
A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons




Abstract
1. Introduction
2. Raman Spectroscopy of Carbon Solids: Basics
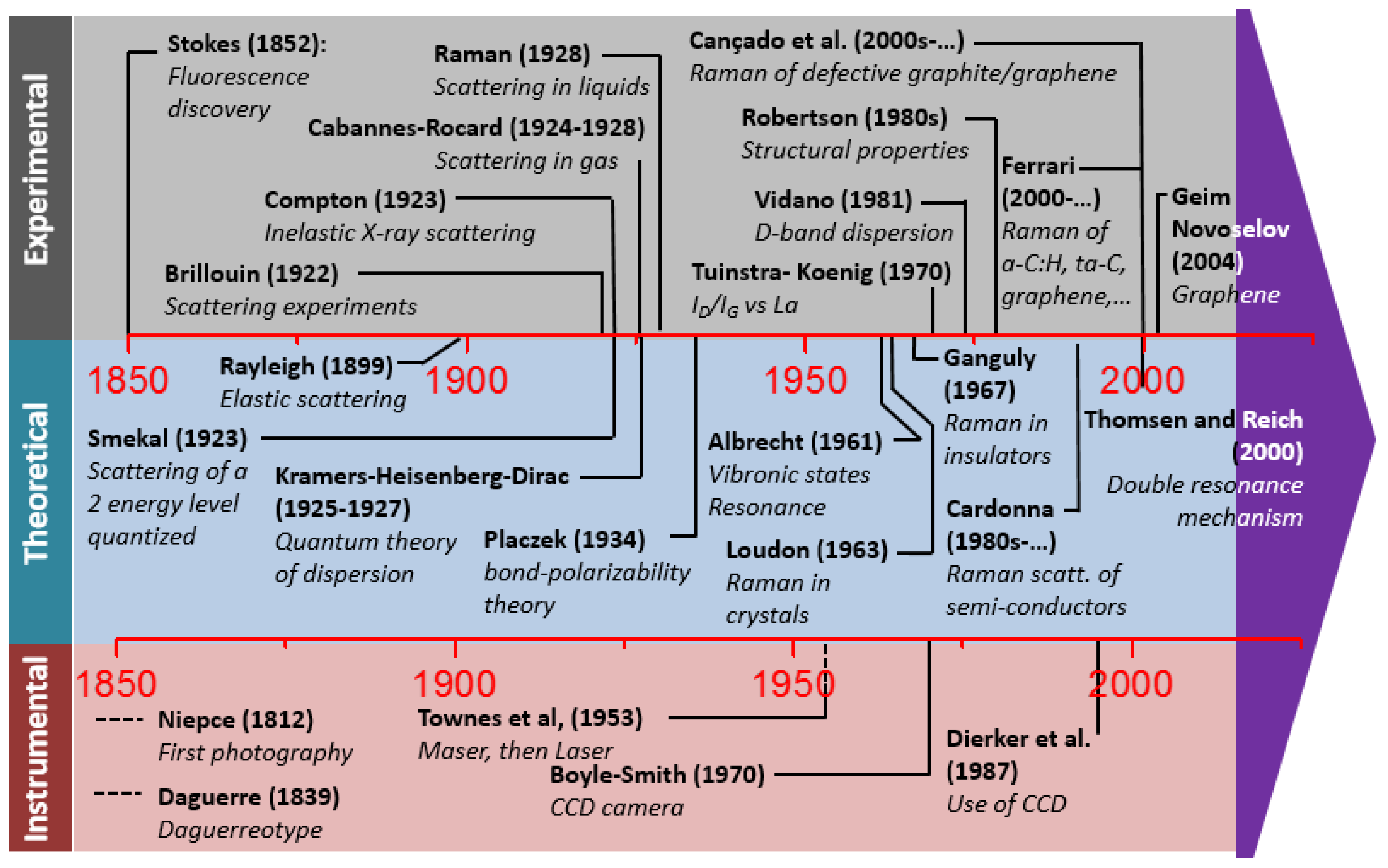
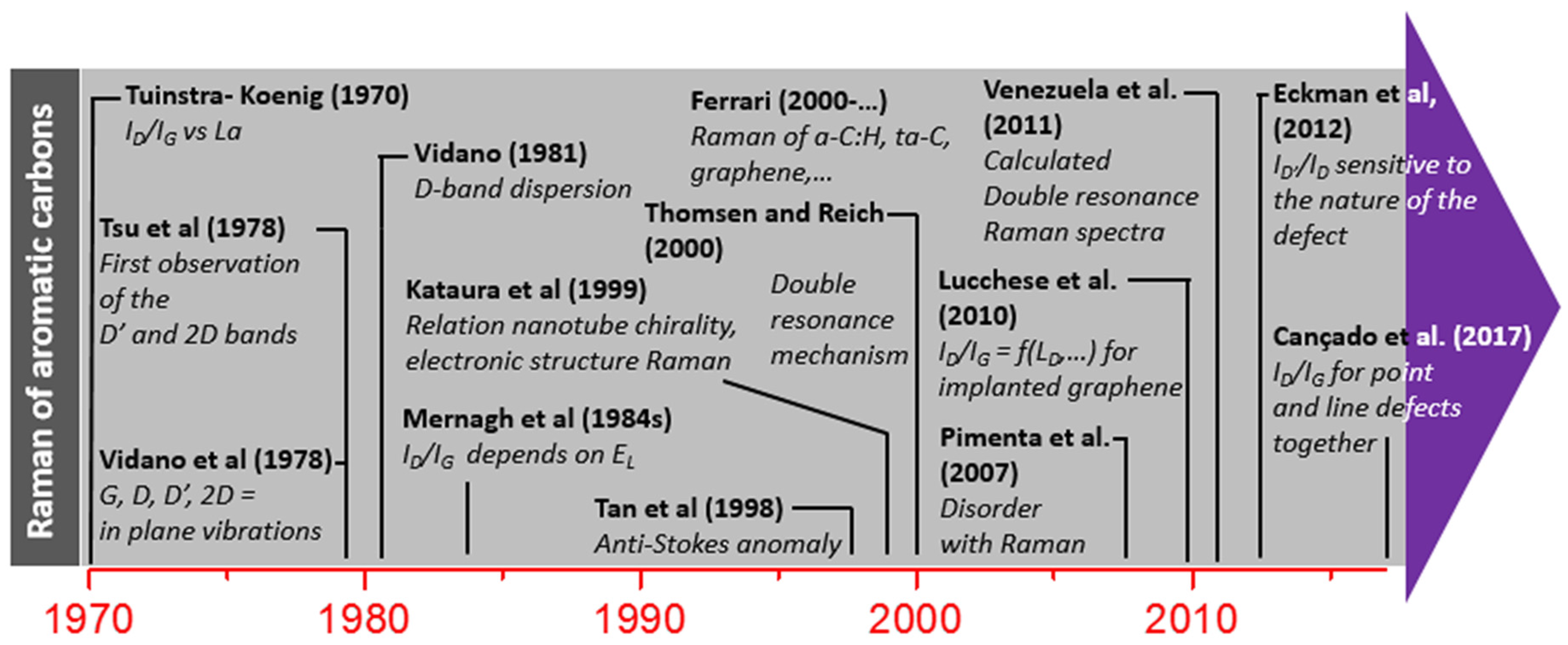
2.1. A Brief History of Raman Spectroscopy
2.2. Basic Knowledge on Raman and Resonance Raman Spectroscopy
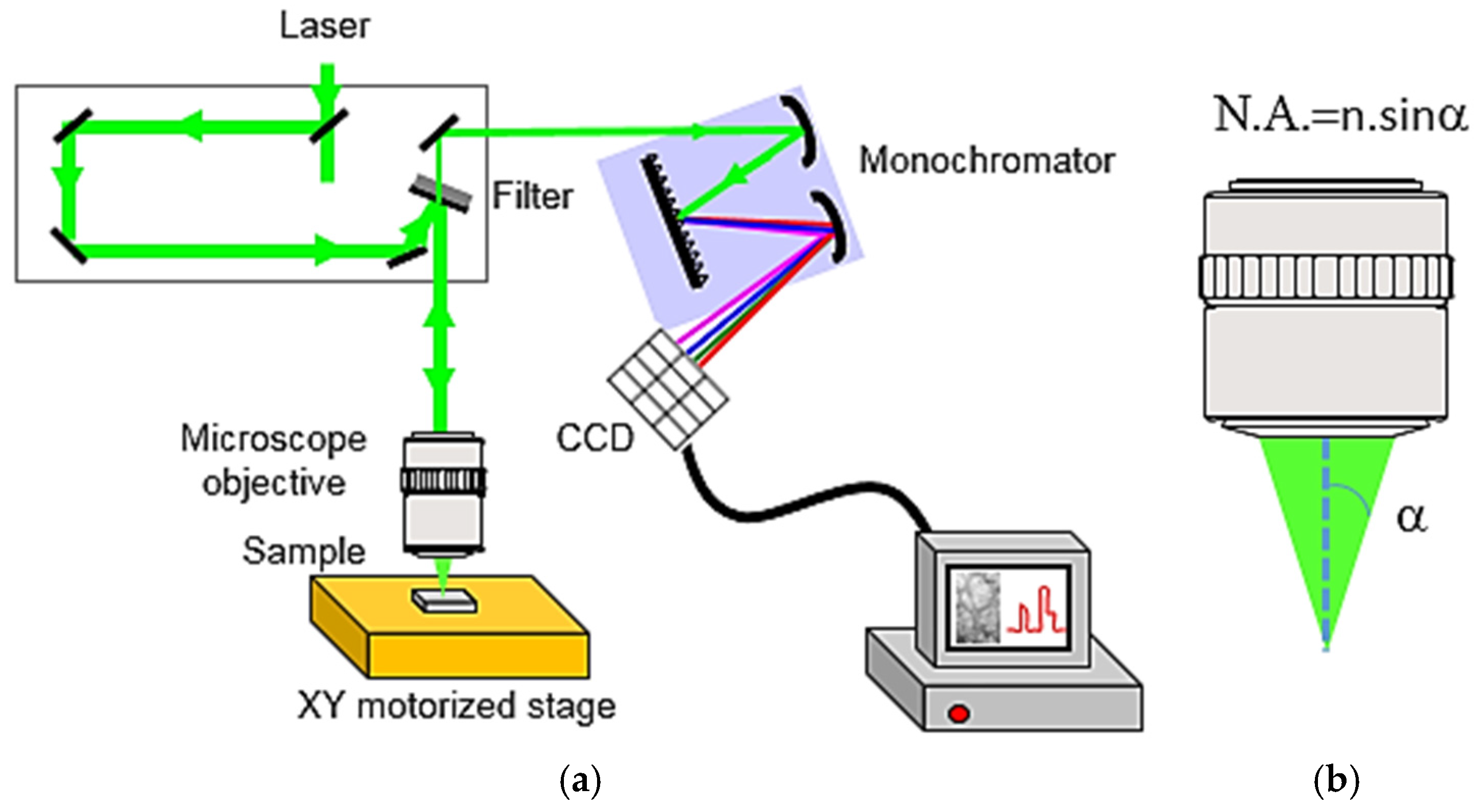
2.2.1. Experimental Set-Up
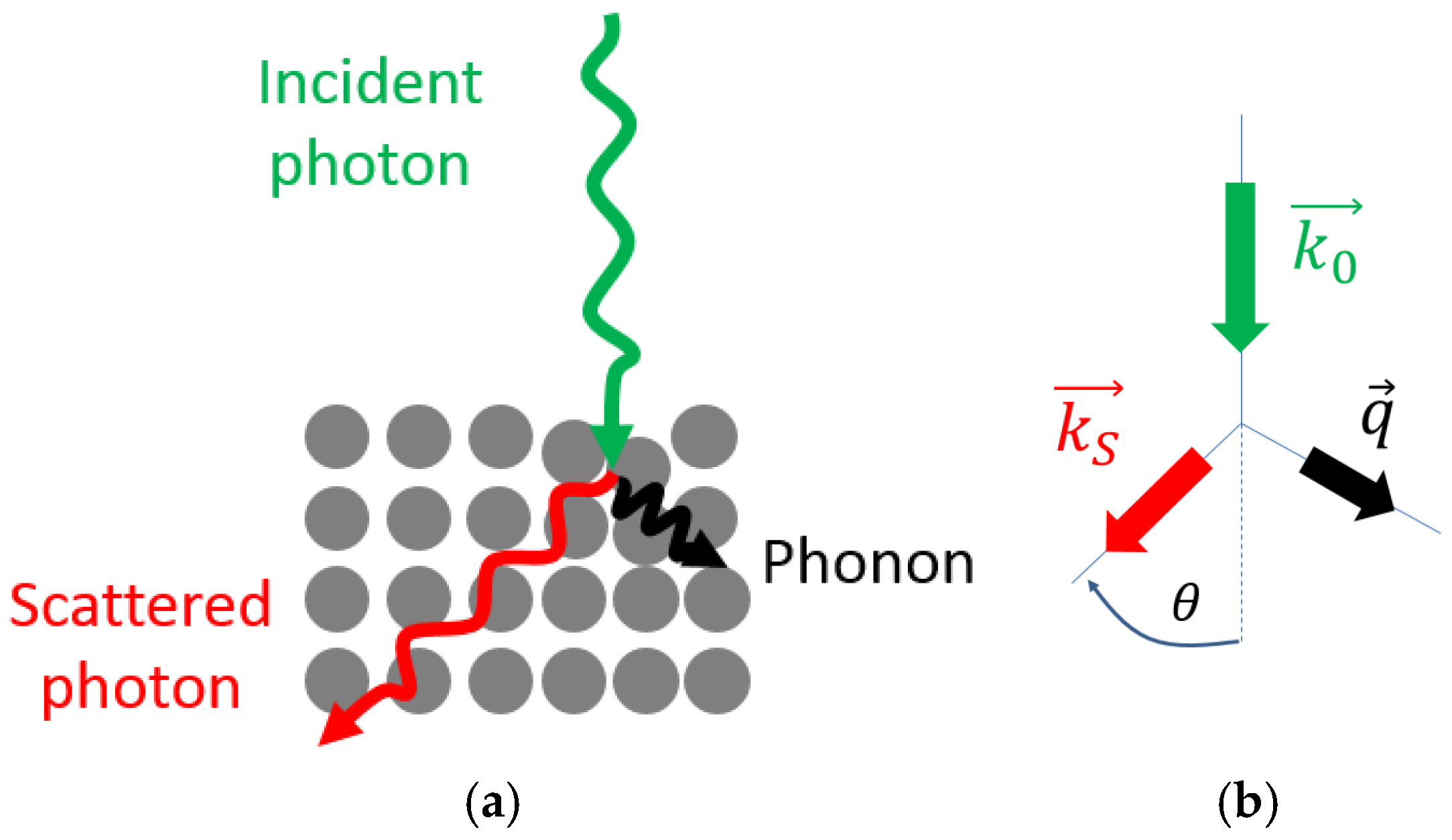
2.2.2. Conservation Rules
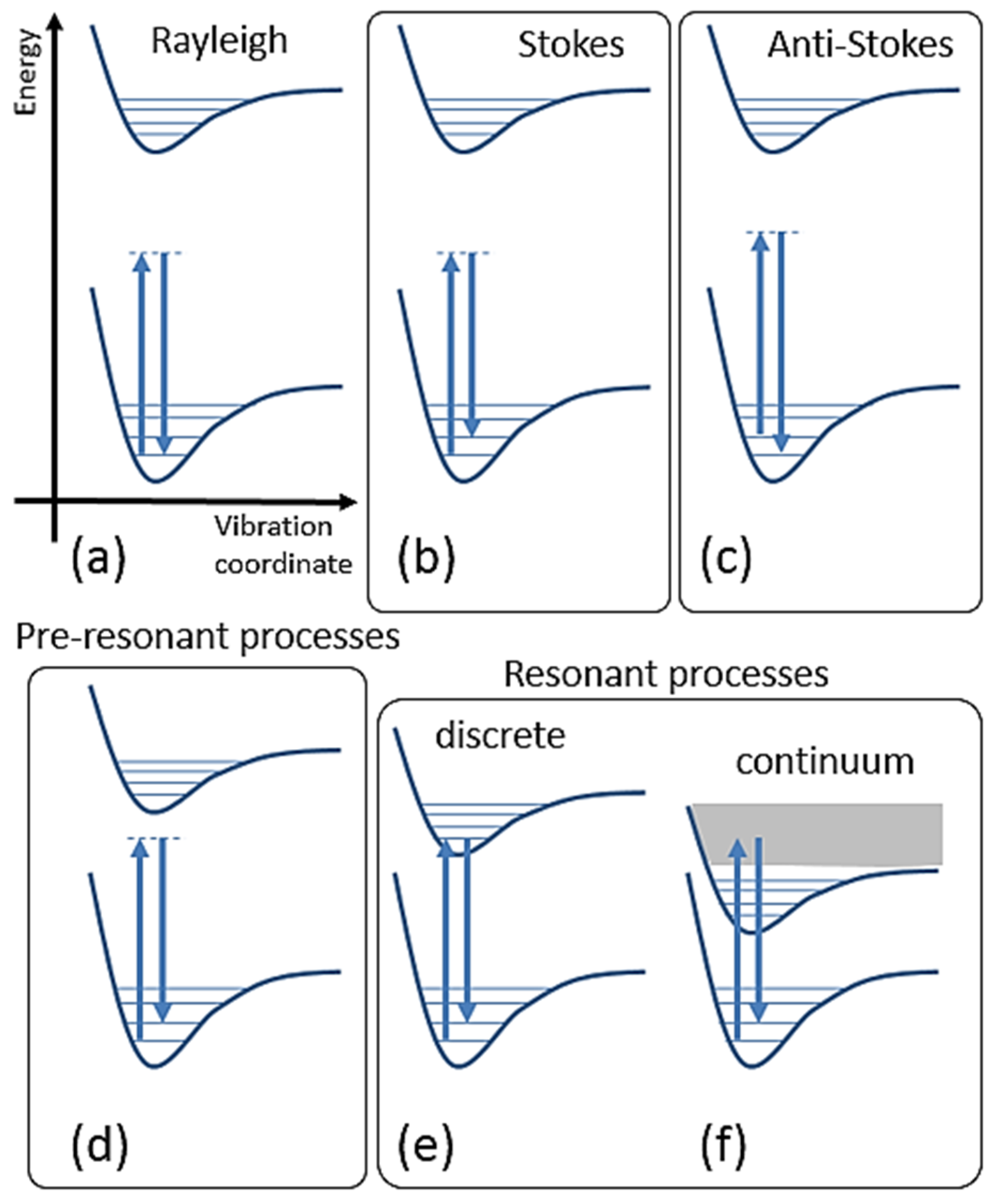
2.2.3. Classical Expressions for Molecules
2.2.4. Semi-Classical Expression for Molecules
2.2.5. Raman Effect in Crystals
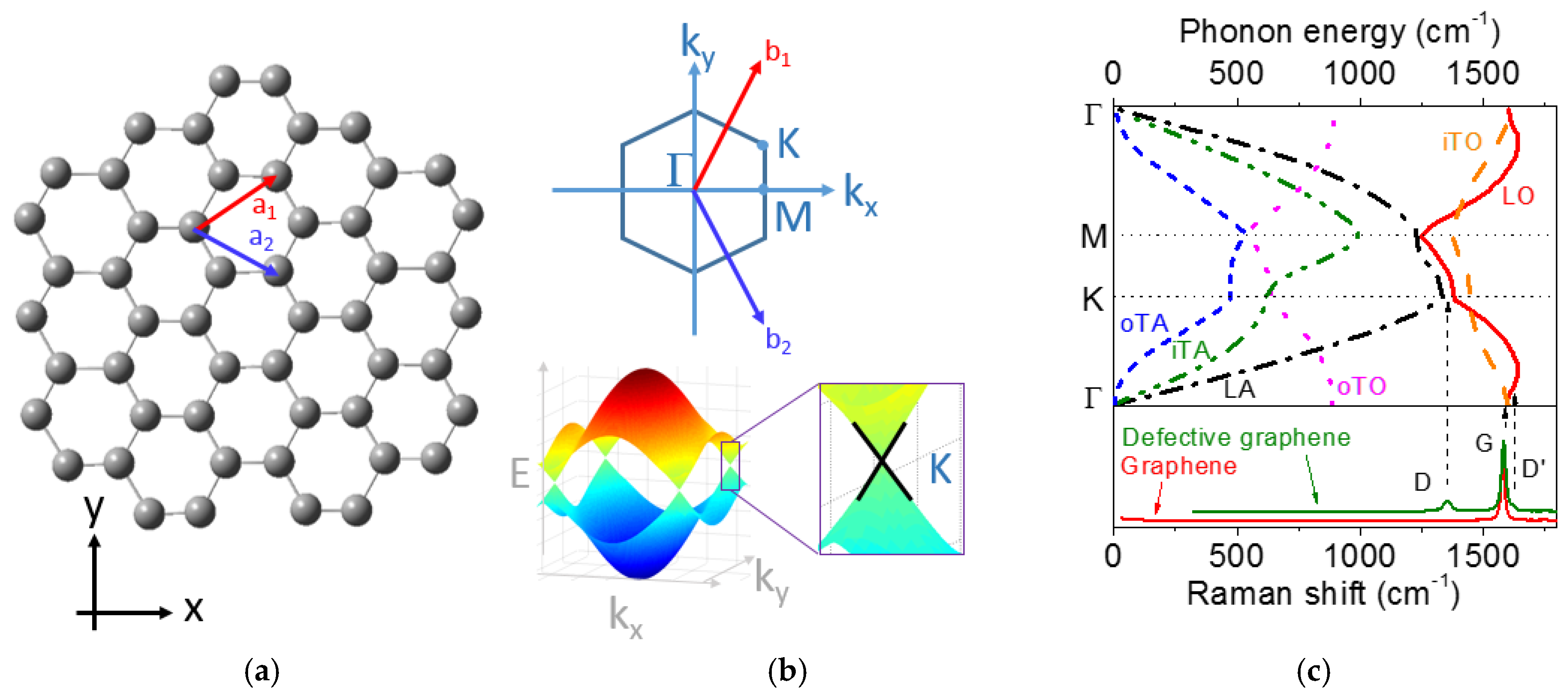
2.3. Basic Properties of Graphene and Related Materials
2.4. Raman Spectra of Graphene, Graphite, and Disordered Carbons
2.4.1. Basics of Raman Spectroscopy for Graphene and Graphite
2.4.2. Historical Aspects
