3.1. Electrolyte Speciation
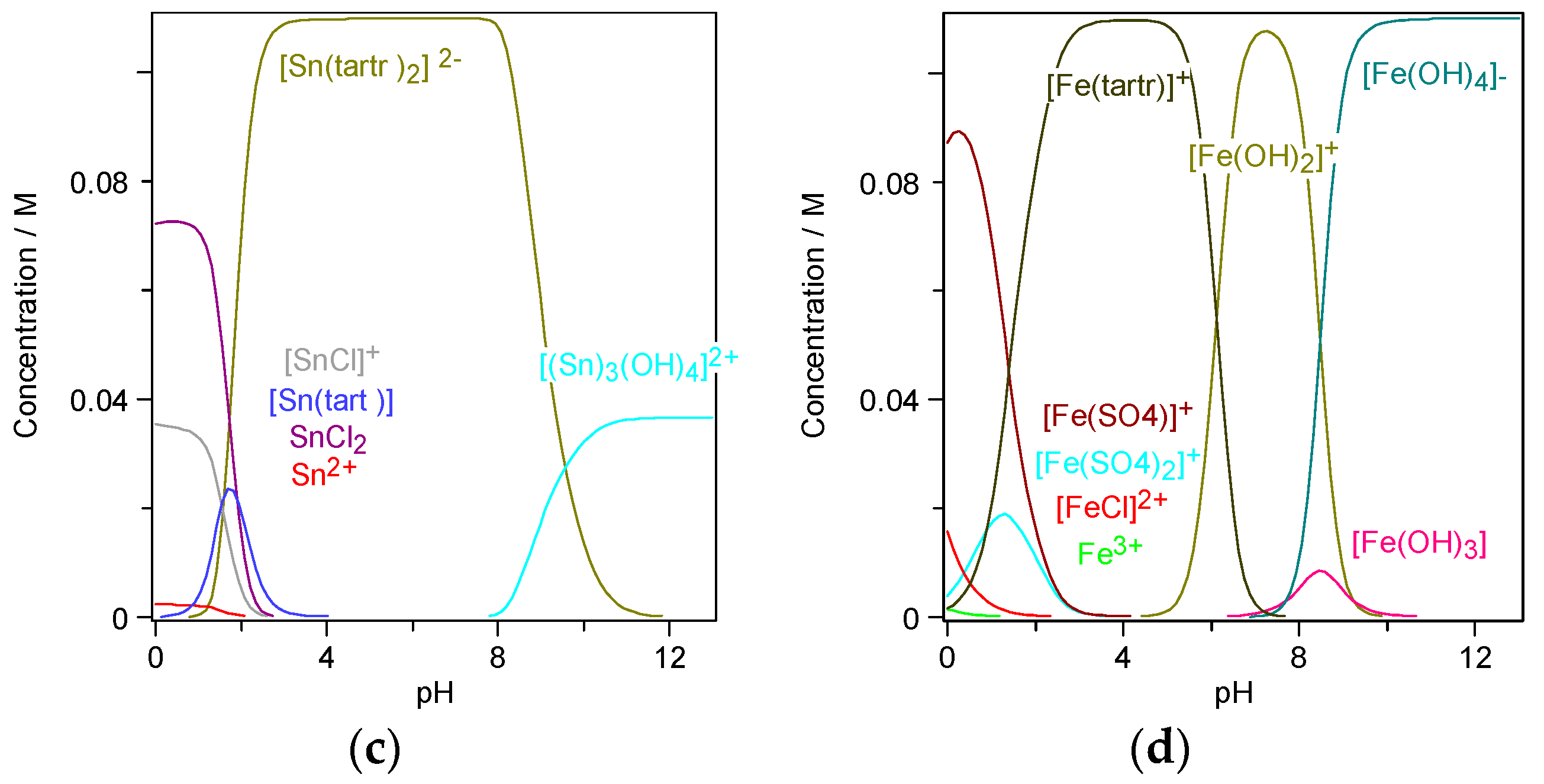
In aqueous solutions, both stannous and ferric ions can be present as free ions as well as in the form of complexes with tartrate, chloride, and sulphate. Based on the chemical equilibrium, the ferric-stannous redox reaction was not considered. The calculation of the metal speciation was performed with the Software Visual MINDTEQ 3.1 [
12]. The pH-dependent speciation of the electrolytes was calculated by the HySS software [
13]. The results are depicted in
Figure 1. In the acidic region of mixed Fe–Sn electrolytes stannous ions are present either as free ions, charged or neutral chloride complexes, or as neutral Sn-tartrate complexes. In the pH region from 3.0 to 8.0, all tin molecules are present in the stable form of [Sn(tartr)
2]
2− complexes. In the weak alkaline region, tin(II) hydroxides are observed (
Figure 1c). Ferric ions in the mixed Fe–Sn acidic electrolytes are present as free ions or charged chloride and sulfate complexes. In the pH range from 3.0 to 5.0, all ferric molecules are present in the stable complex [Fetart]
+. Soluble [Fe(OH)
2]
+ starts to appear around pH 5.0, while insoluble hydroxides Fe(OH)
3 and [Fe(OH)
4]
− are formed in the weak alkaline region (pH ~8.0) (
Figure 1d). Considering the pH dependence of electrolyte speciation, the pH range for stable ferric-stannous electrolytes was assumed to be between the values of 3.0 and 5.0.
