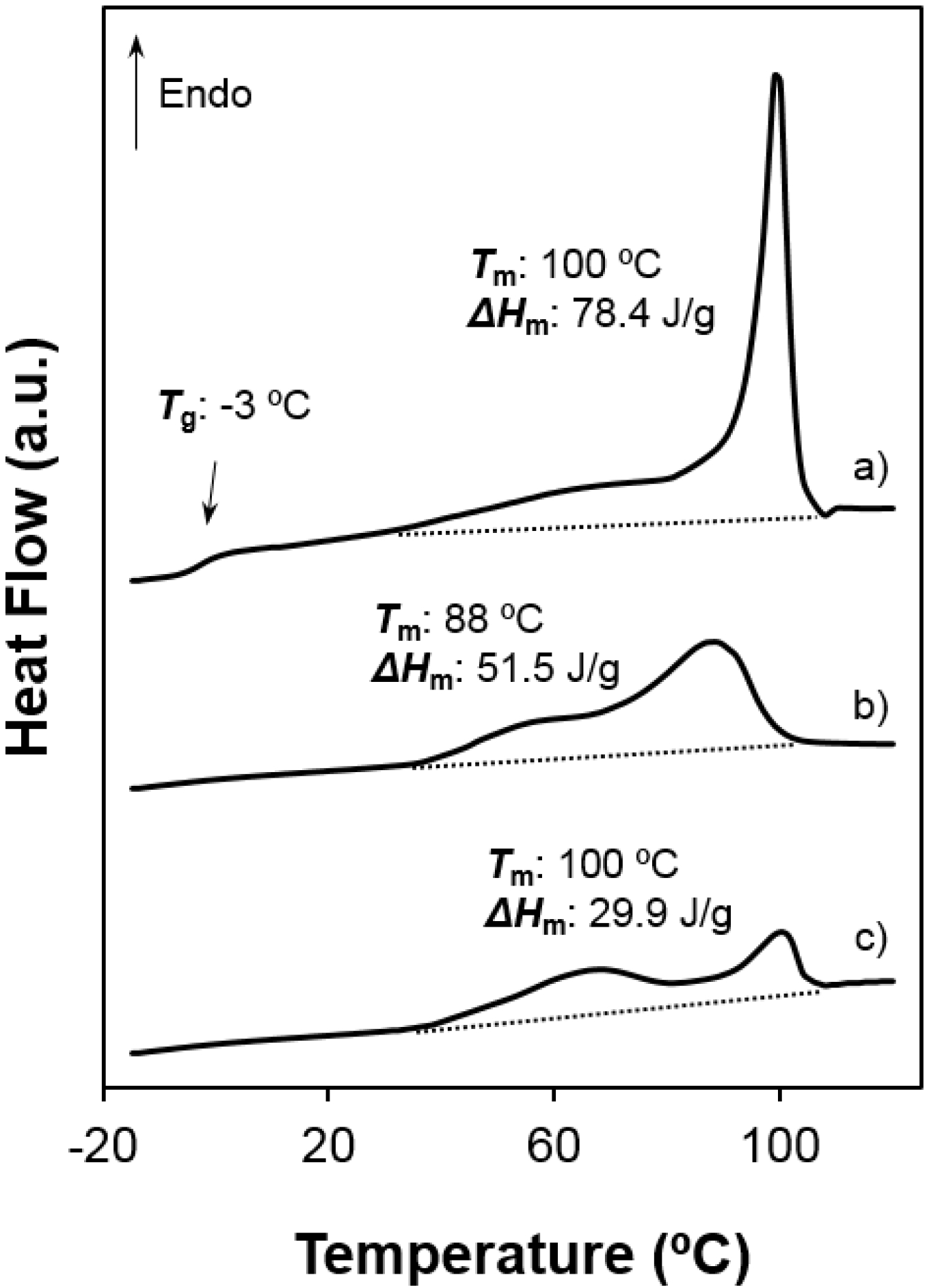
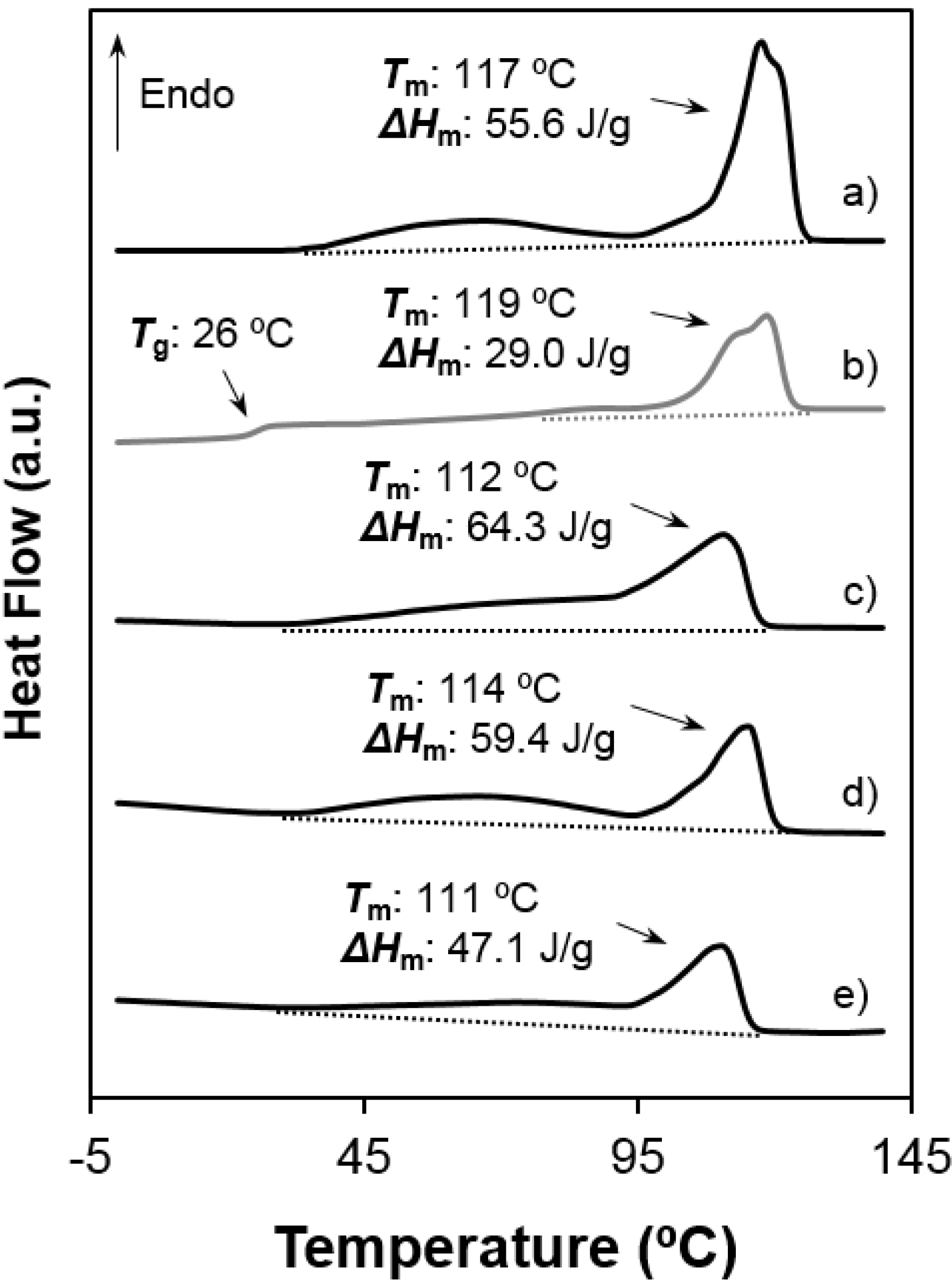
- Both samples were semicrystalline and had a relatively high degree of crystallinity. Note the high melting enthalpies (78.4 J/g and 55.6 J/g) determined for samples precipitated from solution. The amorphous character was defined by intermediate glass transition temperature values (i.e., −3 °C and 26 °C for 1,5-pentenediamine and 1,6-hexanediamine derivatives, respectively) with respect to the ones observed for aliphatic polyamides and polyesters.