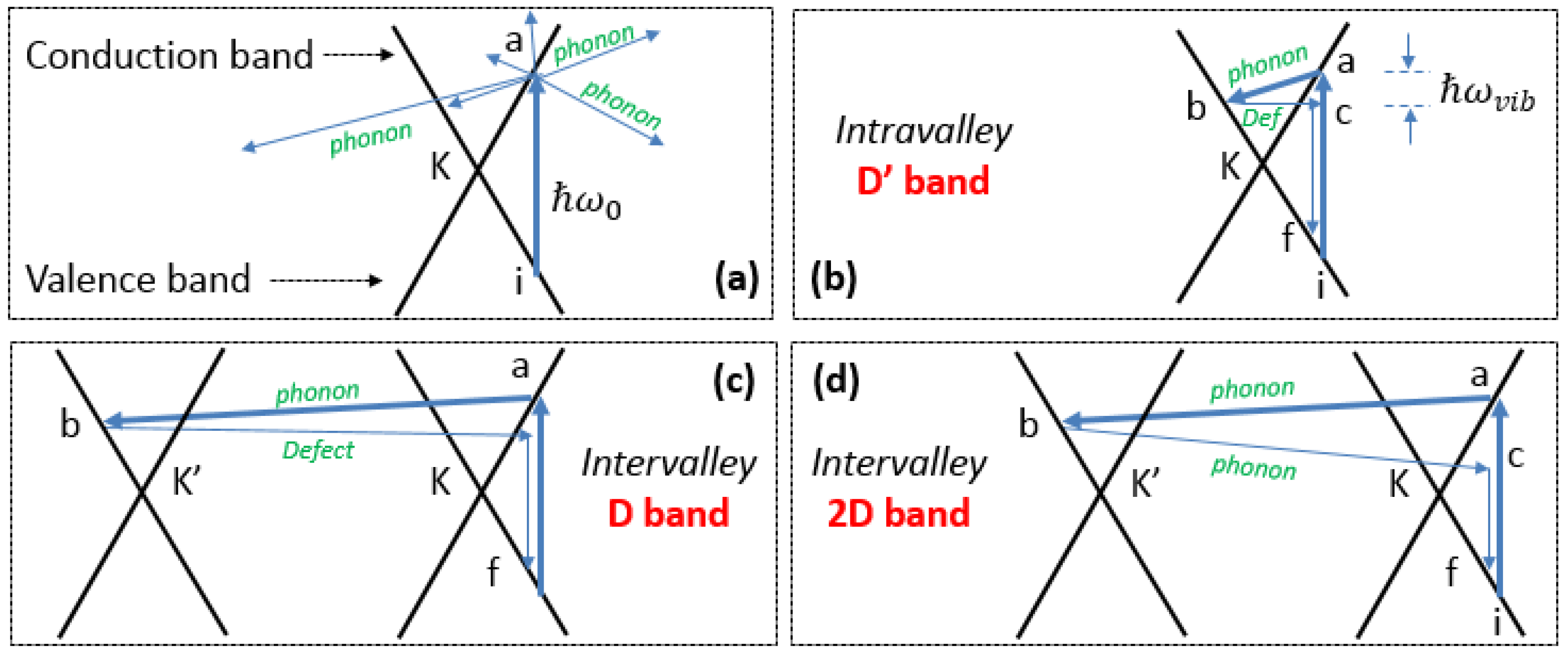
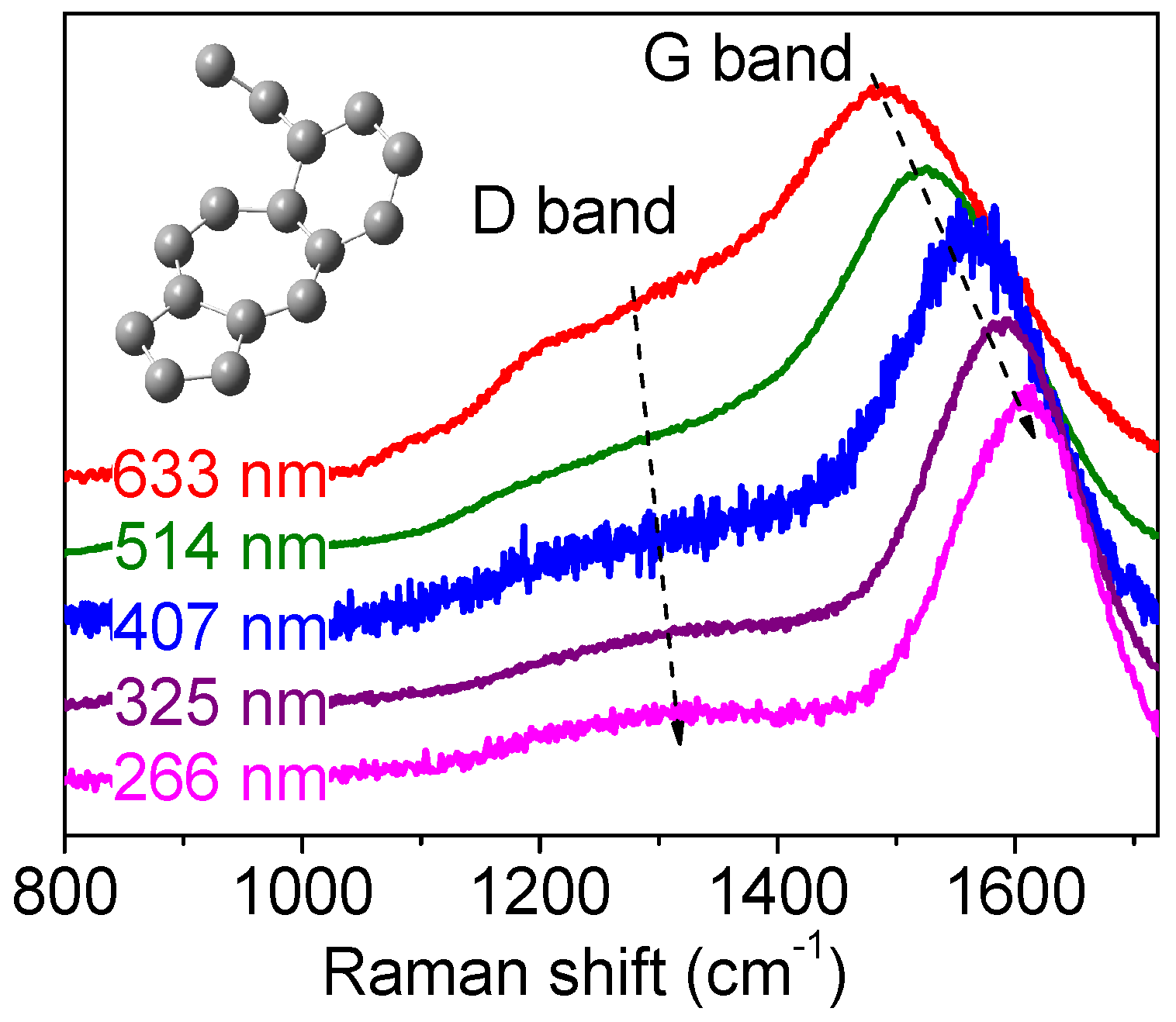
D Band and Combination Band Dispersions
2.5. Brief Introduction to the Double Resonance Mechanism
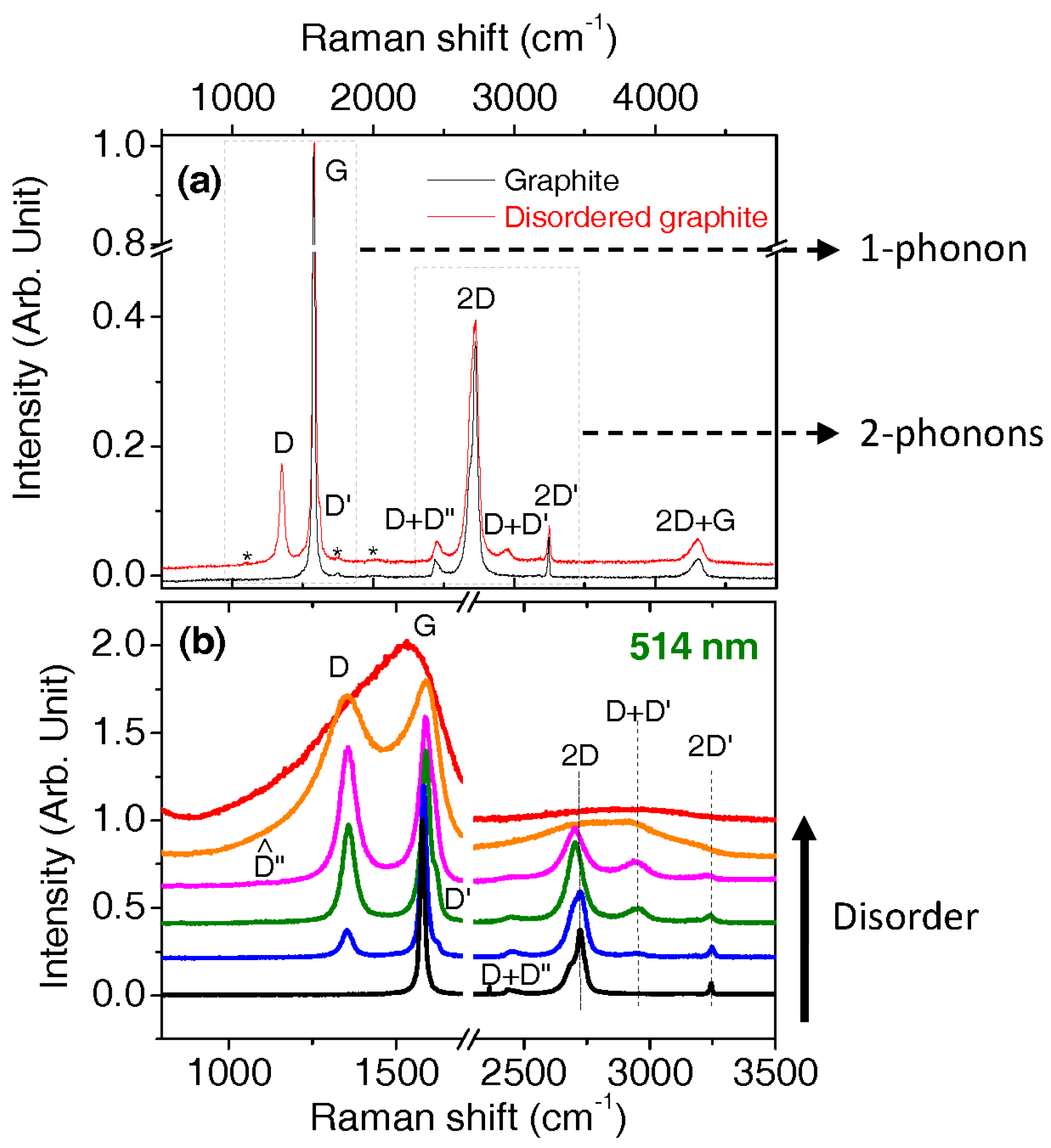
2.6. Intensity, Band Profiles, and Models for Fitting Spectra of Aromatic Carbons
2.7. Procedures for Fitting of the First Order Region
- One band: The G band is fitted by a Lorentzian if symmetric, and by a BWF if not symmetric.
- Three bands: The G, D, and D’ bands are fitted by Lorentzians. D and D’ bands are sometimes labelled D1 and D2, respectively. The D’ band is less intense than the D band by an order of magnitude and can be forgotten when the D and is much less intense than the G band.
- Four bands: The G, D, and D’ bands are fitted by Lorentzians and a Gaussian band is added in the redshift wing of the G band (close to 1500 cm−1). This band is sometimes called the D3 band, in other cases it is called the A band.
- Five bands: Same as the four bands model, but adding another band around 1200 cm−1, which is sometimes found Lorentzian, otherwise found Gaussian. This band is called the D4 band, or D” since the theoretical work of Venezuela et al. [69].
- Six bands: Same as the five bands model, but with another distinct band close to 1150 cm−1. This band is generally more easily seen using red laser (633/785 nm) instead of green/blue lasers.
- Occasionally, no D’ band is observed (possibly merged with the G band so that authors do not try to decompose each component).
- Sometimes, the D and G bands which are Lorentzian are accompanied by two other broader bands (Gaussian or of different line shape) that are red-shifted compared to the D and G bands. The term amorphous component can often be found as well.