3.2. Electrochemical Characterization
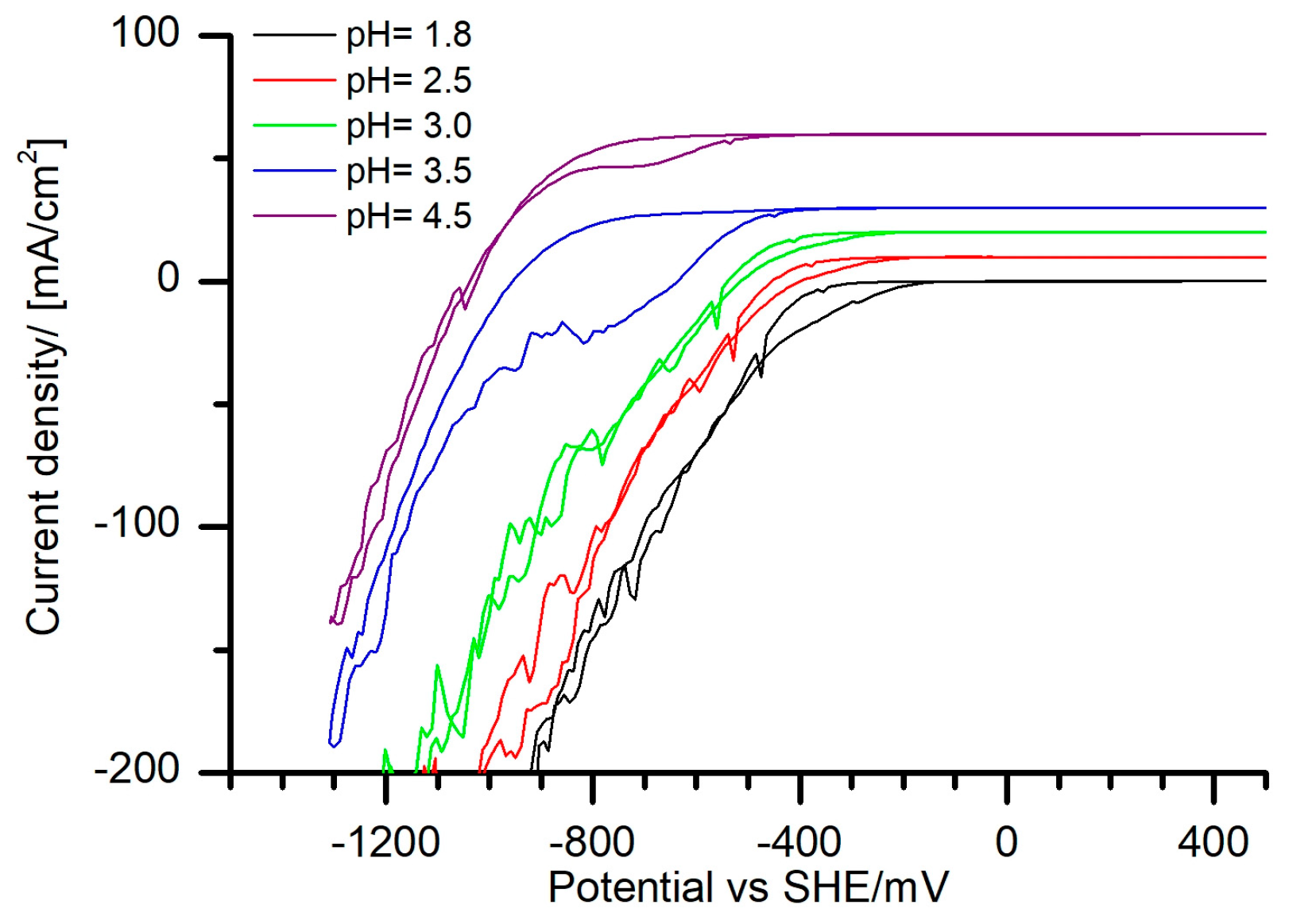
As a first step, CVs in the supporting electrolyte, without any metal ions, at different pH values were measured (
Figure 2). For all measured CVs, reduction peaks attributed to hydrogen reduction were observed in the cathodic sweeps, independent of the pH values of the electrolytes. At higher pH, with the beginning at a pH value of 3.0, a reduction peak around −800 mV started to appear. This peak, reaching its maximum intensity at the pH value of 3.5, was not related to any oxidation peak (
Figure 2). According to the speciation of the supporting electrolyte (
Figure 1a) within the pH range of 2.0 to 4.0 the most of the tartrate ligands were present in the form of [H
2(tart)] and [H(tart)]
−. Predominant species around pH value of 3.5 was [H(tart)]
−. Therefore, the cathodic peak, observed around −800 mV (
Figure 2), could be related to the reduction of hydrogen ions from the [H(tart)]
− ion [
15]. During the anodic sweep of the CVs, no oxidation peaks were detected (
Figure 2). Therefore, it can be assumed that no oxidation of tartaric acid or its anions occurred.