- (2)
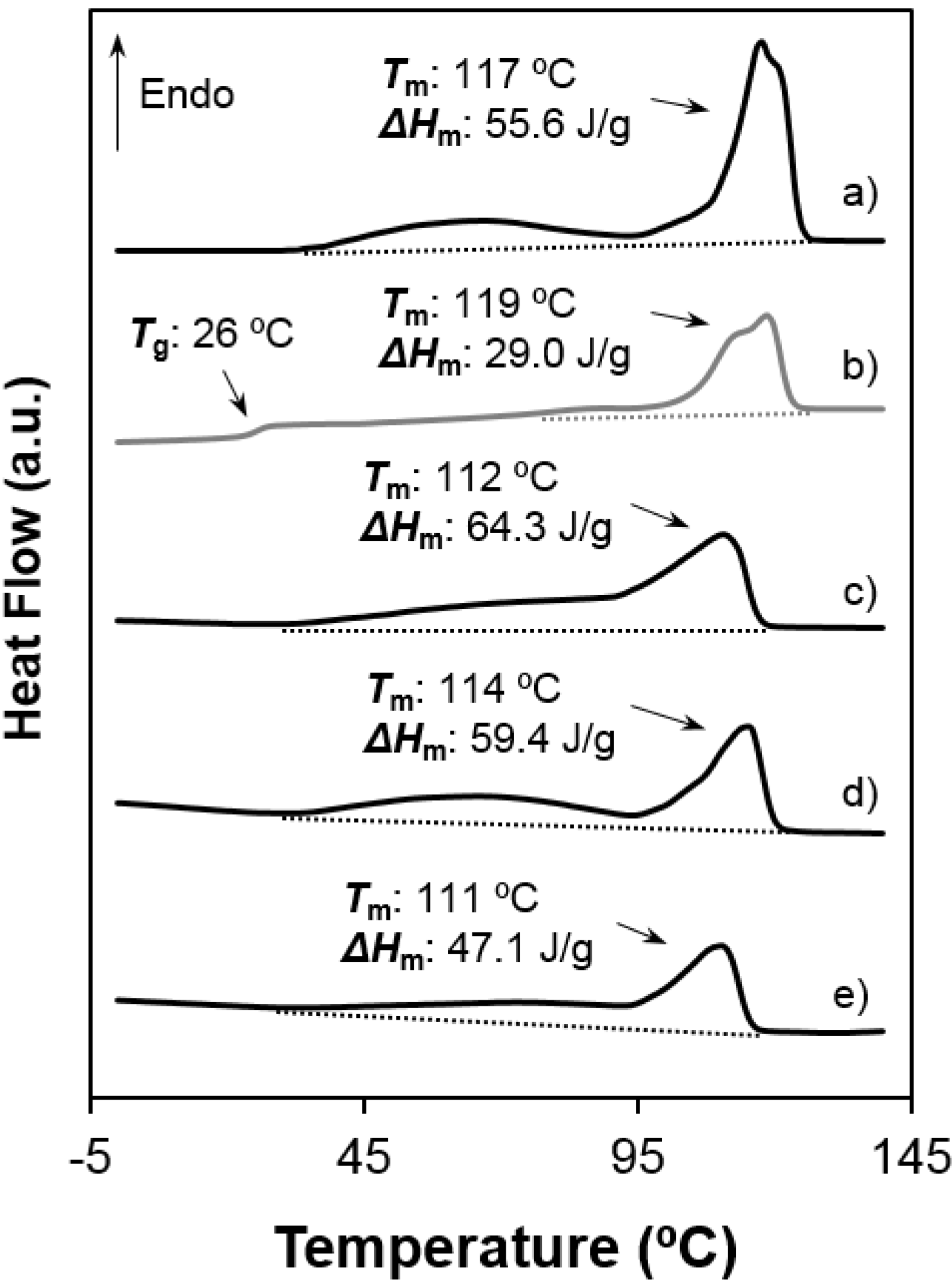
- Melting behaviour was complex since different peaks were always detected. The predominant one appeared at 100 °C and 117–119 °C, the higher value corresponding to the 1,6-hexanediamine derivative. These differences are in agreement with the well-known odd-even effect that reflects structural differences according to the parity and that is characteristic of nylons and aliphatic polyesters. Note that an increase on the number of methylene groups should give a decrease on the melting temperature if crystalline structure was not changed. Both samples showed also a broad peak at a lower temperature (i.e., 60–70 °C) that strongly depends on the preparation method (e.g., solution crystallization, solvent casting or electrospinning) and that should be associated with very defective crystals. The predominant melting peak of the 1,6-hexanediamine derivative appears aplitted up (117 ºC and 119 ºC) in some conditions probably as a consequence of the existence of different lamellar populations that experiment reordering processes during heating. Note, for example, that reordering should be more important for melt-quenched samples and, consequently, the intensity of the corresponding peak at 119 °C became the highest.
- (3)
- Enthalpy of the main melting peak clearly diminished when samples were prepared by solvent casting from 1,1,1,3,3,3,-hexafluoroisopropanol solution and chiefly when they were electrospun from the same solvent. The change was dramatic for the 1,5-pentanediamine derivative whose electrospun fibers had the highest solvent retention capability. Specifically, Figure 6 and Figure 7 clearly showed the higher decrease of the area of the melting peak for the electrospun 1,5-pentanediamine derivative, which means a clearly lower capability of this sample to crystallize during electrospinning regarding the even derivative.