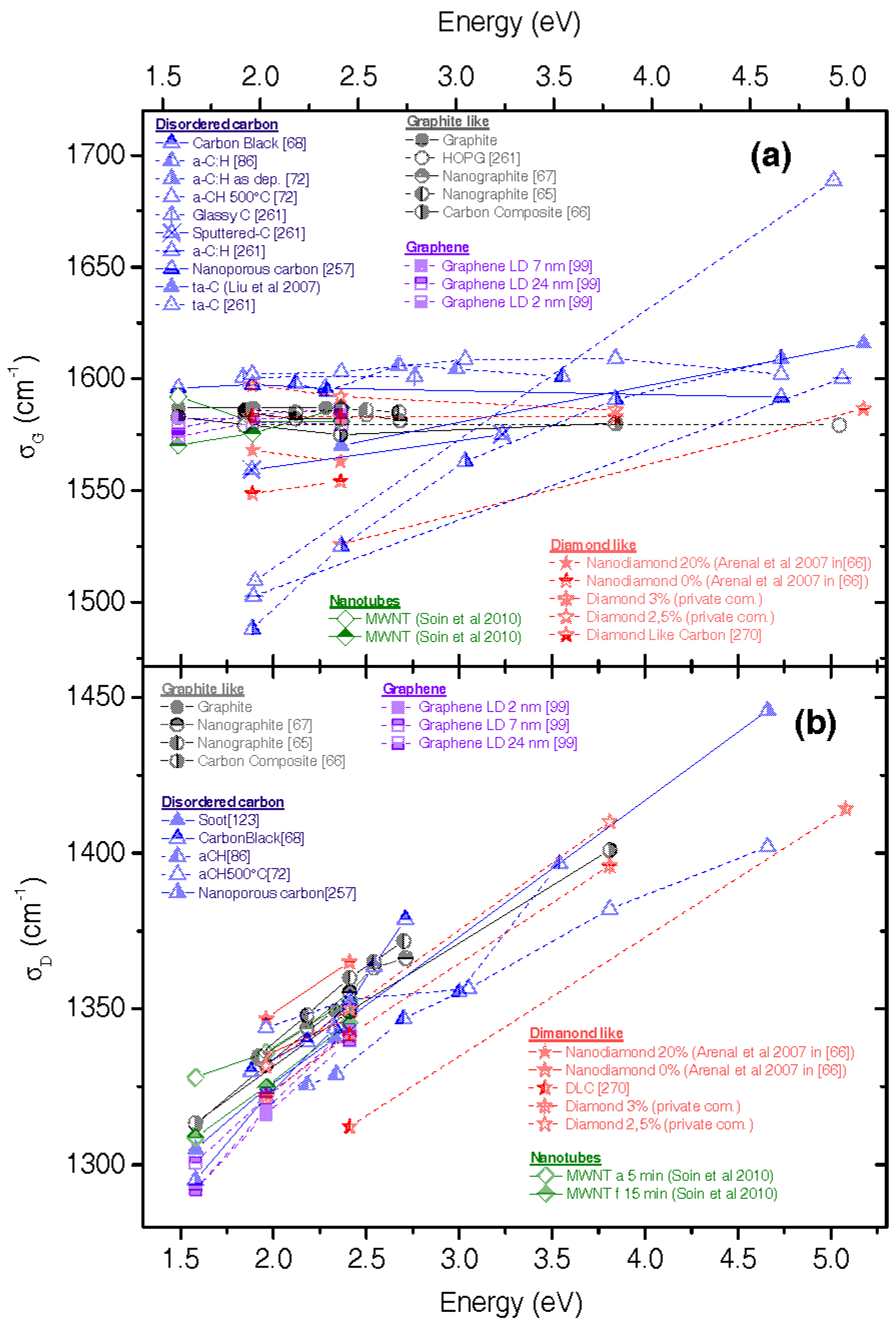
2.8. Examples of How Comparison with First-Principle Calculations Can Help
3. Raman Spectroscopy of Different Aromatic Carbons
3.1. Graphene
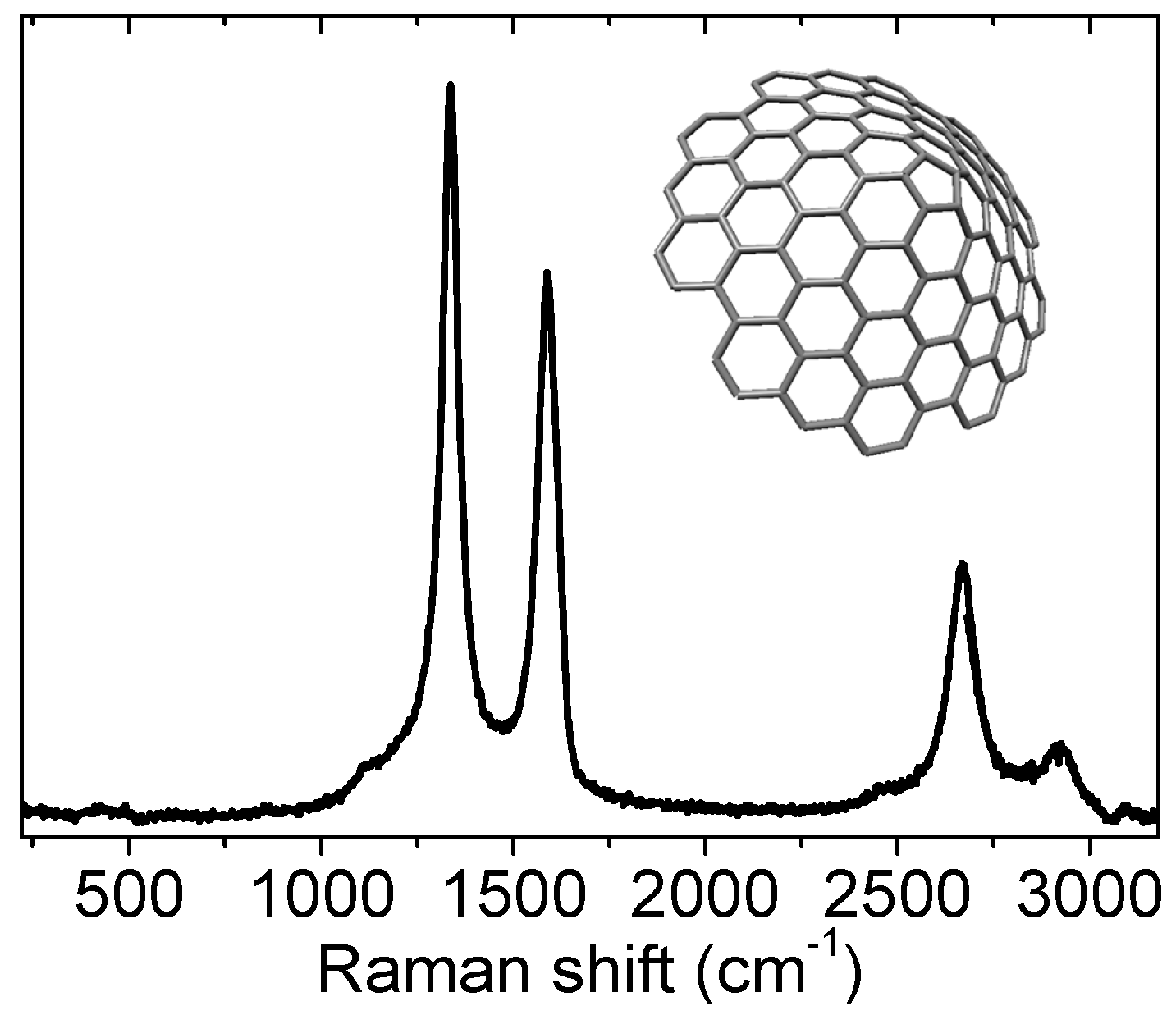
3.2. Graphene based Nanoforms
3.2.1. Nomenclature
3.2.2. Nanodomains, Nanoribbons
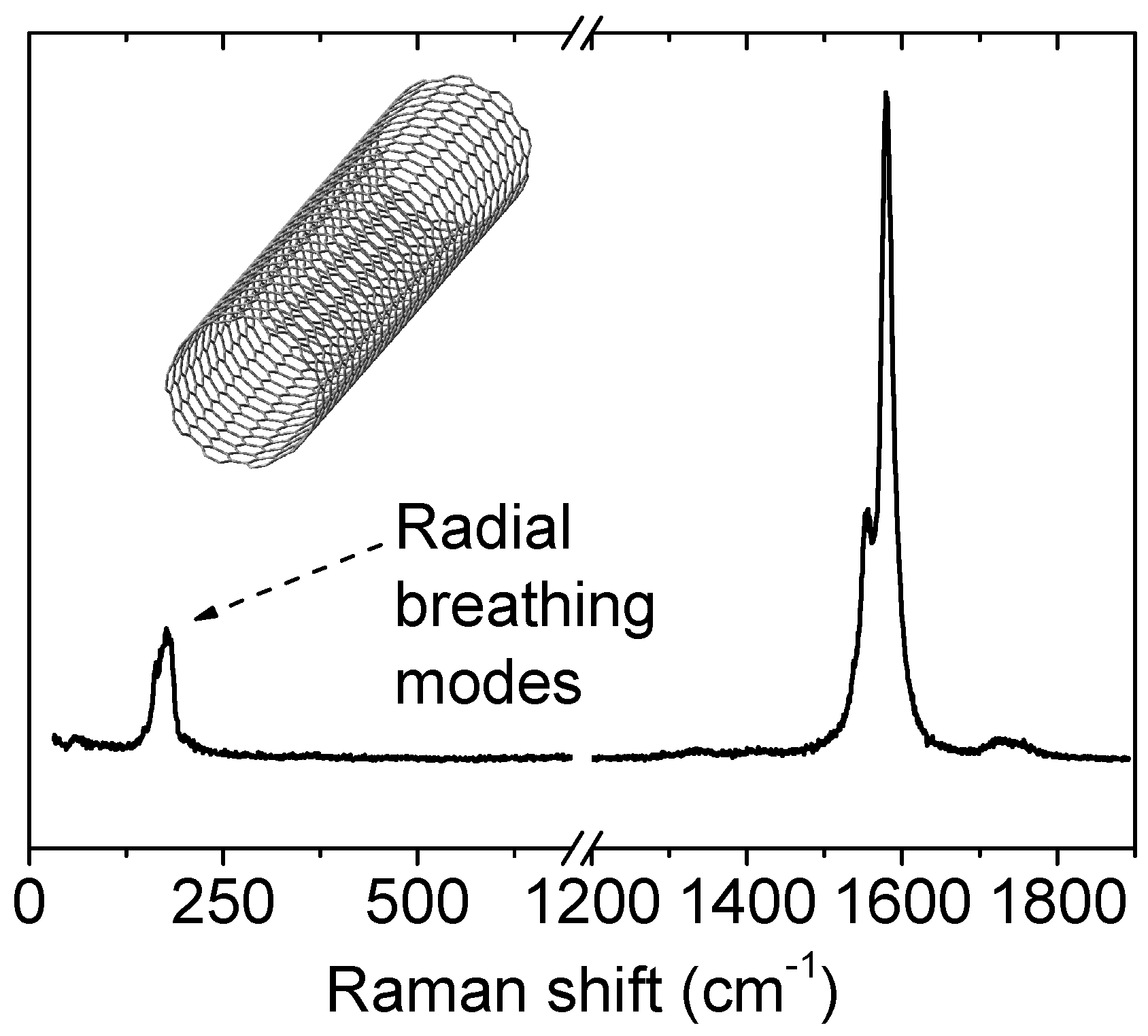
3.2.3. Nanotubes
- The low frequency region (typically between 100 and 300 cm−1), which is associated to radial breathing modes (RBM), see Figure 13. The frequency of these modes is directly related to the diameter of the tubes. One can find a review on RBM not only limited to nanotubes published recently by Ghavanloo et al. [209];