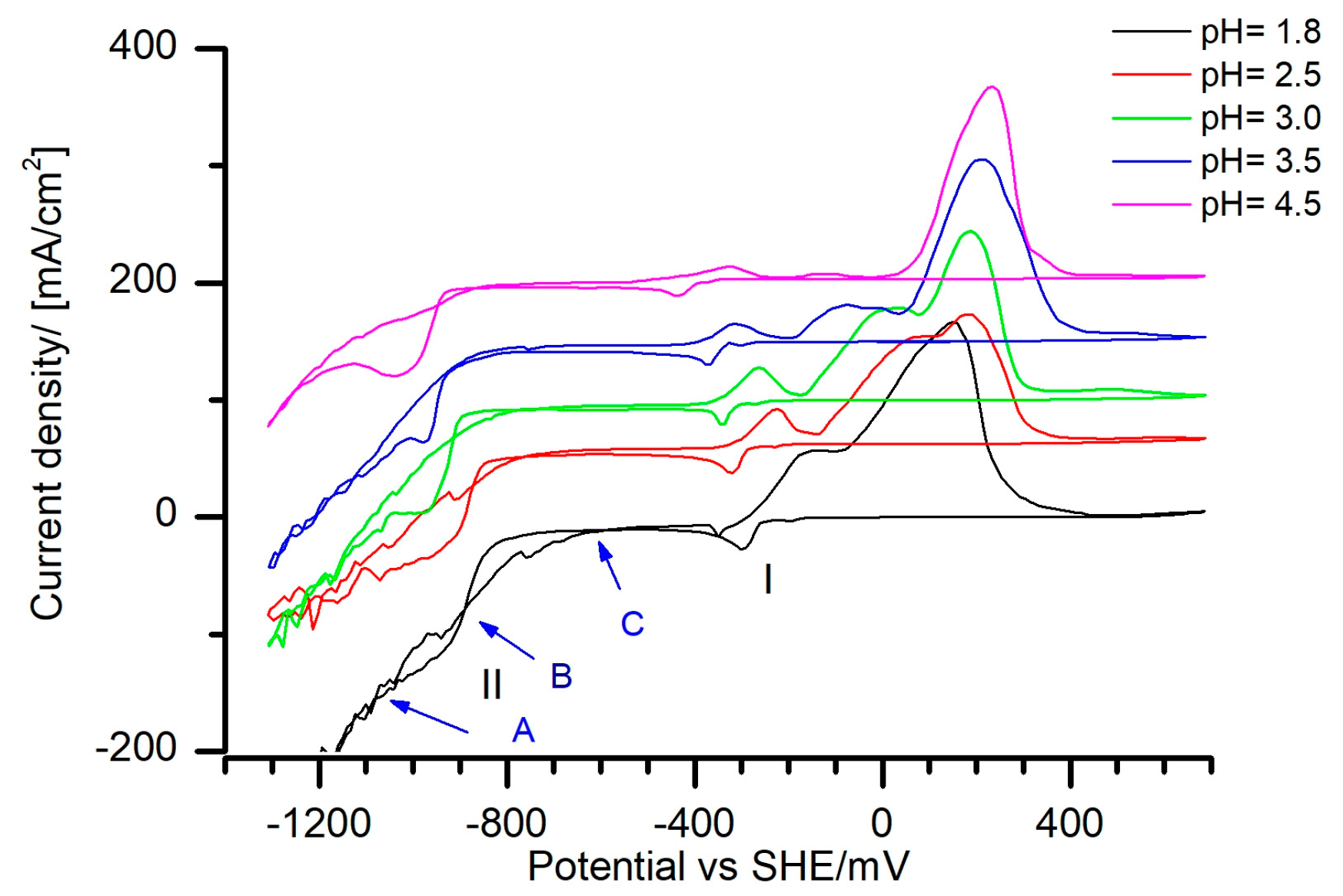
Results of the cyclic voltammograms (CVs) in the Fe(III)–Sn(II) electrolyte for various pH values are depicted in the
Figure 3. In the cathodic branch, two reduction peaks (I, II) and three crossovers (A, B, C) were detected. The peak Nr. I was assigned to the Sn reduction and it is strongly pH dependent. By increasing the pH, the Sn reduction peak was shifted towards more negative potentials. Such a shift was also observed by Lingane [
16] in the Sn(II)-tartrate electrolytes. The author explained it with the fundamental difference in the composition of Sn(II)-tartrate complexes in the acidic and alkaline media. Moreover, according to the polarographic measurements, the presence of ferric ions in the tartrate electrolyte containing a 1:1 ratio of Sn(II) and Fe(III) did not change the reduction potential of Sn(II) [
17]. According to the electrolyte speciation (
Figure 1c), in the case of Fe(III)–Sn(II) tartrate electrolyte, the character of the stannous complexes changed from the [SnCl]
−, [SnCl], and [Sn(tart)] in the acidic media through [Sn(tart)]
+ in weak acidic-neutral towards [Sn
3(OH)
4]
2+ in the alkaline media. The concentration of the [Sn(tart)
2]
2− complexes within the pH values of 3.0 and 7.0 is almost constant. Moreover, the pK
s2 value of tartaric acid is 4.34 [
18]. Due to the dissociation of [H(tart)]
−, the change of concentration of [tart]
2− reaches a maximum around this pH value (
Figure 1a). Therefore, it is reasonable to assume that the shift of the Sn reduction potential in this pH region is due to the increased stability of the [Sn(tart)
2]
2− complex.