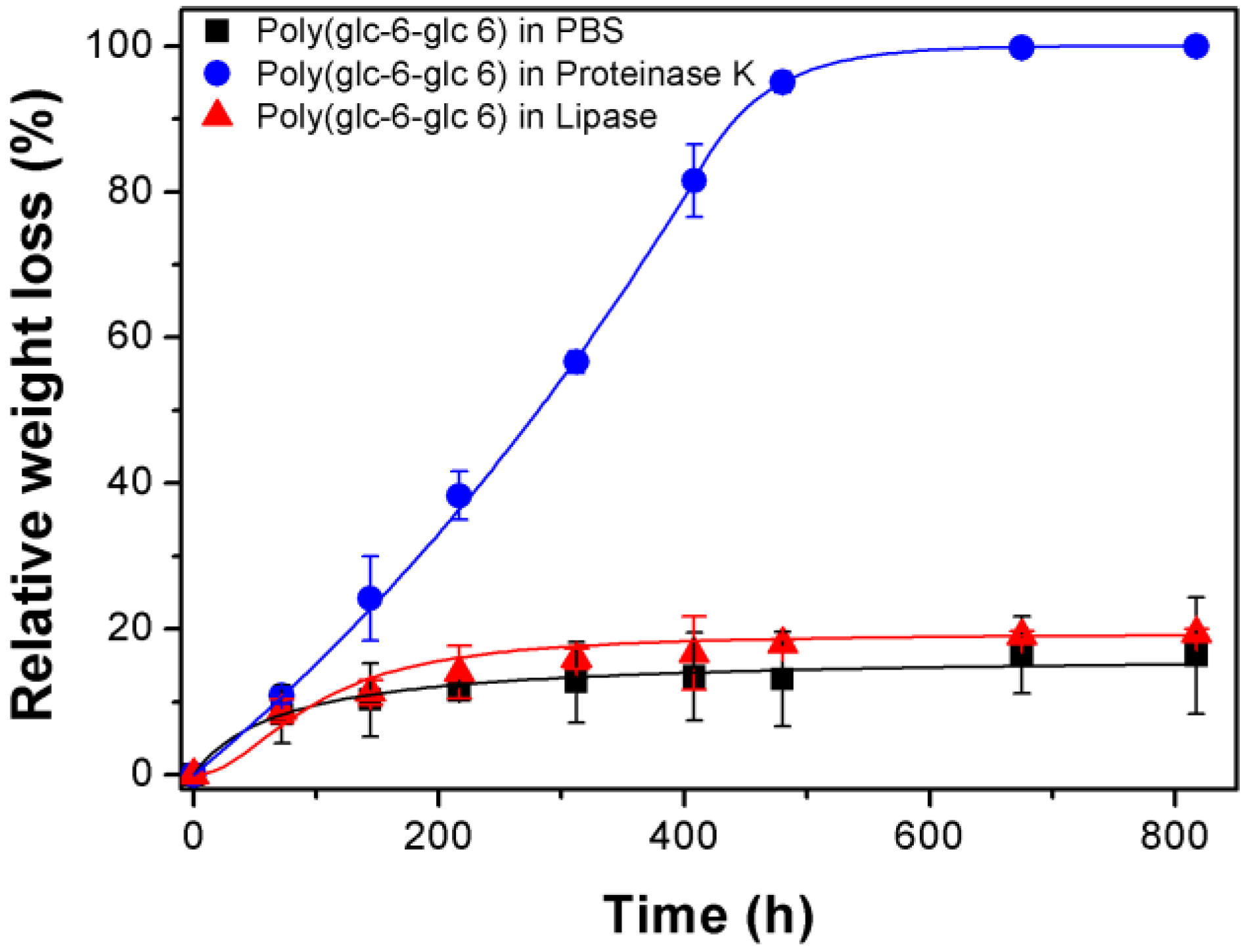
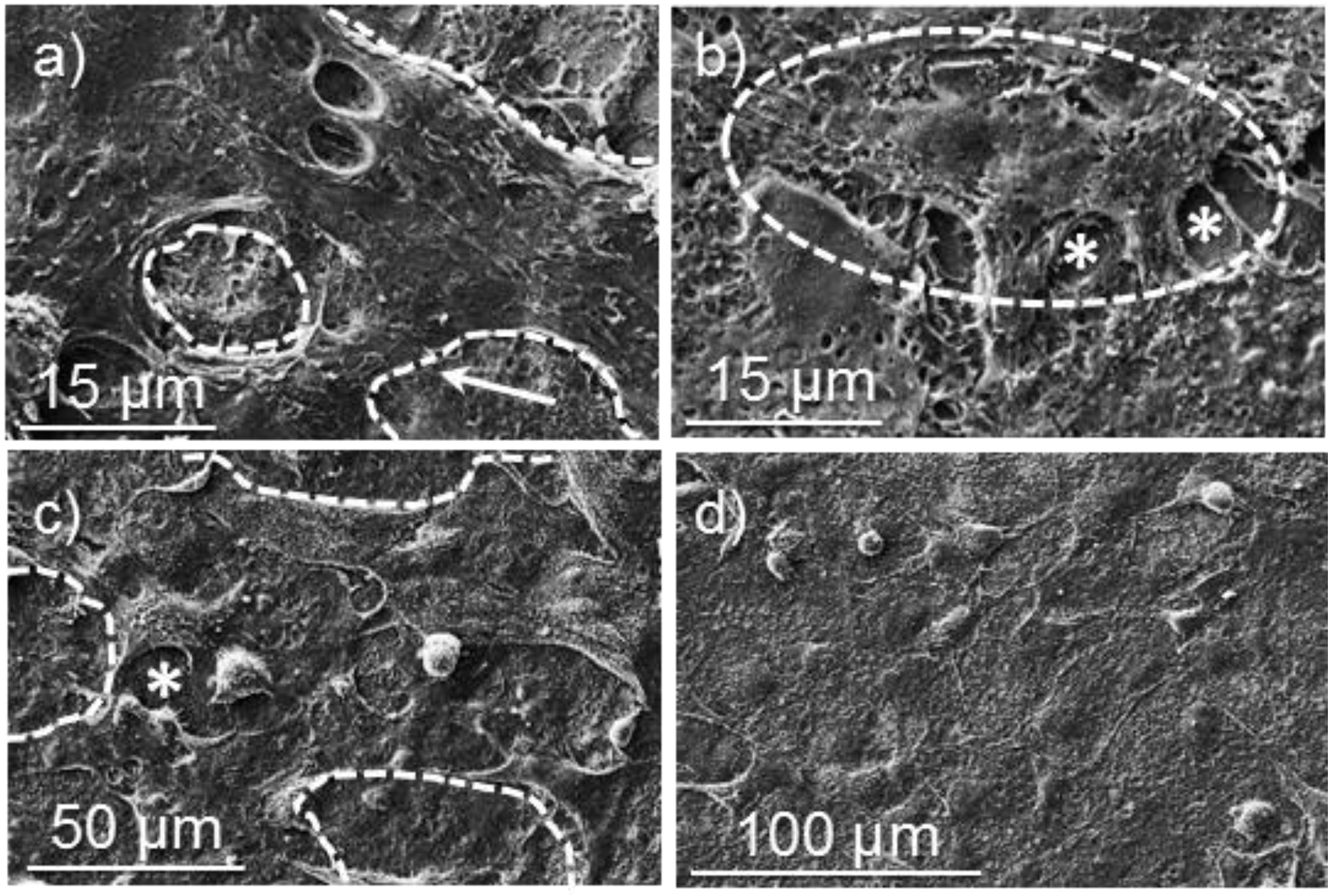
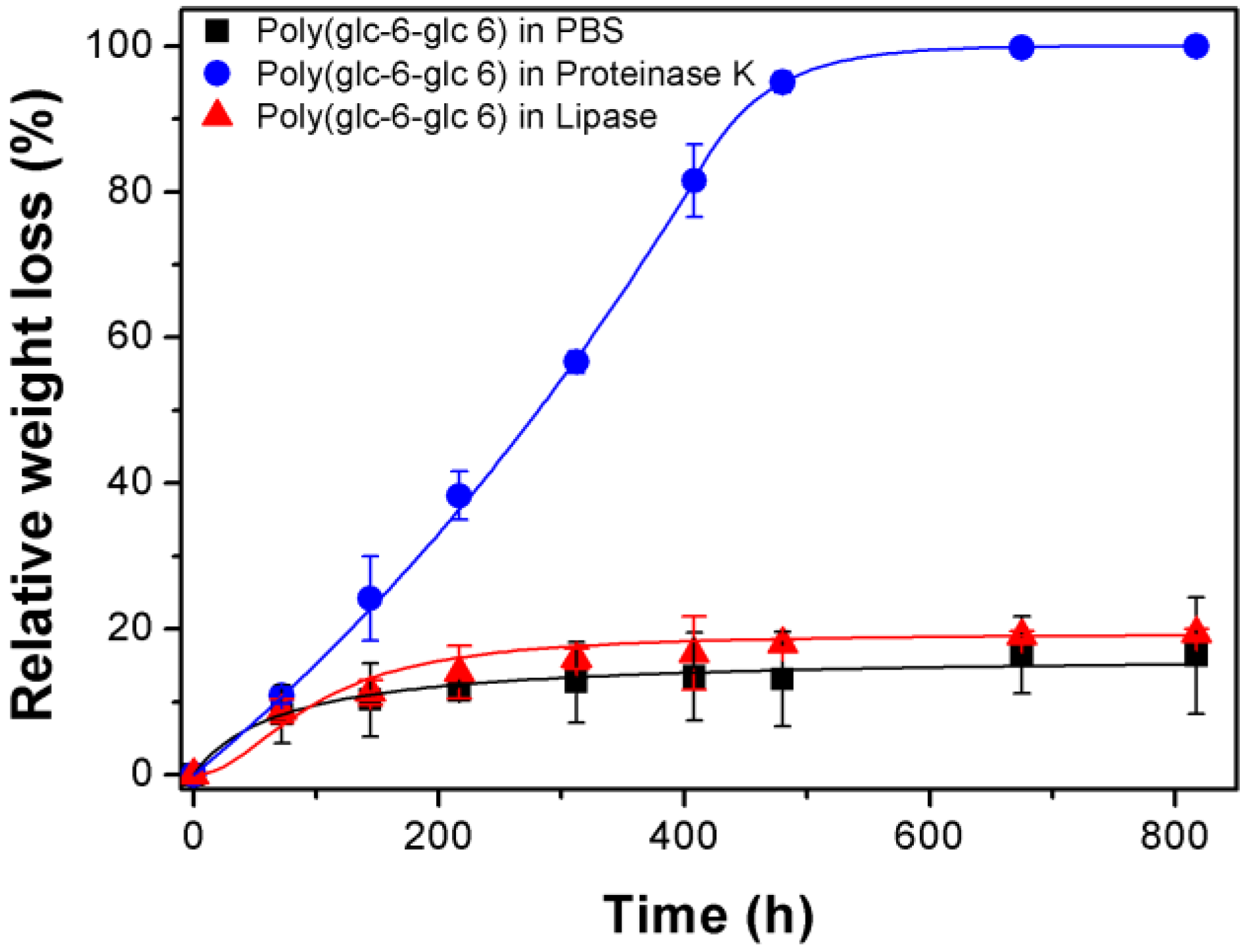
3.3. Degradation of Poly(glc-6-glc 6) Film Samples

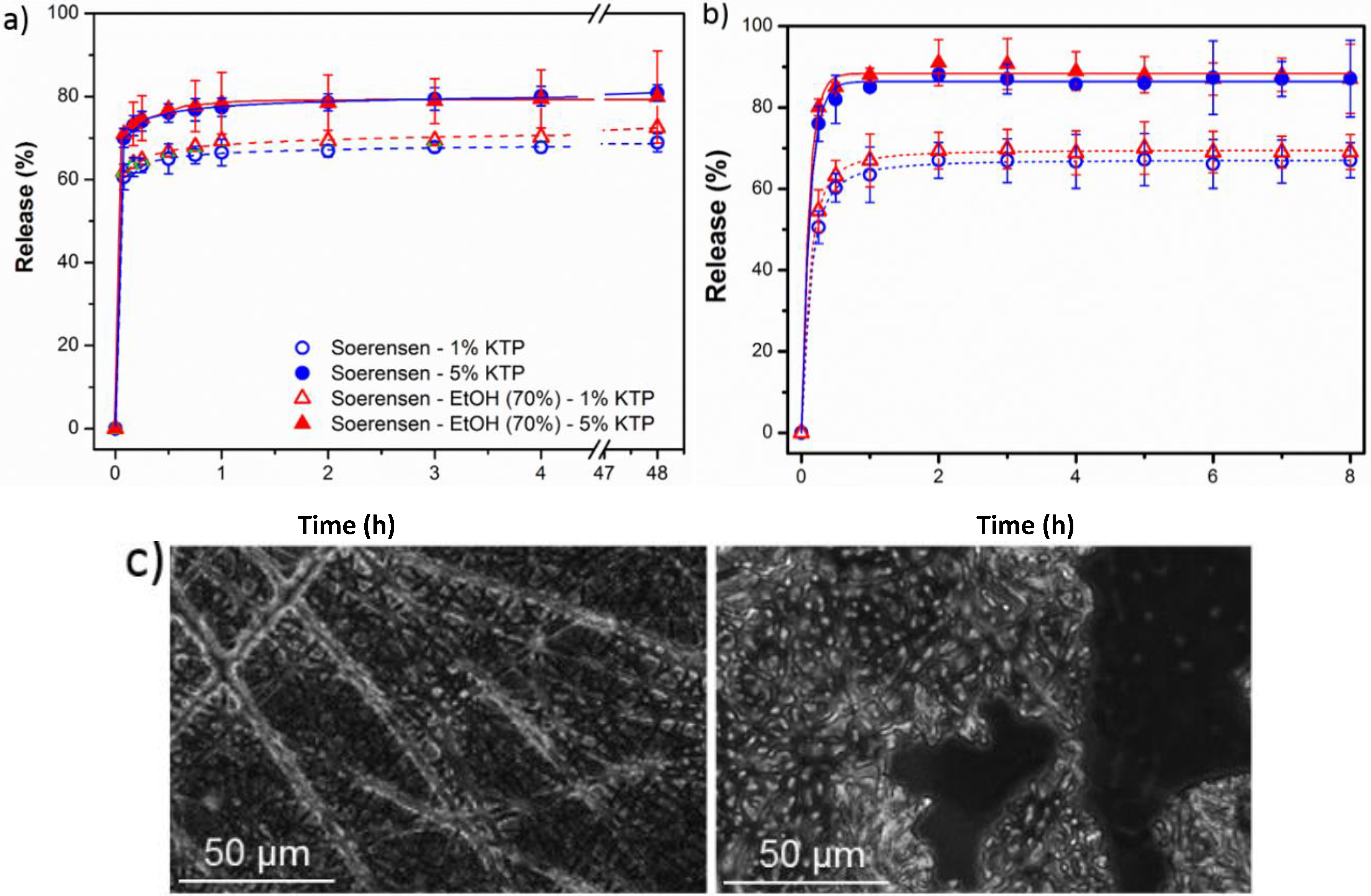
3.4. Load and Release of Ketoprofen in Poly(glc-5-glc 6) and Poly(glc-6-glc 6) Electrospun Samples
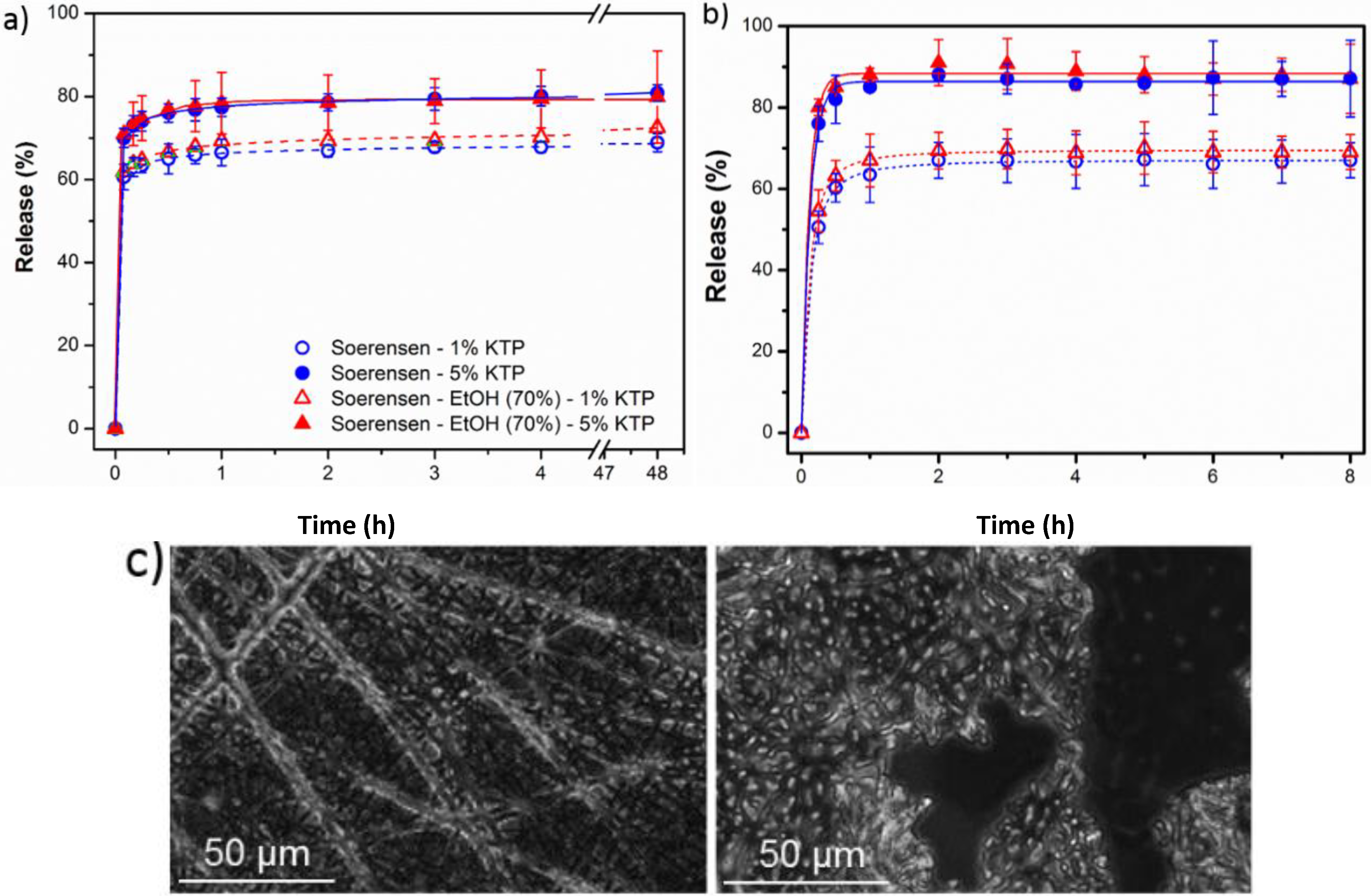
- (1)
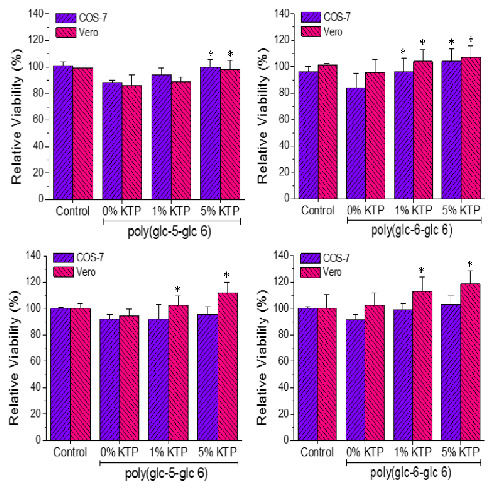
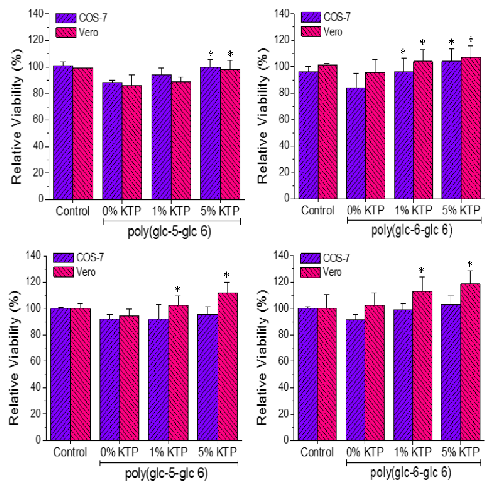
- A small percentage of the loaded drug was effectively trapped into the electrospun fibers, being the value higher when the drug loaded percentage was lower (e.g., 28%–30% and 13%–17% were determined for samples loaded with 1% and 5%, respectively). Probably some molecules were incorporated and effectively trapped in the crystalline domains, being the amount of KTP that could be incorporated in such domains limited. Therefore the release percentage increased with the drug load. Note also that diffusion of KTP from the crystalline arrangement is expected to be hindered. The increase in hydrophobicity of the medium (i.e., addition of ethanol) had a slight influence on the release behaviour, being only detected a small increase of the release percentage when samples were loaded with the lower amount of KTP (i.e., when the percentage of trapped molecules was higher).
- (2)
- Release behaviour was similar for the two matrices despite having different morphologies (e.g., fiber size and cross-sections), molecular weights and chemical structures.