- The D band (around 1300 cm−1), related to defects (as for graphite and graphene);
- The G band (around 1550 cm−1), also similar to the G band of graphite and graphene.
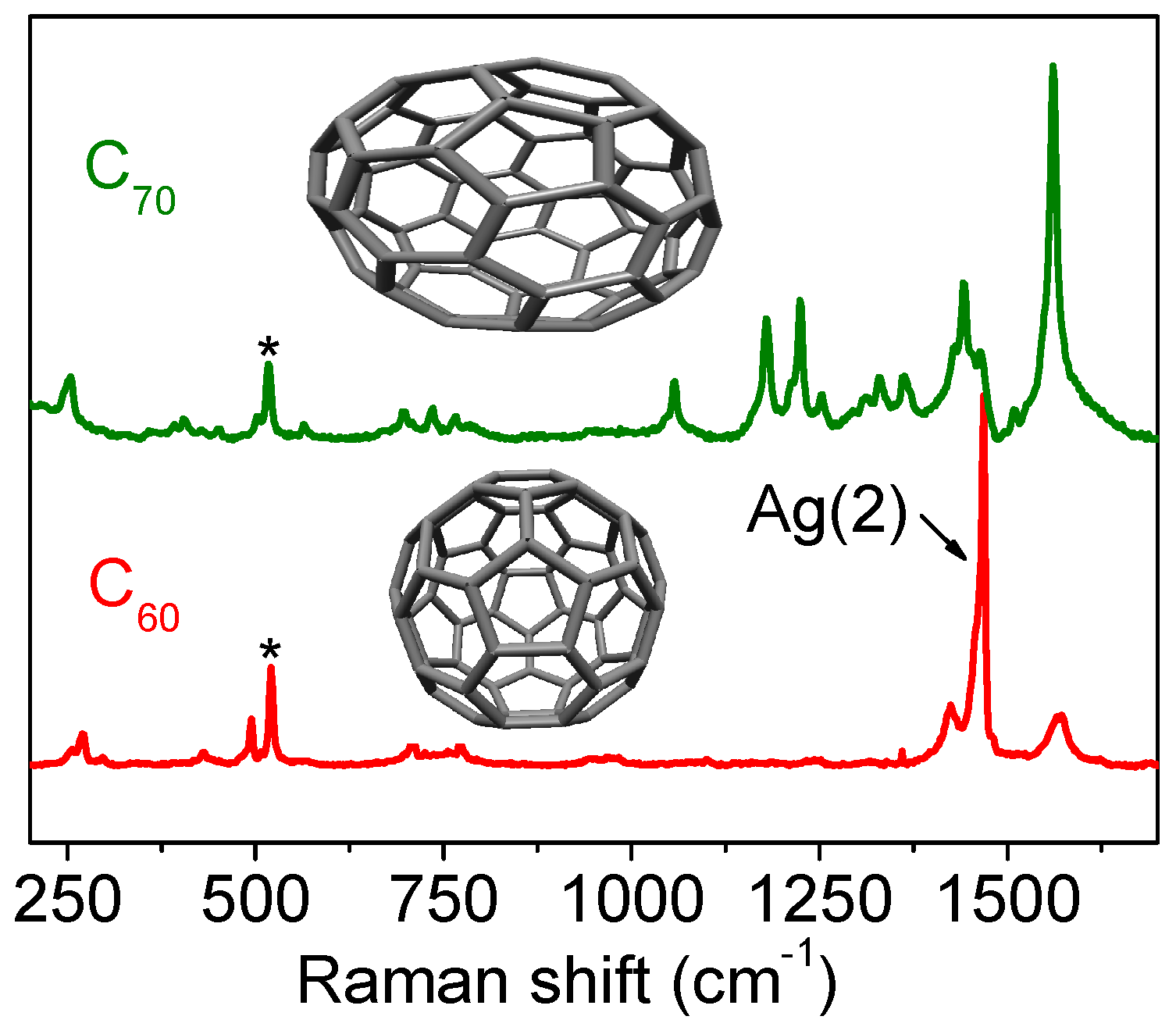
3.2.4. Fullerenes
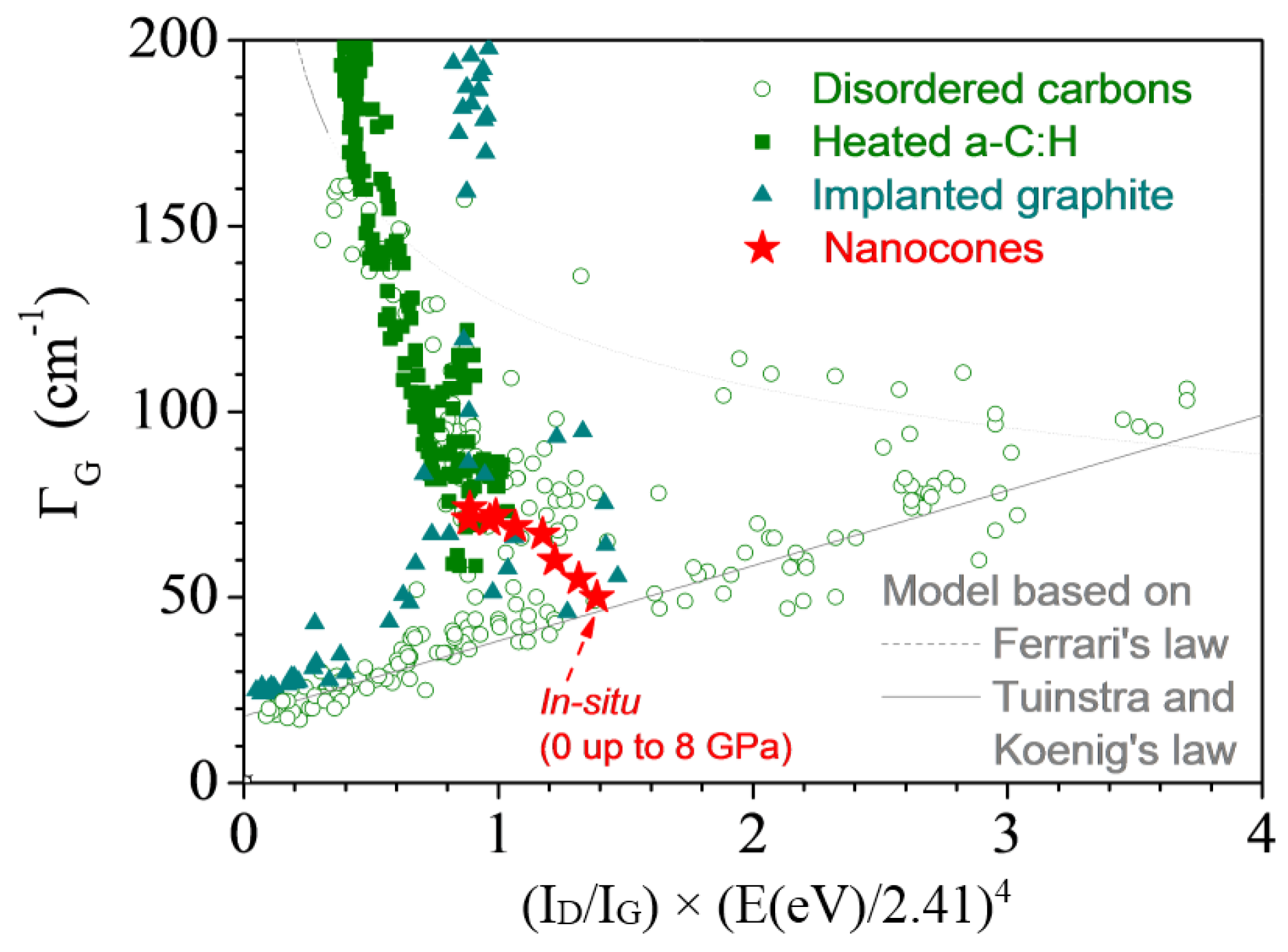
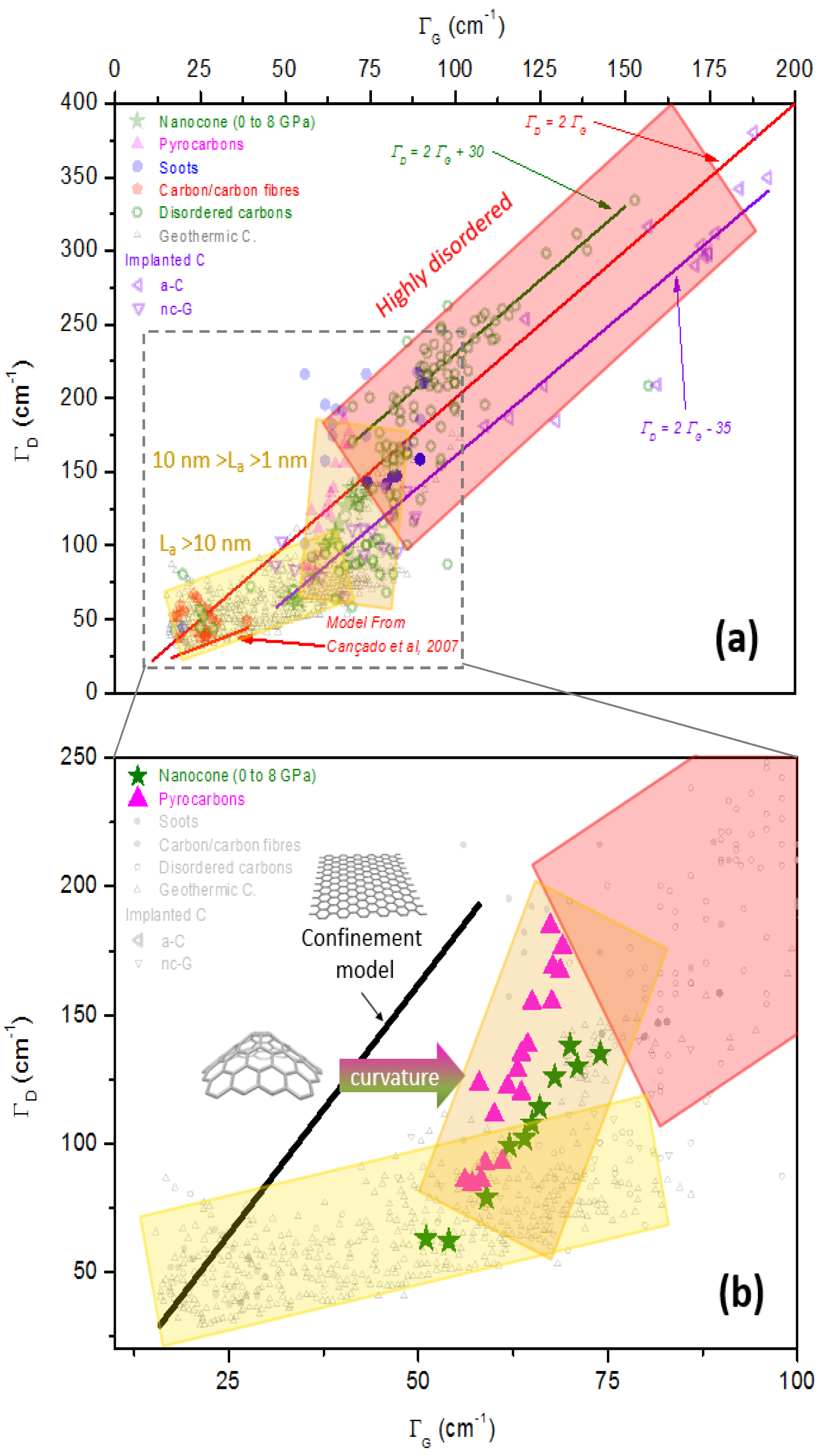
3.2.5. Nanocones
- both the Ferrari relation and Tuinstra and Koenig’s law meet, and/or
- a new set of bands close to 1200 and 1500 cm−1 appears, and/or
- the D bands broaden more than excepted.
3.3. Disordered Graphene as a Reference for More Disordered Carbons