A decrease of the Sn reduction peak intensity as a function of a pH was detected (
Figure 3). A possible explanation could be the suppression of Sn reduction due to the presence of ferric ions. A similar effect was observed in the Ni–Fe anomalous co-deposition [
19] where the electrodeposition of the more noble Ni was suppressed by the presence of Fe at relatively low overpotentials. The suppression of Ni began when Fe-hydroxides where formed on the cathode surfaces. According to the Fe(III)–Sn(II) electrolyte speciation, soluble Fe-hydroxides are starting to form around pH value of 4.0 (
Figure 1d). Due to the local rise of the pH on the cathode surface, Fe-hydroxides might also be present in electrolytes with a bulk pH below 4.0. This finding indicates, that with the increase of the electrolyte pH, Fe–Sn might undergo anomalous deposition.
The Sn reduction peak was followed by the diffusion limiting current density plateau in all of the studied electrolytes (
Figure 3). According to the Nernst equation [
20], the standard hydrogen evolution potential (
E0) is shifted from −104 to −261 mV for pH values of 1.8 and 4.5 respectively. This indicates that hydrogen evolution (HER) is present as a side reaction over the whole deposition potential range [
21]. The presence of various additives and complexing agents in the electrolyte can shift the HER to higher overpotentials than
E0. In the case of the Fe(III)–Sn(II) electrolyte used, the HER potential was shifted to values of −900 mV and below, and a linear increase of the current density was observed (
Figure 3).
The reduction peak Nr. II was shifted from −890 towards −962 mV for the pH values of 1.8 and 4.5, respectively. In the vicinity of peak Nr. II two crossovers (A, B) of cathodic and anodic scans were observed (
Figure 3). Generally, for single metal electrodeposition, the presence of a crossover indicates the existence of a nucleation process. The first crossover corresponds to the nucleation overpotential, since the potential of metal deposition on itself is more positive than for the metal deposition on a foreign substrate. The second crossover corresponds to the equilibrium potential of Metal/Metal
n+ couple [
22]. On the other hand, the reduction peak around −800 mV in the supporting electrolyte was assigned to the reduction of the hydrogen from the [H(tart)]
− and [H
2(tart)] species (
Figure 1a and
Figure 2). For these reasons, the cathodic peak Nr. II in the Fe(III)–Sn(II) electrolyte (
Figure 3) might be related either to the reduction of some metal phase or to hydrogen reduction from [H(tart)]
− and [H
2(tart)] complexes.
The anodic sweeps of the CVs exhibited a rather complex behavior. Two different current peaks were detected at pH values of 1.8 and 2.5. Starting from the pH value of 3.0, three different current peaks appeared (