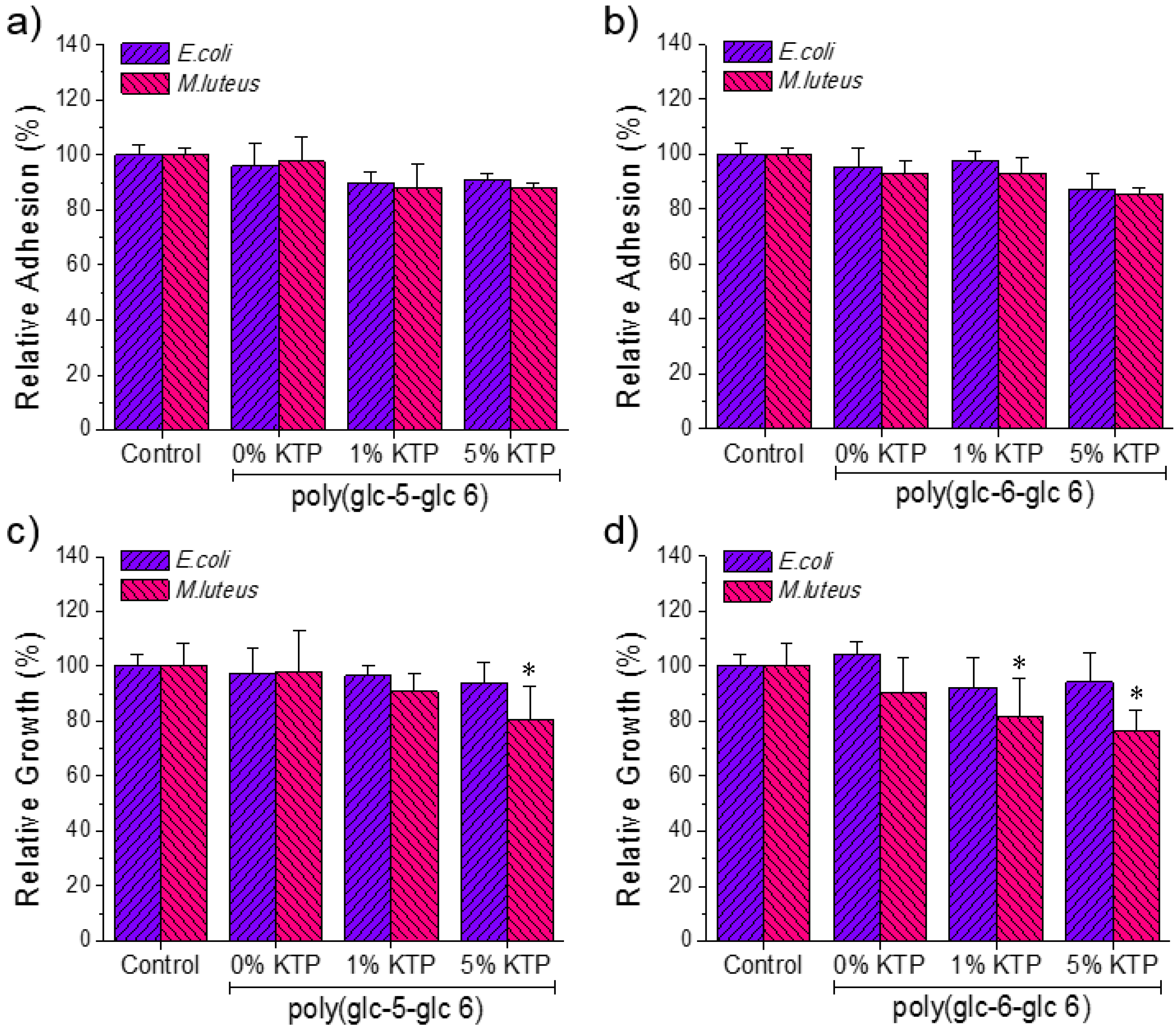
3.5. Bactericide Activity of Ketoprofen Loaded Poly(glc-5-glc 6) and poly(glc-6-glc 6) Electrospun Samples

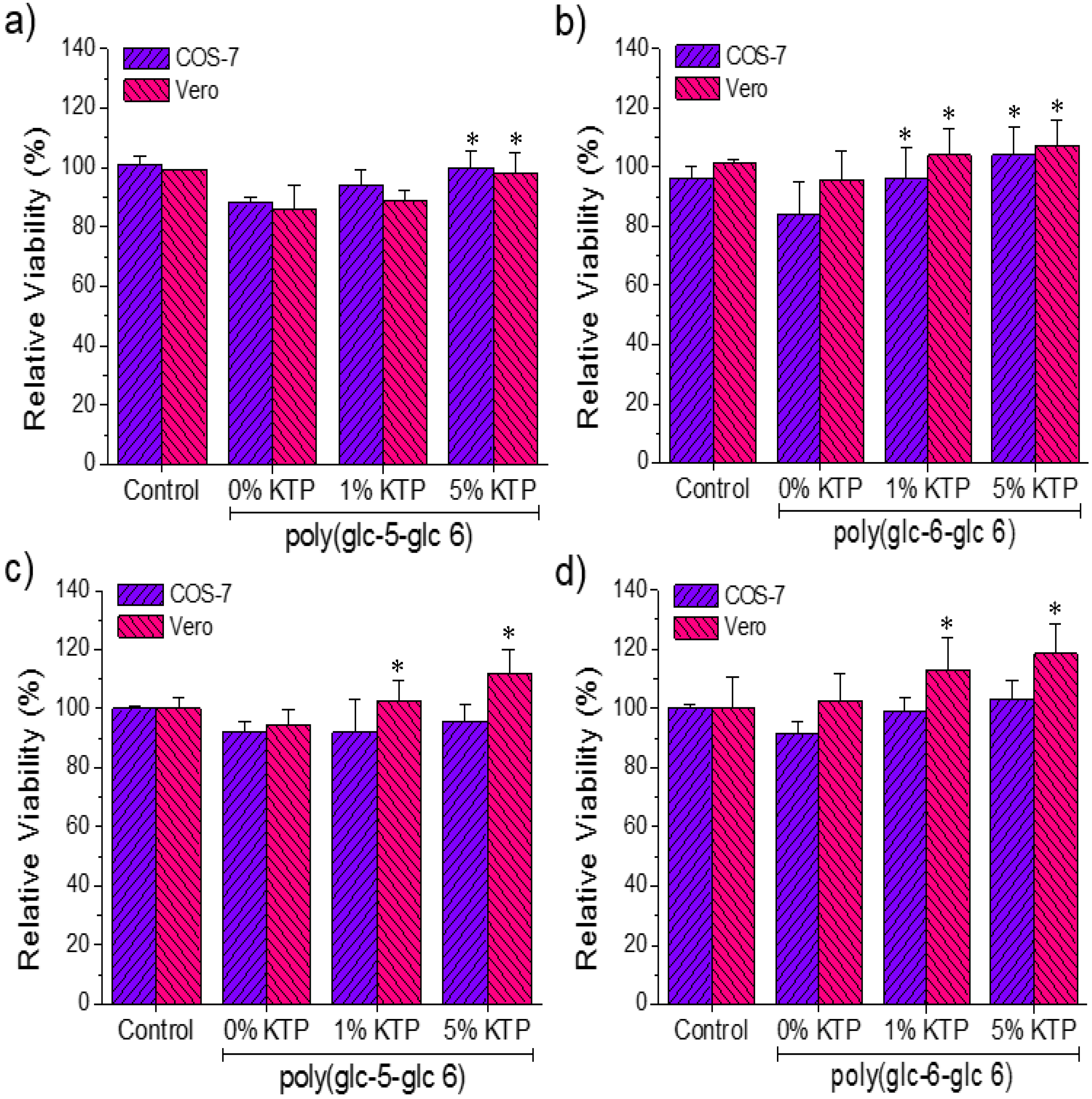
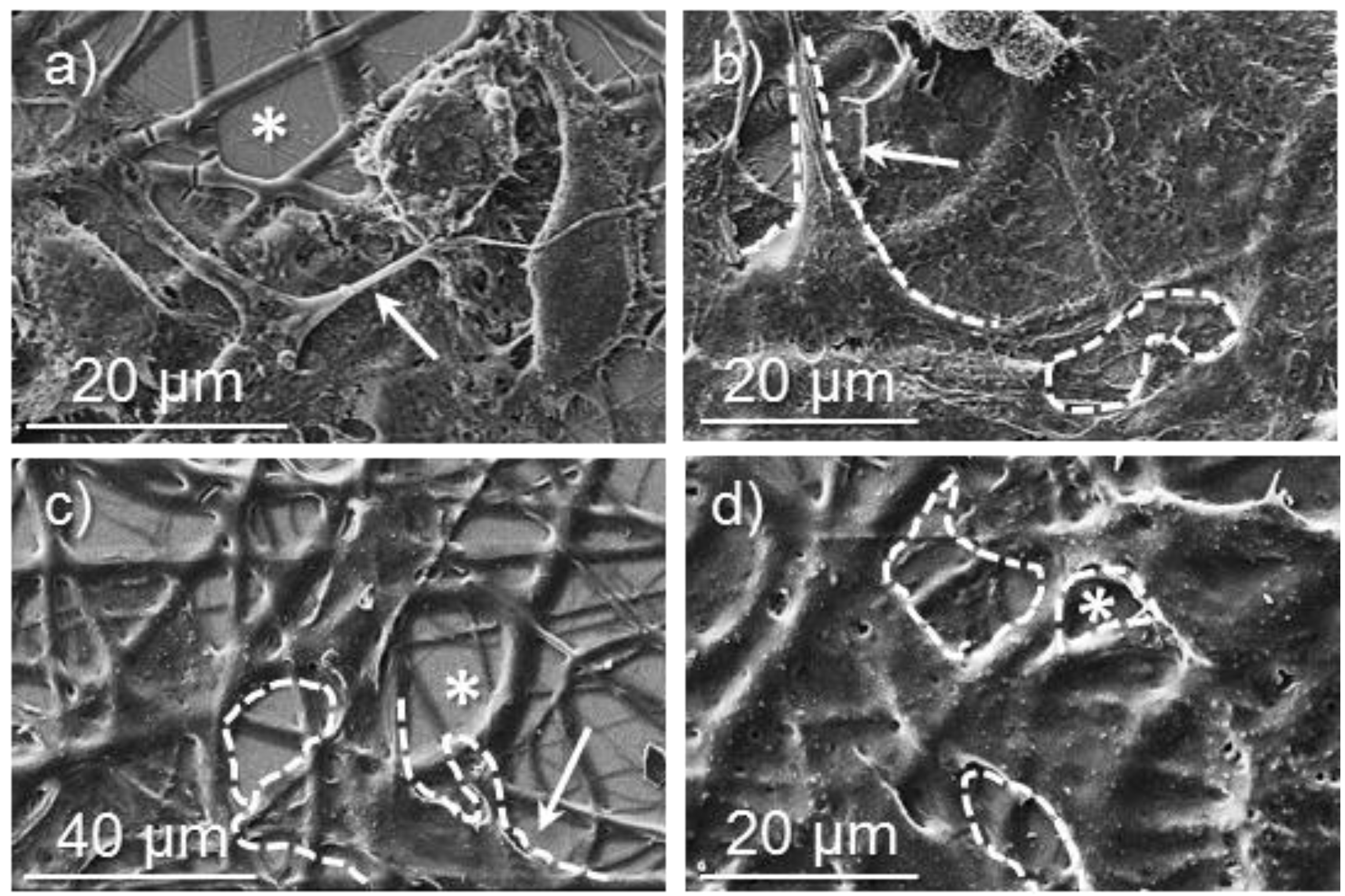
3.6. Biocompatibility of Poly(glc-5-glc 6) and Poly(glc-6-glc 6) Electrospun Samples
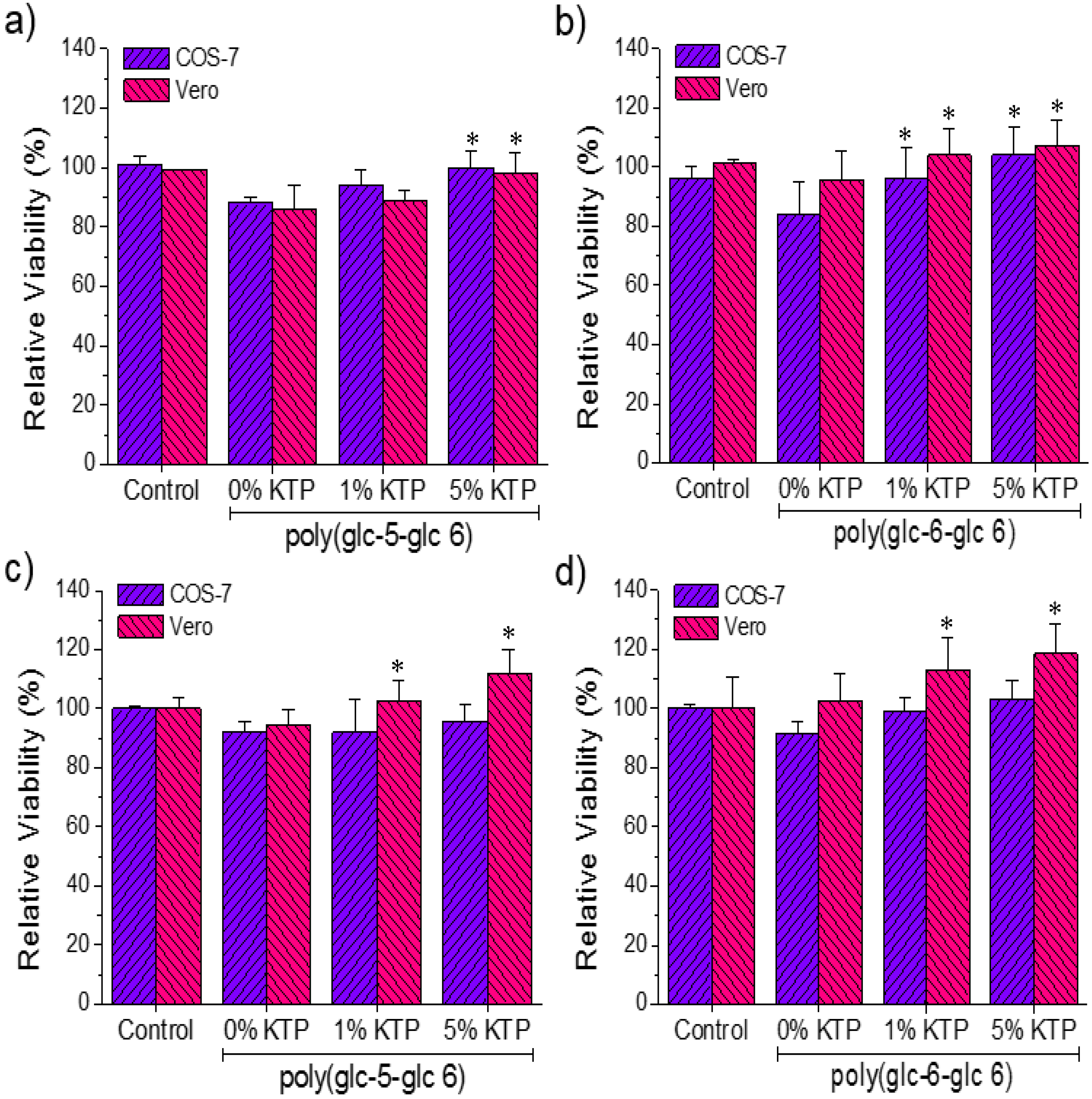


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qian, Z.; Li, S.; He, Y.; Zhang, H.; Liu, X. Preparation of biodegradable polyesteramide microspheres. Colloid Polym. Sci. 2004, 282, 1083–1088. [Google Scholar] [CrossRef]
- Ostacolo, L.; Russo, P.; de Rosa, G.; La Rotonda, M.I.; Maglio, G.; Nese, G.; Spagnuolo, G.; Rengo, S.; Oliva, A.; Quaglia, F. Poly(ether ester amide) microspheres for protein delivery: Influence of copolymer composition on technological and biological properties. Macromol. Biosci. 2008, 8, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Chu, C.C. Biodegradable and injectable paclitaxel-loaded poly(ester amide)s microspheres: fabrication and characterization. J. Biomed. Mater. Res. B. Appl. Biomater. 2009, 89, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-H.; Purevdorj, O.; Castano, O.; Planell, J.A.; Kim, H.-W. A short review: Recent advances in electrospinning for bone tissue regeneration. J. Tissue Eng. 2012, 3. [Google Scholar] [CrossRef]
- Han, D.K.; Hubbell, J.A. Synthesis of polymer network scaffolds from L-lactide and poly(ethylene glycol) and their interaction with cells. Macromolecules 1997, 30, 6077–6083. [Google Scholar] [CrossRef]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol-gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162. [Google Scholar] [CrossRef]
- Feng, Y.; Behl, M.; Kelch, S.; Lendlein, A. Biodegradable multiblock copolymers based on oligodepsipeptides with shape-memory properties. Macromol. Biosci. 2009, 9, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, H.; Jia, L.; Ling, J.; Tang, R.; Qiao, J.; Zhang, Z. Polyesteramides used for hot melt adhesives: Synthesis and effect of inherent viscosity on properties. J. Appl. Polym. Sci. 2001, 81, 2696–2701. [Google Scholar] [CrossRef]
- Ohya, Y.; Toyohara, M.; Sasakawa, M.; Arimura, H.; Ouchi, T. Thermosensitive biodegradable polydepsipeptide. Macromol. Biosci. 2005, 5, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Roby, M.S.; Jiang, Y. Polyesteramides with Amino Acid-Derived Groups Alternating with α-Hydroxiacid-Derived Groups and Surgical Articles Made Therefrom. U.S. Patent 5,914,387, 22 June 1999. [Google Scholar]