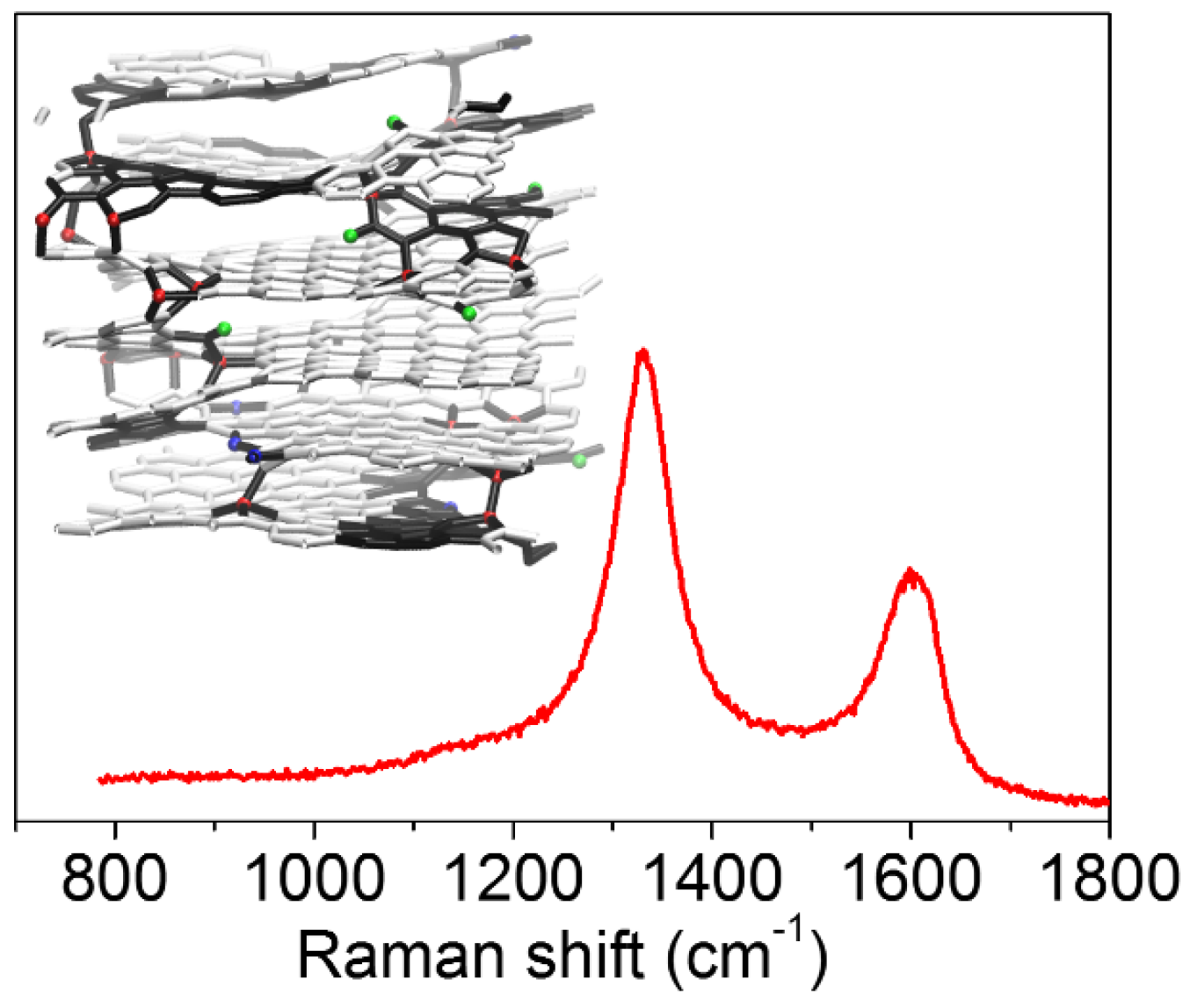
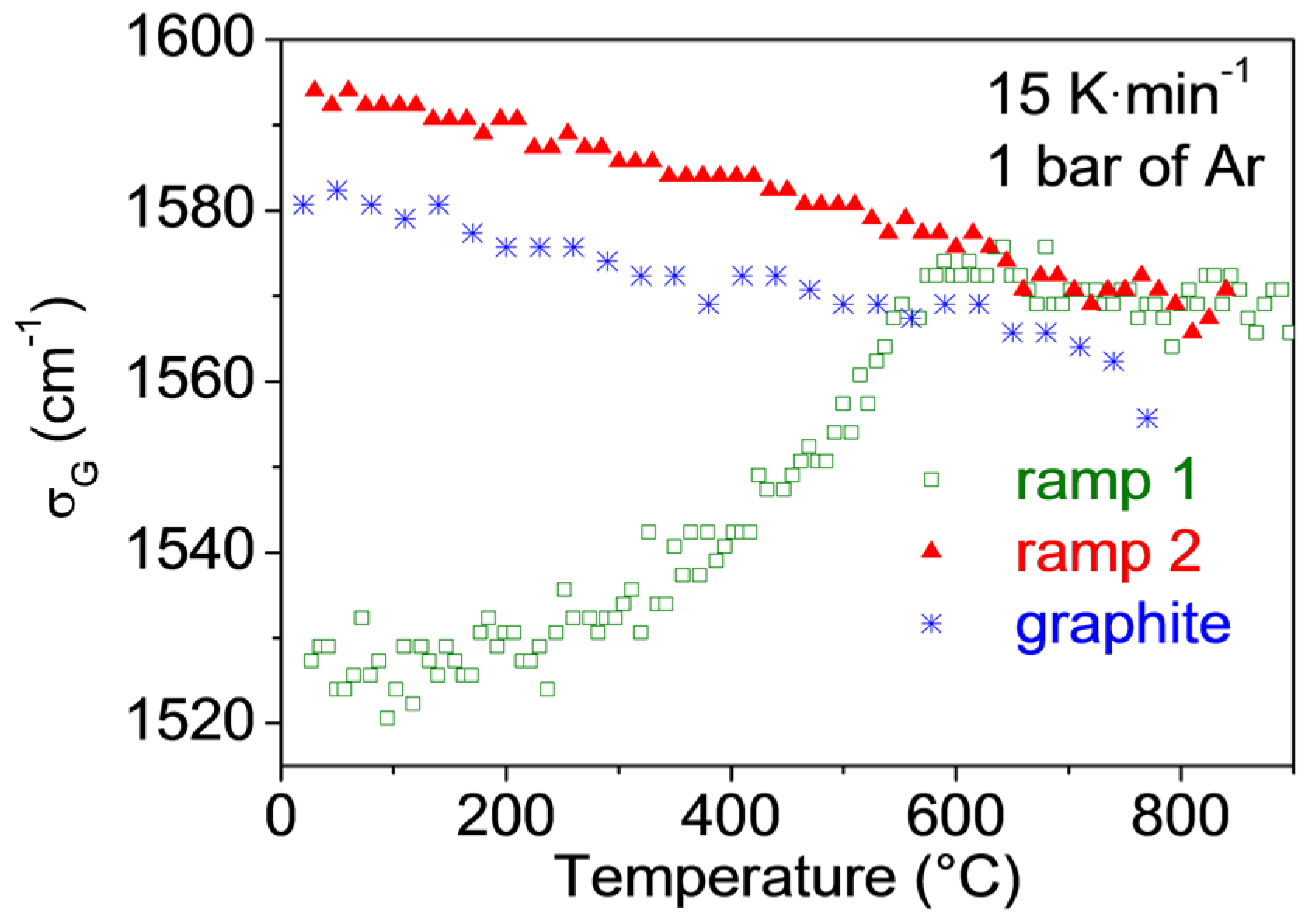
3.4. Very Defective Carbons: Pyrocarbons, Coals, and Soots
3.5. Graphite Intercalated Compounds
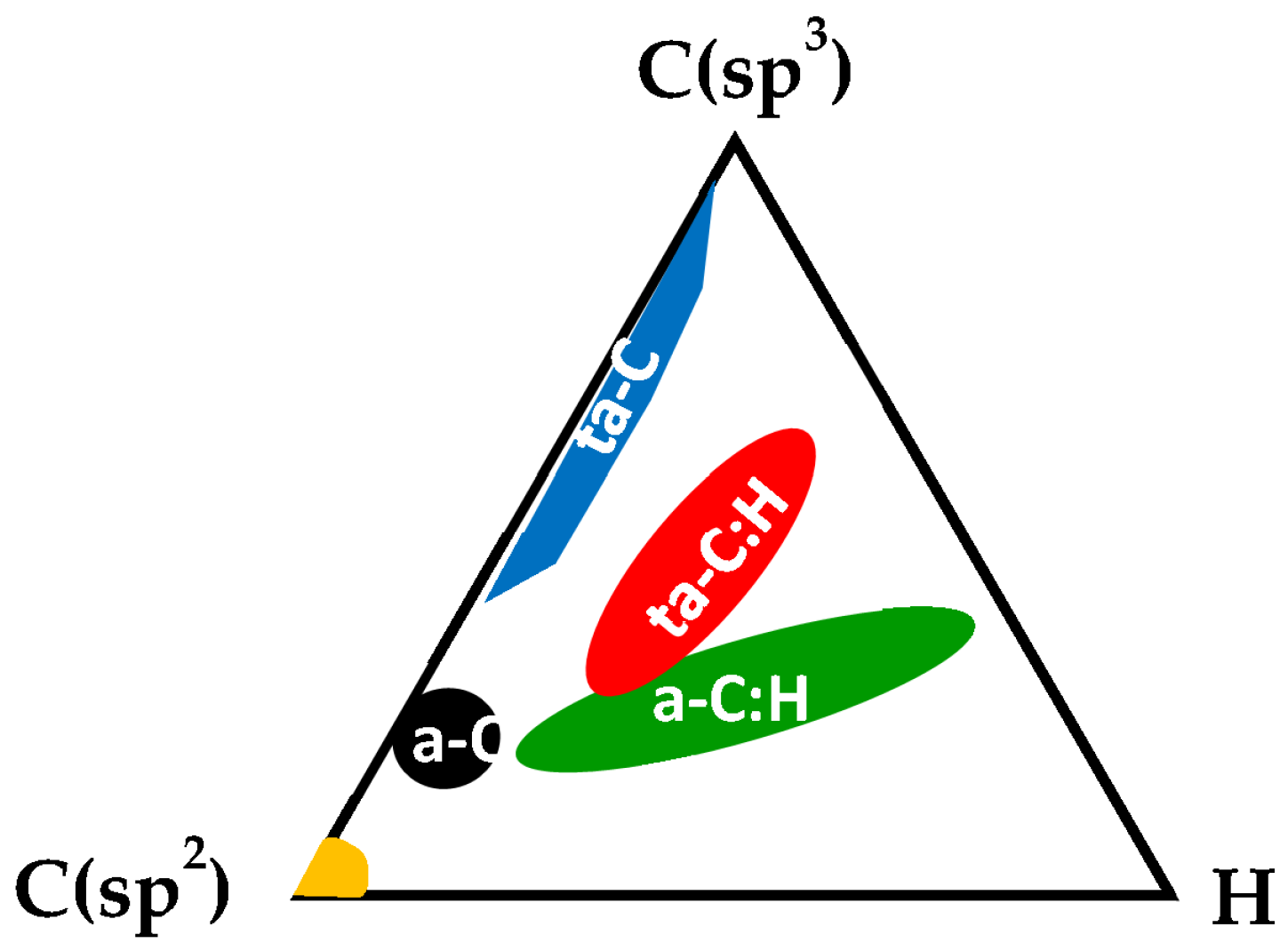
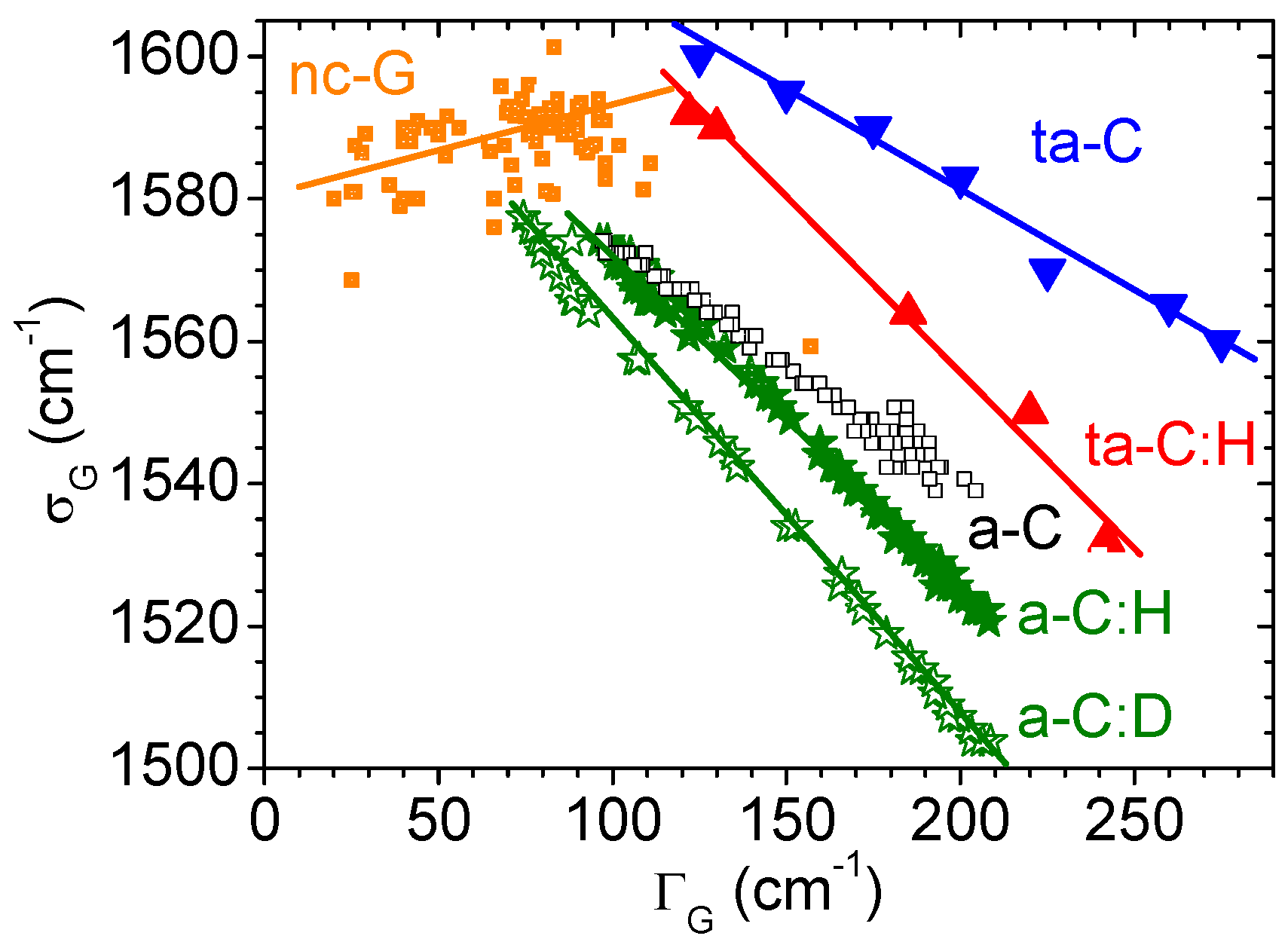
3.6. Amorphous and Diamond-Like Carbons
4. Discussion
4.1. Role of Resonance
4.2. Role of Defects
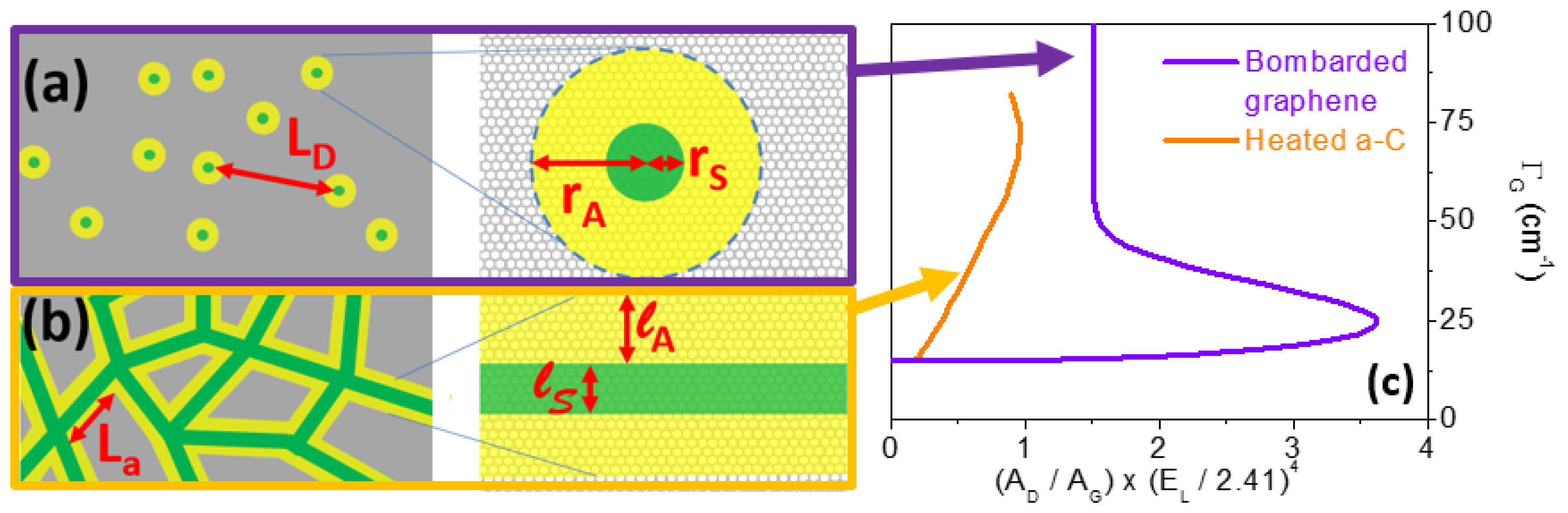
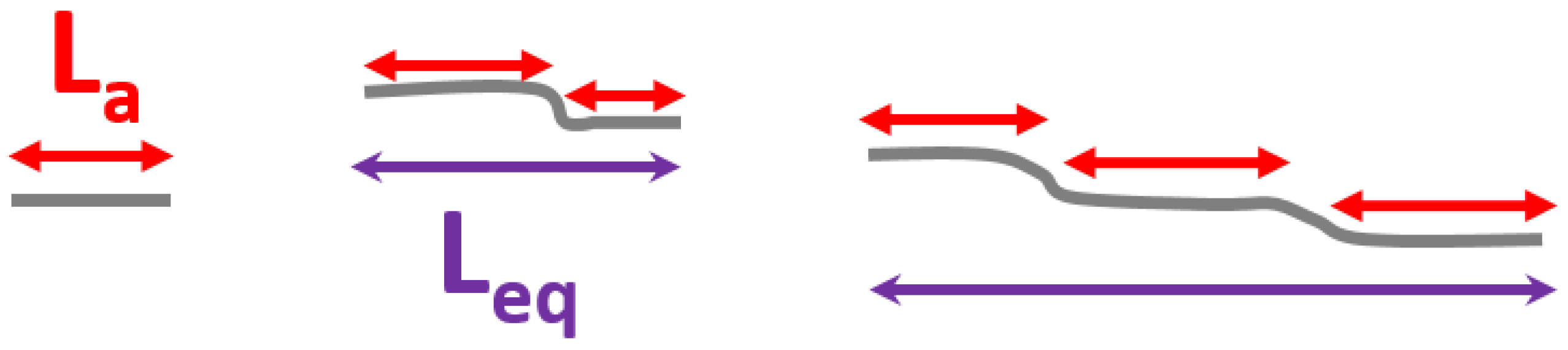
4.3. Other Effects: Our Propositions
- curvature effects;
- phonon confinement effects (due to poor coupling between different aromatic planes, porosities, etc.);
- and combinations thereof.