Figure 3). Considering the observed pH influence on the Sn reduction potential and the increased number of the stripping peaks, it was assumed that starting from the pH value of 3.0 additional Fe–Sn metal phases could be deposited.
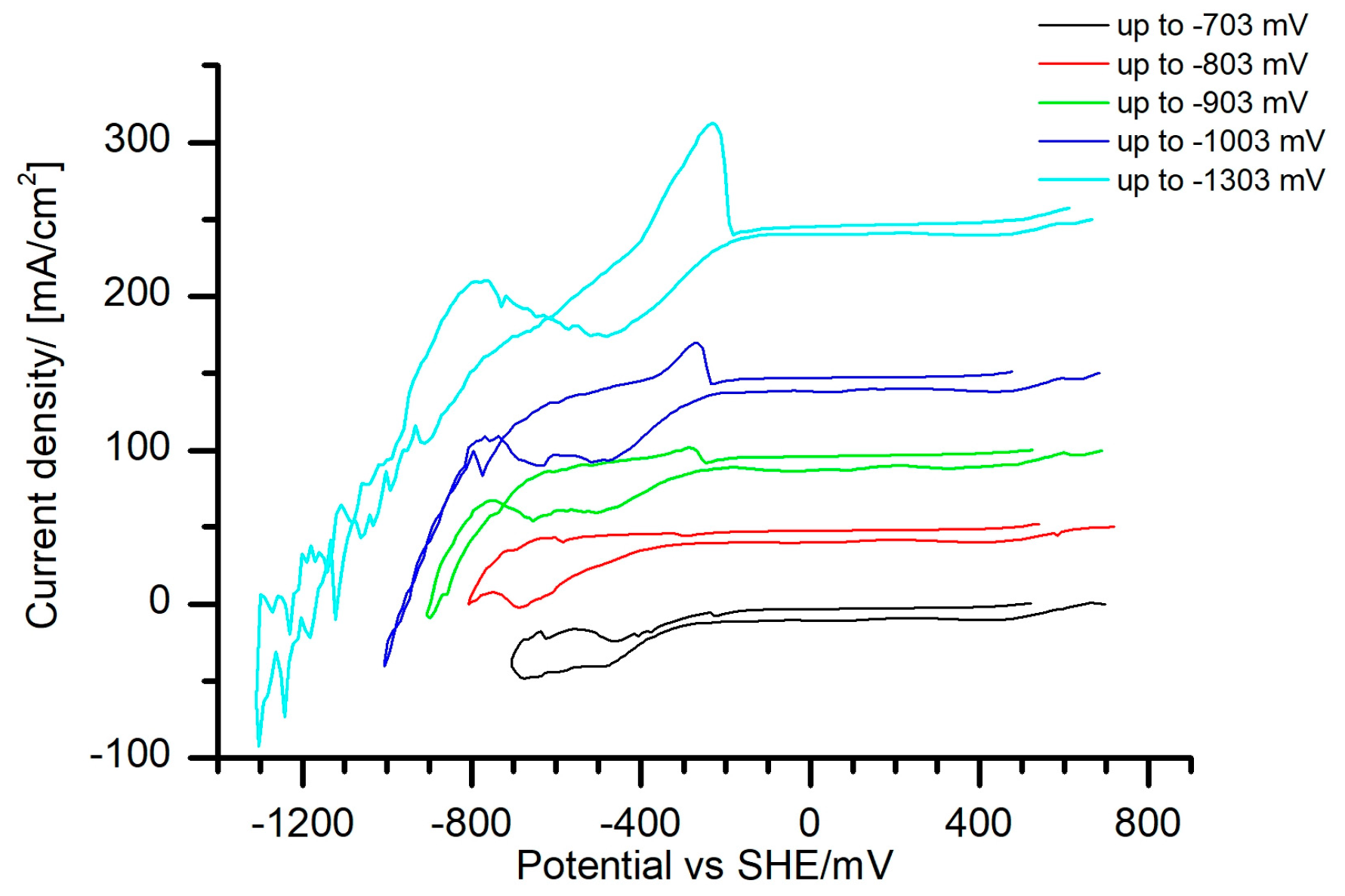
In order to relate the peak Nr. II (
Figure 3) with hydrogen or with metal phase reduction, the iron deposition potential was studied. For this purpose, CVs with different cathodic reverse potentials in the single metal Fe(III)-tartrate electrolyte were measured (
Figure 4). An anodic dissolution peak was first observed when the electrode was polarized cathodically up to −903 mV. On the other hand, no dissolution peak was observed when the electrode was polarized to −803 mV. Hence, the position of Fe deposition potential at the pH value of 1.8 was located between −803 and −903 mV. This finding confirms that the cathodic peak Nr. II in the mixed Fe–Sn electrolyte (
Figure 3) could be also related to the deposition of Fe or an Fe–Sn phase.
3.3. Structural Characterization
In order to study the Fe–Sn electrodeposition in the current density range from 1 to 80 mA/cm
2, Hull cells were used. SEM topography images and deposit stoichiometries are listed in
Table 1. The surface of the Hull cell cathodes, deposited from electrolytes in the pH range of 1.8 to 3.0, was covered with dendrites over the whole current density range. At higher pH values, 3.5 and 4.5, the character of the dendrites changed to powder (not shown). In order to analyze the compact layer below the dendrites and the powder, these were removed from the samples by a cotton pad with isopropanol. In all studied samples, a high amount of oxygen was observed (
Table 1). The oxygen content varied from 48.7 to 21 at % for the samples deposited at pH value 1.8 with 20 mA/cm
2 and the pH value of 4.5 with the current density of 80 mA/cm
2, respectively. Samples deposited at the pH value of 1.8 had the highest surface roughness and therefore, also the biggest surface area. Additionally, no Fe or Sn hydroxides should be present at such an acidic pH (
Figure 1c,d). Therefore, it could be assumed that the high oxygen content is a surface phenomenon dependent on the deposit surface roughness. From the EDX analyses, it is obvious that, with the increase of the electrolyte pH, the amount of Sn in the deposit decreased. Iron-rich deposits were obtained with both 20 mA/cm
2 (pH values of 3.5 and 4.5) and 80 mA/cm
2 (pH range from 3.0 to 4.5) (
Table 1). This observation agrees with the potentiodynamic results, in which suppression of the reduction of the more noble Sn with the increasing pH was detected (
Figure 3).