- Murase, S.K.; Franco, L.; del Valle, L.J.; Puiggalí, J. Synthesis and characterization of poly(ester amides)s with a variable ratio of branched odd diamide units. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Vera, M.; Admetlla, M.; Rodríguez-Galán, A.; Puiggalí, J. Synthesis, characterization and degradation studies on the series of sequential poly(ester amide)s derived from glycolic acid, 1,6-hexanediamine and aliphatic dicarboxylic acids. Polym. Degrad. STable 2005, 89, 21–32. [Google Scholar] [CrossRef]
- Ajinomoto Co., Inc.; Toray Industries, Inc. Ajinomoto and Toray to Conduct Joint Research on Biobased Nylon. Availalable online: http://www.ajinomoto.com/en/presscenter/press/detail/g2012_02_13.html (accessed on 7 January 2015).
- Rennovia Rennovia produces RENNLON nylon, a 100% bio-based nylon-6,6 polymer. Available online: http://www.rennovia.com/LinkClick.aspx?fileticket=UKm3fT8QZ-c=&tabid=62 (accessed on 7 January 2015).
- Cooley, J.F. Apparatus for Electrically Dispersing Fluids. U.S. Patent 692,631 A, 4 February 1902. [Google Scholar]
- Cooley, J.F. Electrical Method of Dispersing Fluids. US Patent 745,276 A, 24 November 1903. [Google Scholar]
- Hagiwara, K. Process for Manufacturing Artificial Silk and other Filaments by Applying Eletric Current. US Patent 1,699,615 A, 22 January 1929. [Google Scholar]
- Formhals, A. Process ad Apparatus for Preparing Artificial Threads. US Patent 1,975,504 A, 2 October 1934. [Google Scholar]
- Li, D.; Xia, Y. Electrospinning of nanofibers: Reinventing the wheel? Adv. Mater. 2004, 16, 1151–1170. [Google Scholar] [CrossRef]
- Dhakate, S.R.; Singla, B.; Uppal, M.; Mathur, R.B. Effect of processing parameters on morphology and thermal properties of electrospun polycarbonate nanofibers. Adv. Mater. Lett. 2010, 1, 200–204. [Google Scholar] [CrossRef]
- Llorens, E.; del Valle, L.J.; Ferrán, R.; Rodríguez-Galán, A.; Puiggalí, J. Scaffolds with tuneable hydrophilicity from electrospun microfibers of polylactide and poly(ethylene glycol) mixtures: Morphology, drug release behavior, and biocompatibility. J. Polym. Res. 2014, 21. [Google Scholar] [CrossRef]
- Llorens, E.; Calderón, S.; del Valle, L.J.; Puiggalí, J. Polybiguanide (PHMB) loaded in PLA scaffolds displaying high hydrophobic, biocompatibility and antibacterial properties. Mater. Sci. Eng. C 2015, 50, 74–84. [Google Scholar] [CrossRef]