- heated C60;
- bombarded nanoribbons of different shapes and sizes deposited on deformable surfaces;
- in situ measurements of nanocones in high pressure/temperature cells.
5. Conclusions
- the ratio intensity between the D and G bands;
- the presence of additional bands (e.g., 2D, D’, etc.);
- the width of all bands.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| σx (expressed in cm−1): Band position of the band labelled x (x could be G, D, 2D, D’, …). |
| Γx (expressed in cm−1): Full width at half maximum of the band labelled x. |
| Ix (expressed in arbitrary units related to the number of counts on the detector): height of the band labelled x. |
| Ax (expressed in arbitrary units related to the number of counts on the detector): integrated area of the band labelled x. |
References
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef] [PubMed]
- Elias, D.C.; Nair, R.R.; Mohiuddin, T.M.G.; Morozov, S.V.; Blake, P.; Halsall, M.P.; Ferrari, A.C.; Boukhvalov, D.W.; Katsnelson, M.I.; Geim, A.K.; et al. Control of graphene’s properties by reversible hydrogenation: Evidence for graphane. Science 2009, 323, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, M.S.; Jorio, A.; Saito, R. Characterizing graphene, graphite, and carbon nanotubes by Raman spectroscopy. Annu. Rev. Condens. Matter Phys. 2010, 1, 89–108. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamondlike carbon. Phys. Rev. B 2001, 64, 075414. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 2477–2512. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, C.; Ferrari, A.C.; Robertson, J. Raman spectroscopy of hydrogenated amorphous carbons. Phys. Rev. B 2005, 72, 085401. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Roubin, P.; Giacometti, G.; Hopf, C.; Schwarz-Selinger, T.; Jacob, W. Raman spectroscopy investigation of the H content of heated hard amorphous carbon layers. Diam. Relat. Mater. 2013, 34, 100–104. [Google Scholar] [CrossRef]
- Mohiuddin, T.M.G.; Lombardo, A.; Nair, R.R.; Bonetti, A.; Savini, G.; Jalil, R.; Bonini, N.; Basko, D.M.; Galiotis, C.; Marzari, N.; et al. Uniaxial strain in graphene by Raman spectroscopy: G peak splitting, Grüneisen parameters, and sample orientation. Phys. Rev. B 2009, 79, 205433. [Google Scholar] [CrossRef]
- Piscanec, S.; Mauri, F.; Ferrari, A.C.; Lazzeri, M.; Robertson, J. Ab initio resonant Raman spectra of diamond-like carbons. Diam. Relat. Mater. 2005, 14, 1078–1083. [Google Scholar] [CrossRef]
- Brillouin, L. Diffusion de la lumière et des rayons X par un corps transparent homogène. Influence de l’agitation thermique. Ann. Phys. 1922, 17, 88–122. (In French) [Google Scholar] [CrossRef]
- Compton, A. A quantum theory of the scattering of X-rays by light elements. Phys. Rev. 1923, 21, 483. [Google Scholar] [CrossRef]
- Krishnan, R.S.; Shankar, R.K. Raman effect: History of the discovery. J. Raman Spectrosc. 1981, 10, 1–8. [Google Scholar] [CrossRef]
- Raman, C.V. A new radiation. Ind. J. Phys. 1928, 2, 387–398. [Google Scholar]
- Stokes, G.G. On the change of refrangibility of Light. Philos. Trans. R. Soc. 1852, 142, 463–562. [Google Scholar] [CrossRef]
- Ramaseshan, S. The Raman effect. Curr. Sci. 1998, 75, 6. [Google Scholar]
- Singh, R.; Riess, F. Seventy years ago—The discovery of the Raman effect as seen from German physicists. Curr. Sci. 1998, 74, 1112–1115. [Google Scholar]
- Singh, R.C.V. Raman and the discovery of the Raman effect. Phys. Perspect. 2002, 4, 399–420. [Google Scholar] [CrossRef]
- Smekal, A. The quantum, theory of dispersion. Naturwissenschaften 1923, 11, 873–878. [Google Scholar] [CrossRef]
- Kramers, H.A.; Heisenberg, W. Über die streuung von strahlung durch atome. Z. Phys. 1925, 31, 681–708. (In German) [Google Scholar] [CrossRef]
- Dirac, P.A.M. The quantum theory of dispersion. Proc. R. Soc. Lond. A 1927, 114, 710–728. [Google Scholar] [CrossRef]
- Breit, G. Quantum theory of dispersion. Rev. Mod. Phys. 1932, 4, 504. [Google Scholar] [CrossRef]
- Placzek, G. Rayleigh-Streuung und Raman-Effekt. In Handbuch der Radiologie; Marx, E.A., Ed.; Akademische Verlagsgesellschaft: Leipzig, Germany, 1934; Volume 6, p. 205. (In German) [Google Scholar]
- Albrecht, A.C. On the theory of Raman intensities. J. Chem. Phys. 1961, 34, 1476–1484. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; The International Series of Monographs on Physics; Oxford University Press: Oxford, UK, 1956. [Google Scholar]
- Loudon, R. The Raman effect in crystals. Adv. Phys. 1964, 13, 423–482. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Birman, J.L. Theory of Lattice Raman Scattering in Insulators. Phys. Rev. 1967, 162, 806–816. [Google Scholar] [CrossRef]
- Cardona, M.; Güntherodt, G. Light-Scattering in Solids; Topics in Applied Physics; Springer-Verlag: Berlin, Germany, 1989; Volume 66, pp. 2–12. [Google Scholar]
- Nafie, L.A. Recent advances in linear and non-linear Raman spectroscopy. Part X. J. Raman Spectrosc. 2016, 47, 1548–1565. [Google Scholar] [CrossRef]
- Gouadec, G.; Colomban, P. Raman spectroscopy of nanomaterials: How spectra relate to disorder, particle size and mechanical properties. Prog. Cryst. Growth Charact. Mater. 2007, 53, 1–56. [Google Scholar] [CrossRef]
- Rocard, Y. Role des vibrations des atomes dans les molécules dans le phénomène de diffusion de la lumière. Compt. Rend. 1927, 185, 1026–1028. (In French) [Google Scholar]
- Rocard, Y. Les nouvelles radiations diffusées. Compt. Rend. 1928, 186, 1107–1109. (In French) [Google Scholar]
- Raman, C.V. The scattering of light in crystals and the nature of their vibration spectra. Proc. Indian Acad. Sci. 1951, 34, 61–71. [Google Scholar]
- Poulet, H.; Mathieu, J.P. Détermination des vibrations fondamentales du sulfure de cadmium cristallisé. Ann. Phys. 1964, 13, 549–552. (In French) [Google Scholar] [CrossRef]
- Landsberg, G.; Mandelstam, L. Über die lichtzerstreuung in kristallen. Z. Phys. 1928, 50, 769–780. (In German) [Google Scholar] [CrossRef]
- Boyle, W.S.; Smith, G.E. Charge coupled semiconductors devices. Bell Syst. Tech. J. 1970, 49, 587–593. [Google Scholar] [CrossRef]
- Dierker, S.B.; Murray, C.A.; Legrange, J.D.; Schlotter, N.E. Characterization of order in langmuir-blodgett monolayers by unenhanced raman-spectroscopy. Chem. Phys. Lett. 1987, 137, 453–457. [Google Scholar] [CrossRef]
- Rabolt, J.F.; Santo, R.; Swalen, J.D. Raman measurements on thin polymer-films and organic monolayers. Appl. Spectrosc. 1980, 34, 517–521. [Google Scholar] [CrossRef]
- Adar, F.; Delhaye, M.; DaSilva, E. Evolution of instrumentation for detection of the Raman effect as driven by available technologies and by developing applications. J. Chem. Educ. 2007, 84, 50–60. [Google Scholar] [CrossRef]
- Long, D.A. Early history of the Raman effect. Int. Rev. Phys. Chem. 1988, 7, 317–349. [Google Scholar] [CrossRef]
- Langeluddecke, L.; Singh, P.; Deckert, V. Exploring the nanoscale: Fifteen years of tip-enhanced Raman spectroscopy. Appl. Spectrosc. 2015, 69, 1357–1371. [Google Scholar] [CrossRef] [PubMed]
- Long, D.A. The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; John Wiley & Sons, Ltd.: Chichester, UK, 2002. [Google Scholar]
- Cardona, M. Resonance phenomena. Top. Appl. Phys. 1982, 50, 19–178. [Google Scholar]