Anomalous co-deposition, enhanced deposition of the less noble metal, and suppression of the reduction of the more noble one, has been reported for various iron alloys [
19,
23,
24,
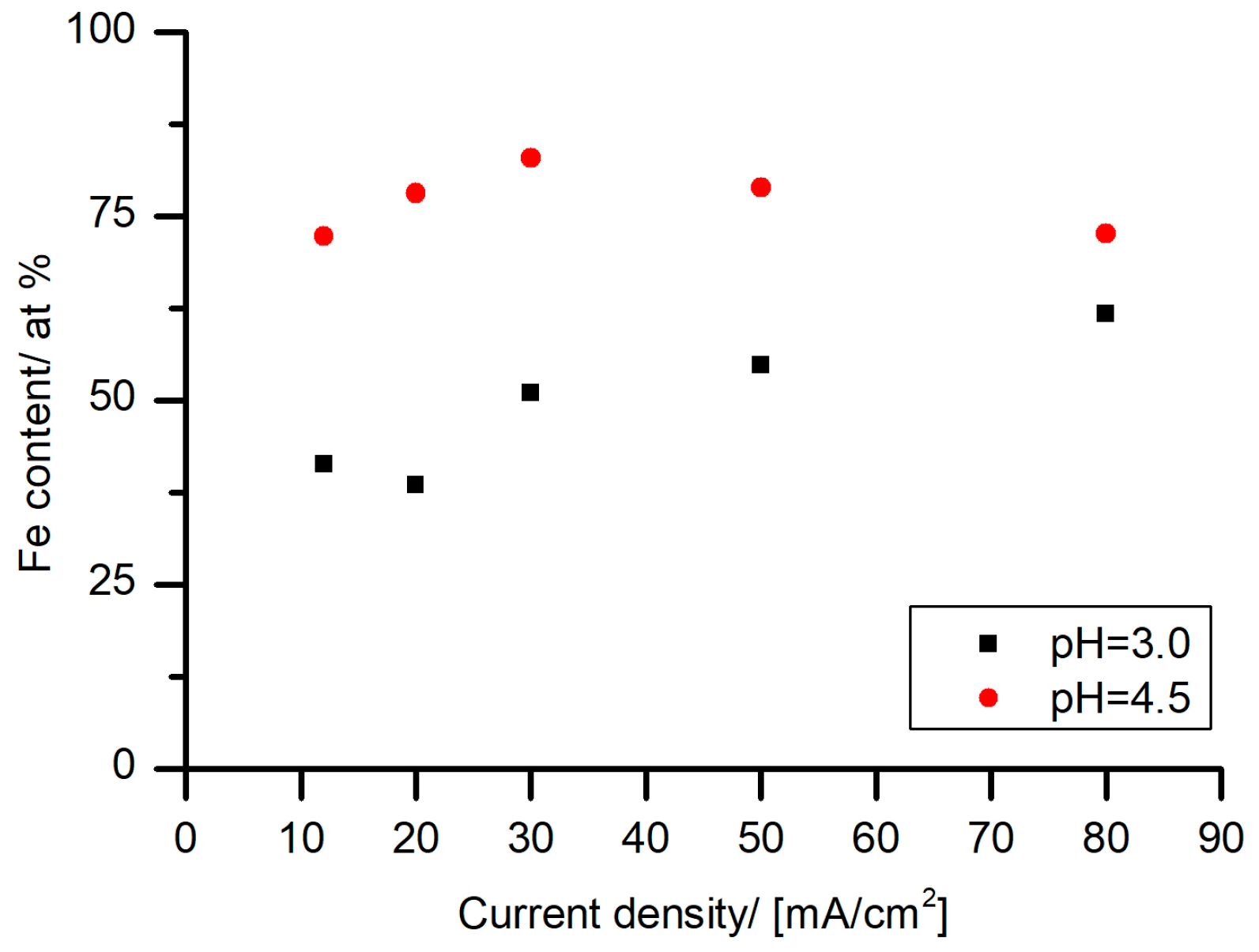
25]. A literature overview showed no report of an anomalous codeposition in the Fe–Sn system. The iron content, as a function of current density in the Hull cells, was studied (
Figure 5). The assumptions, based on the CVs results (
Figure 3), of the suppression of the Sn reduction and Fe–Sn anomalous co-deposition were confirmed. In the samples deposited at the pH value of 3.0, Fe-rich deposits were obtained starting with 30 mA/cm
2. In the case of the electrolyte with the pH value of 4.5, Fe-rich deposits were obtained over the whole studied current density range. These findings indicate that anomalous deposition of Fe–Sn from tartrate electrolyte starts to appear at a current density of around 30 mA/cm
2 and a pH value of 3.0. Moreover, the Fe–Sn anomalous deposition is a pH- and current density-dependent phenomenon. One of the possible explanations for the anomalous co-deposition is blocking of the cathode surface with hydroxides [
19,
21]. According to the species simulation (
Figure 1c,d), Fe hydroxides starts to form from around the pH value of 4 and Sn hydroxides start to appear around the pH value of 8.0. Consequently, it can be assumed that the possible reason for the Fe–Sn anomalous co-deposition is the presence of Fe hydroxides on the cathode surface.