- Planellas, M.; Pérez-Madrigal, M.M.; del Valle, L.J.; Kobauri, S.; Katsarava, R.; Alemán, C.; Puiggalí, J. Microfibres of conducting polythiophene and biodegradable poly(ester urea) for scaffolds. Polym. Chem. 2014, 6, 925–937. [Google Scholar] [CrossRef]
- Dhanalakshmi, M.; Jog, J.P. Preparation and characterization of electrospun fibers of Nylon 11. Express Polym. Lett. 2008, 2, 540–545. [Google Scholar] [CrossRef]
- Celebioglu, A.; Uyar, T. Electrospun porous cellulose acetate fibers from volatile solvent mixture. Mater. Lett. 2011, 65, 2291–2294. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, Y.; Lee, K.-R.; Lai, J.-Y. A three-dimensional dual-layer nano/microfibrous structure of electrospun chitosan/poly(d,l-lactide) membrane for the improvement of cytocompatibility. J. Memb. Sci. 2014, 450, 224–234. [Google Scholar] [CrossRef]
- Koski, A.; Yim, K.; Shivkumar, S. Effect of molecular weight on fibrous PVA produced by electrospinning. Mater. Lett. 2004, 58, 493–497. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Ganguly, K.; Chaudhuri, K.; Chakrabarty, A.N. The anti-bacterial action of diclofenac shown by inhibition of DNA synthesis. Int. J. Antimicrob. Agents 2000, 14, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Abaas, H.A.; Serry, F.M.; El-Masry, E.M. Combating pseudomonas aeruginosa biofilms by potential biofilm inhibitors. Asian J. Res. Pharm. Sci. 2012, 2, 66–72. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murase, S.K.; Del Valle, L.J.; Puiggalí, J. Electrospun Scaffolds from Low Molecular Weight Poly(ester amide)s Based on Glycolic Acid, Adipic Acid and Odd or Even Diamines. Fibers 2015, 3, 151-172. https://doi.org/10.3390/fib3020151
Murase SK, Del Valle LJ, Puiggalí J. Electrospun Scaffolds from Low Molecular Weight Poly(ester amide)s Based on Glycolic Acid, Adipic Acid and Odd or Even Diamines. Fibers. 2015; 3(2):151-172. https://doi.org/10.3390/fib3020151
Chicago/Turabian StyleMurase, Sara Keiko, Luís Javier Del Valle, and Jordi Puiggalí. 2015. "Electrospun Scaffolds from Low Molecular Weight Poly(ester amide)s Based on Glycolic Acid, Adipic Acid and Odd or Even Diamines" Fibers 3, no. 2: 151-172. https://doi.org/10.3390/fib3020151
APA StyleMurase, S. K., Del Valle, L. J., & Puiggalí, J. (2015). Electrospun Scaffolds from Low Molecular Weight Poly(ester amide)s Based on Glycolic Acid, Adipic Acid and Odd or Even Diamines. Fibers, 3(2), 151-172. https://doi.org/10.3390/fib3020151