The layers on the Hull cell samples deposited with 20 and 80 mA/cm
2 at different pH values were characterized by XRD diffraction. The XRD-diffraction patterns were recorded in the angular range of 20°–100°. For the phase identification, the most relevant was the 2θ range 27°–47° (
Figure 6). Three intermetallic phases, FeSn
2 (PDF-04-006-5524), Fe
5Sn
3 (PDF-03-065-7052), Fe
3Sn (PDF-03-065-3524), and Sn (PDF-04-008-4977) were observed in all of the electrodeposited samples, independent of current density and the electrolyte pH. Additionally, α-Fe (PDF-01-089-4186) was detected at the samples deposited with 20 mA/cm
2 with the pH values between 3.0 and 4.5 as well as with 80 mA/cm
2 with the pH values of 3.0 and 3.5. A decrease in the intensity and broadening of the peaks (30.7°, 32° and 45°) related to the diffraction of β-Sn was observed with the increasing electrolyte pH for both studied current densities (
Figure 6). The same tendency was observed for the Fe
5Sn
3 diffraction peak at 30° for the samples deposited with 20 mA/cm
2. These findings indicate that with an increase of the pH of the electrolyte, the amount of the electrodeposited β-Sn and Fe
5Sn
3 decreased and the crystallite size got smaller. The detected Fe-rich intermetallic phases Fe
5Sn
3 and Fe
3Sn have hexagonal structure (P6
3/mmc) and exhibit ferromagnetic properties [
2,
3]. In the thermodynamic Fe–Sn phase diagram, both appear at temperatures above 1000 K [
3]. Experimentally, they were prepared by DC-magnetron sputtering [
6] and solid state reaction [
2,
3]. The available literature on electrodeposition in the Fe–Sn system showed no results for the preparation of Fe
5Sn
3 and Fe
3Sn [
10,
26,
27].
The Rietveld method was applied to the recorded XRD-diffraction patterns in order to quantify the amount of detected metal phases. The results, together with the calculated size of the crystallites (
L-vol), are listed in the
Table 2. The recorded diffraction patterns were complex and with overlapping diffraction maxima (
Figure 6). Hence, in the calculated values of the mass fractions and crystallite sizes, a significant error might occur. These values are marked with an asterisk in
Table 2. Generally, based on the calculated values, several conclusions could be made. The results of the crystallite size and mass fraction calculations (
Table 2) confirmed the assumptions based on the diffractograms observation (
Figure 6). By increasing the pH of the electrolyte at both current densities, the amount of the electrodeposited β-Sn, and its crystallite size decreased. The same tendency was observed in the case of the Fe
5Sn
3 phase deposited with a current density of 20 mA/cm
2. In contrast, the amount of α-Fe, electrodeposited with a current density of 20 mA/cm
2, increased as a function of the electrolyte pH. It also seems that the iron-rich phase Fe
5Sn
3 is preferentially deposited with a current density of 20 mA/cm
2. On the other hand, the tin-rich phase FeSn
2 is preferentially deposited with a current density of 80 mA/cm
2. The obtained results (
Table 2) also suggest that the amount of Fe
3Sn is not depending on the electrolyte pH nor on the current density.
3.4. Electrolyte Stability
In the electrolyte speciation’s simulation, several unstable or insoluble species such as [Sn(tart)] or the iron hydroxides appeared (
Figure 1). For the chemical equilibria calculation, the redox reaction of Fe(III)–Sn(II) and the existence of bimetallic Fe–Sn-tartrate complexes were not considered. In order to revise the reliability of the species distribution, the stability of the electrolytes was studied. The stoichiometry of the precipitates was studied by EDX and the results are presented in
Table 3.
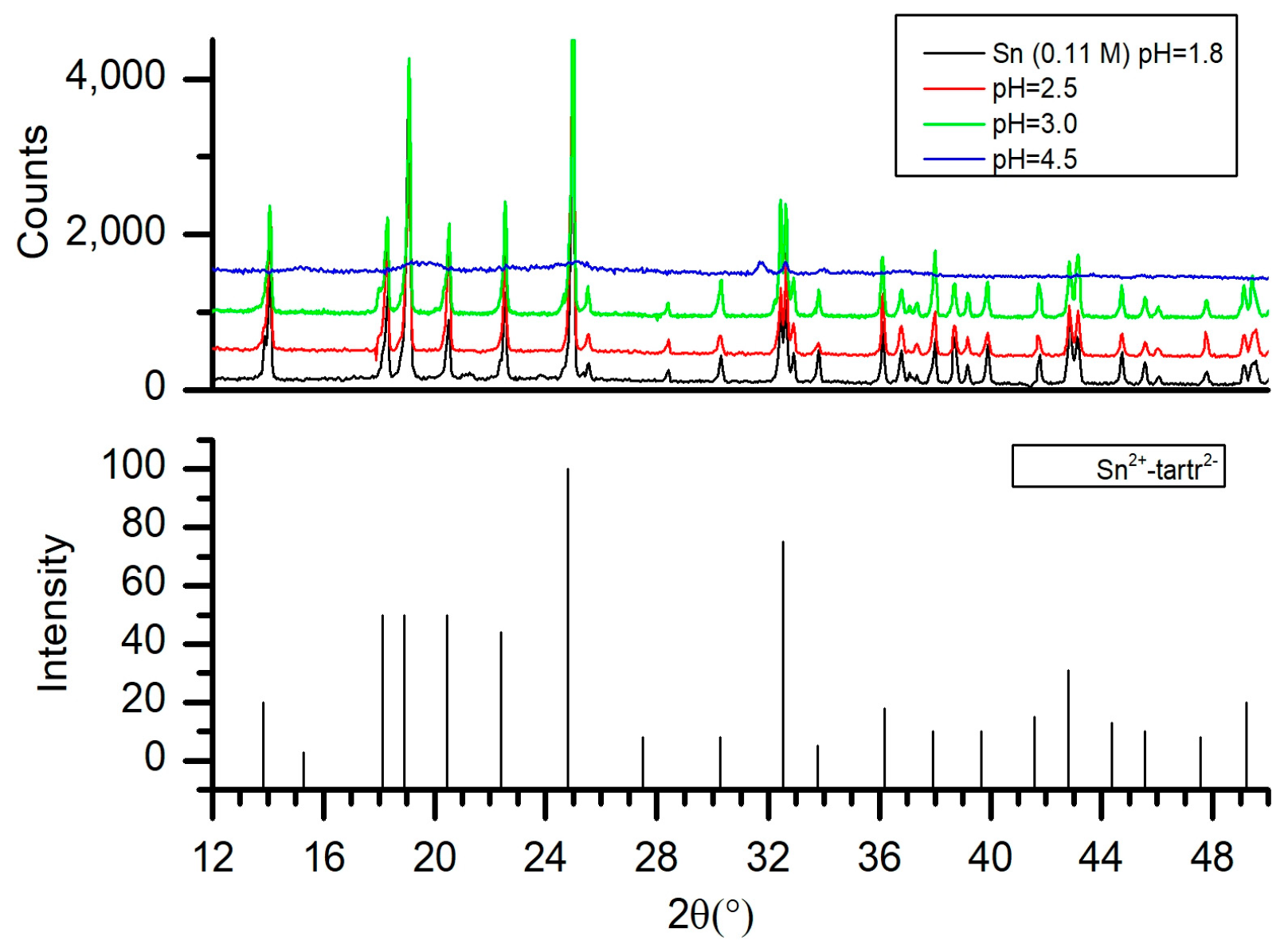
The single metal Sn electrolyte (0.11 M) at the pH value of 1.8 was rapidly precipitating. The XRD diffraction spectra on the Sn precipitate showed rather complex crystalline behavior (
Figure 7). Although not all the diffraction peaks were successfully assigned, most of them (e.g., 13.8°, 15.3°, 18.2°, 18.9°, 24.9°, etc.) were in good correlation with the powder diffractogram of [Sntartr]
0 (PDF-00-001-0529). This finding agrees with the species distribution of the single metal Sn electrolyte (
Figure 1b) where at the pH value of 1.8 approximately 0.04 M of the Sn ions are present as neutral tartrate complex.
The Sn(II) electrolyte (pH = 1.8) was stabilized with the addition of FeCl
3. The mixed Fe(III)–Sn(II) electrolyte with the pH value of 1.8 was stored and observed for a week and during this time, no precipitates or change of color appeared (
Table 3). Two explanations for the stabilization of the Sn(II) electrolyte by the FeCl
3 are possible. The first one is based on the calculated species distribution (
Figure 1b–d). With the addition of the 0.11 M of FeCl
3 into the solution, the concentration of the Cl
− anions increased to 0.55 M. Consequently, the concentration of Sn–chloride complexes in the acidic region rose in respect to the concentration of [Sn(tart)] complexes which dropped to approximately half of its initial value (0.02 M) (
Figure 1b,c) and therefore does not precipitate. The second possible explanation is based on the existence of bimetallic Sn–Fe-tartrate complexes [
17]. In order to decide which explanation is correct, the concentration of Cl
− anions in the single metal Sn electrolyte was adjusted to 0.55 M by adding (NH
4)Cl and the modified electrolyte was aged on a hot plate. After approximately half an hour, large amounts of a Sn-rich precipitate appeared. Based on this observation, we might conclude that single metal Sn electrolyte at the pH value of 1.8 was stabilized by the creation of bimetallic Fe–Sn-tartrate complexes.
As the pH of the mixed Fe(III)–Sn(II) electrolyte increased to the value of 2.5 and 3.0, crystalline [Sn(tart)] precipitates appeared again (
Table 3,
Figure 7). According to the species distribution (
Figure 1c), at the pH value around 3.5, the concentration of the [Sn(tart)] drops down to 0 M. In the aged electrolyte at this pH value, only marginal amount of the precipitation appeared. After the filtration of the initial precipitate, the electrolyte appeared stable, without color change and further precipitation for more than a week. The amount of the filtered precipitate was not sufficient for a detailed analysis and the EDX and XRD analyses were not conclusive. For this reason, the electrolyte with the pH value of 3.5 was considered as stable.
In the electrolyte with a pH value of 4.5, Fe-rich amorphous precipitates were detected (
Table 3,
Figure 7). This finding agrees with the speciation distribution, where Fe-hydroxides start to appear around the pH value of 4.0 (
Figure 1d). During the electrodeposition process, the Fe hydroxides could be formed at bulk electrolyte pH values lower than 4.0 due to the local increase of the pH on the cathode surface. This observation agrees with the assumption that the Fe–Sn anomalous co-deposition is caused by the presence of Fe hydroxides on the cathode surface.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