- Cantarero, A.; Tralleroginer, C.; Cardona, M. Excitons in one-phonon resonant Raman scattering: Deformation-potential interaction. Phys. Rev. B 1989, 39, 8388–8397. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Jorio, A. Applications of Group Theory to the Physics of Solids; Springer: New York, NY, USA, 2008. [Google Scholar]
- Yu, P.Y.; Cardona, M. Fundamentals of Semiconductors, Physics and Materials Properties, 4th ed.; Springer: Berlin, Germany, 2010. [Google Scholar]
- Ramsteiner, M.; Wild, C.; Wagner, J. Interference effects in the raman-scattering intensity from thin-films. Appl. Opt. 1989, 28, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Ni, Z.H.; Yu, T.; Shen, Z.X.; Wang, H.M.; Wu, Y.H.; Chen, W.; Wee, A.T.S. Raman studies of monolayer graphene: The substrate effect. J. Phys. Chem. C 2008, 112, 10637–10640. [Google Scholar] [CrossRef]
- Yoon, D.; Moon, H.; Son, Y.W.; Choi, J.S.; Park, B.H.; Cha, Y.H.; Kim, Y.D.; Cheong, H. Interference effect on Raman spectrum of graphene on SiO2/Si. Phys. Rev. B 2009, 80, 125422. [Google Scholar] [CrossRef]
- Klar, P.; Lidorikis, E.; Eckmann, A.; Verzhbitskiy, I.A.; Ferrari, A.C.; Casiraghi, C. Raman scattering efficiency of graphene. Phys. Rev. B 2013, 87, 205435. [Google Scholar] [CrossRef]
- Hirsch, A. The era of carbon allotropes. Nat. Mater. 2010, 9, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Ostrikov, K.; Neyts, E.C.; Meyyappan, M. Plasma nanoscience: From nano-solids in plasmas to nano-plasmas in solids. Adv. Phys. 2013, 62, 113–224. [Google Scholar] [CrossRef]
- Bernier, P.; Lefrant, S. Le Carbone Dans Tous Ses Etats; Gordon and Breach Science Publishers: Philadelphia, PA, USA, 1998. [Google Scholar]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Wallace, P. The band theory of graphite. Phys. Rev. 1947, 71, 622–634. [Google Scholar] [CrossRef]
- Ivanovskaya, V.V.; Zobelli, A.; Teillet-Billy, D.; Rougeau, N.; Sidis, V.; Briddon, P.R. Hydrogen adsorption on graphene: A first principles study. Eur. Phys. J. B 2010, 76, 481–486. [Google Scholar] [CrossRef]
- Wirtz, L.; Rubio, A. The phonon dispersion of graphite revisited. Solid State Commun. 2004, 131, 141–152. [Google Scholar] [CrossRef]
- Reich, S.; Thomsen, C. Raman spectroscopy of graphite. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 2271–2288. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, M.; Piscanec, S.; Mauri, F.; Ferrari, A.C.; Robertson, J. Phonon linewidths and electron-phonon coupling in graphite and nanotubes. Phys. Rev. B 2006, 73, 155426. [Google Scholar] [CrossRef]
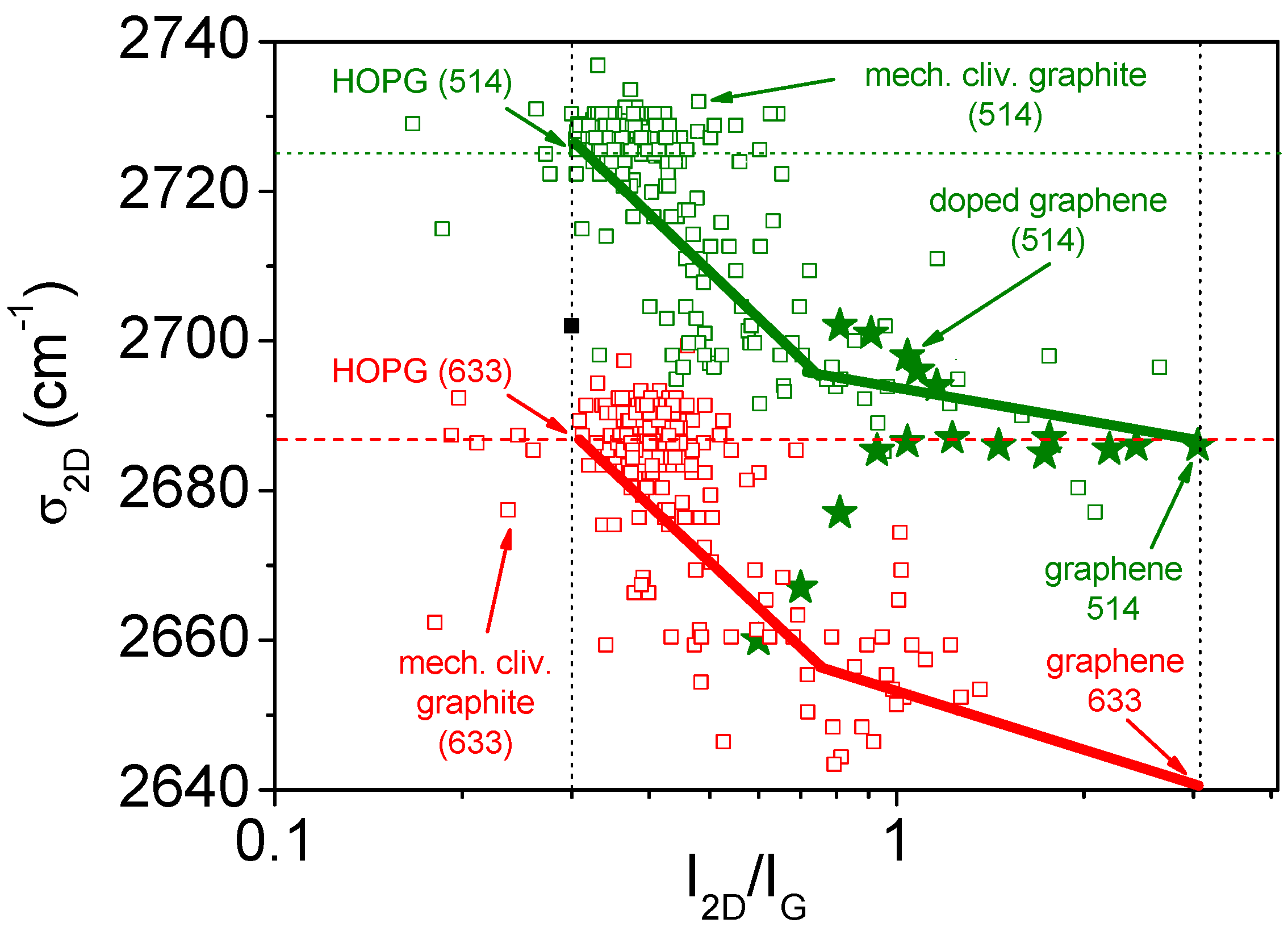
- Cancado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Speziali, N.L.; Jorio, A.; Pimenta, M.A. Measuring the degree of stacking order in graphite by Raman spectroscopy. Carbon 2008, 46, 272–275. [Google Scholar] [CrossRef]
- Kawashima, Y.; Katagiri, G. Fundamentals, overtones, and combinations in the raman-spectrum of graphite. Phys. Rev. B 1995, 52, 10053–10059. [Google Scholar] [CrossRef]
- Couzi, M.; Bruneel, J.L.; Talaga, D.; Bokobza, L. A multi wavelength Raman scattering study of defective graphitic carbon materials: The first order Raman spectra revisited. Carbon 2016, 107, 388–394. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Brar, V.W.; Samsonidze, G.G.; Dresselhaus, M.S.; Dresselhaus, G.; Saito, R.; Swan, A.K.; Unlu, M.S.; Goldberg, B.B.; Souza, A.G.; Jorio, A. Second-order harmonic and combination modes in graphite, single-wall carbon nanotube bundles, and isolated single-wall carbon nanotubes. Phys. Rev. B 2002, 66, 155418. [Google Scholar] [CrossRef]
- Tuinstra, F.; Koenig, J.L. Raman spectrum of graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
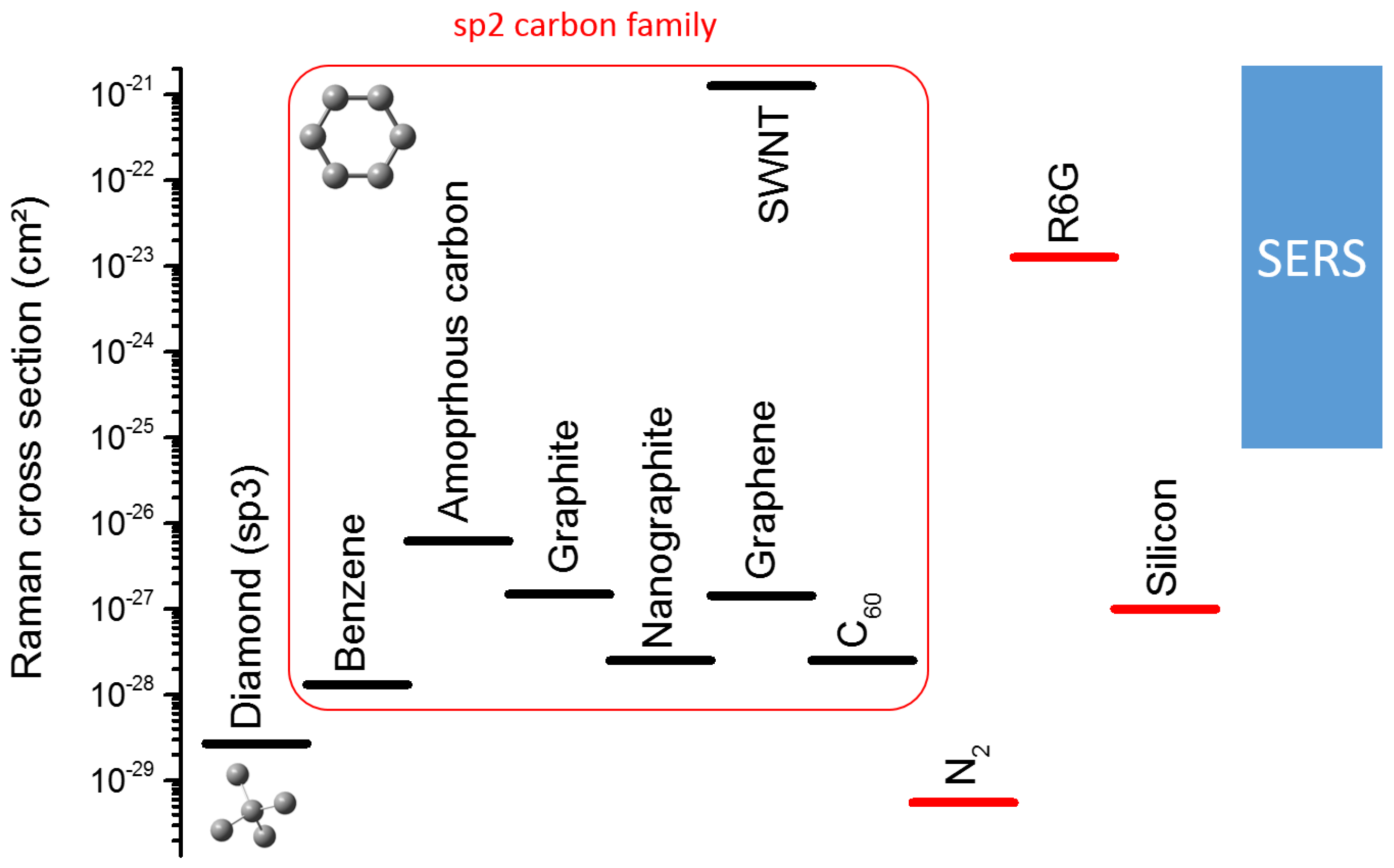
- Cançado, L.G.; Jorio, A.; Pimenta, M.A. Measuring the absolute Raman cross section of nanographites as a function of laser energy and crystallite size. Phys. Rev. B 2007, 76, 064304. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Roubin, P. Multiwavelength Raman spectroscopy analysis of a large sampling of disordered carbons extracted from the Tore Supra tokamak. Vib. Spectrosc. 2014, 70, 187–192. [Google Scholar] [CrossRef]
- Cancado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhaes-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Mernagh, T.P.; Cooney, R.P.; Johnson, R.A. Raman-spectra of graphon carbon-black. Carbon 1984, 22, 39–42. [Google Scholar] [CrossRef]
- Venezuela, P.; Lazzeri, M.; Mauri, F. Theory of double-resonant Raman spectra in graphene: Intensity and line shape of defect-induced and two-phonon bands. Phys. Rev. B 2011, 84, 035433. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef]
- Robertson, J.; Oreilly, E.P. Electronic and atomic-structure of amorphous-carbon. Phys. Rev. B 1987, 35, 2946–2957. [Google Scholar] [CrossRef]
- Lajaunie, L.; Pardanaud, C.; Martin, C.; Puech, P.; Hu, C.; Biggs, M.J.; Arenal, R. Advanced spectroscopic analyses on a:C–H materials: Revisiting the EELS characterization and its coupling with multi-wavelength Raman spectroscopy. Carbon 2017, 112, 149–161. [Google Scholar] [CrossRef]
- Ramaswamy, C. Raman effect in diamond. Nature 1930, 125, 704. [Google Scholar] [CrossRef]
- Solin, S.A.; Ramdas, A.K. Raman spectrum of diamond. Phys. Rev. B 1970, 1, 1687. [Google Scholar] [CrossRef]
- Yoshimori, A.; Kitano, Y. Theory of the lattice vibration of graphite. J. Phys. Soc. Jpn. 1956, 2, 352. [Google Scholar] [CrossRef]
- Young, J.A.; Koppel, J.U. Phonon spectrum of graphite. J. Chem. Phys. 1965, 42, 357. [Google Scholar] [CrossRef]
- Tsu, R.; Gonzalez, J.; Hernandez, I. Observation of splitting of E2g mode and two-phonon spectrum in graphites. Solid State Commun. 1978, 27, 507–510. [Google Scholar] [CrossRef]
- Vidano, R.; Fischbach, D.B. New lines in raman-spectra of carbons and graphite. J. Am. Ceram. Soc. 1978, 61, 13–17. [Google Scholar] [CrossRef]
- Lespade, P.; Marchand, A.; Couzi, M.; Cruege, F. Characterization of carbon materials with Raman microspectrometry. Carbon 1984, 22, 375–385. [Google Scholar] [CrossRef]
- Nakamizo, M.; Kammereck, R.; Walker, P.L. Laser Raman studies on carbons. Carbon 1974, 12, 259–267. [Google Scholar] [CrossRef]
- Nemanich, R.J.; Solin, S.A. 1st-order and 2nd-order raman-scattering from finite-size crystals of graphite. Phys. Rev. B 1979, 20, 392–401. [Google Scholar] [CrossRef]
- Vidano, R.P.; Fischbach, D.B.; Willis, L.J.; Loehr, T.M. Observation of raman band shifting with excitation wavelength for carbons and graphites. Solid State Commun. 1981, 39, 341–344. [Google Scholar] [CrossRef]
- Baranov, A.V.; Bekhterev, A.N.; Bobovich, Y.S.; Petrov, V.I. Interpretation of some singularities in raman-spectra of graphite and glass carbon. Opt. I Spektrosk. 1987, 62, 1036–1042. [Google Scholar]
- Wang, Y.; Alsmeyer, D.C.; McCreery, R.L. Raman-spectroscopy of carbon materials—Structural basis of observed spectra. Chem. Mater. 1990, 2, 557–563. [Google Scholar] [CrossRef]
- Ramsteiner, M.; Wagner, J. Resonant raman-scattering of hydrogenated amorphous-carbon—Evidence for pi-bonded carbon clusters. Appl. Phys. Lett. 1987, 51, 1355–1357. [Google Scholar] [CrossRef]
- Kastner, J.; Pichler, T.; Kuzmany, H.; Curran, S.; Blau, W.; Weldon, D.N.; Delamesiere, M.; Draper, S.; Zandbergen, H. Resonance Raman and infrared-spectroscopy of carbon nanotubues. Chem. Phys. Lett. 1994, 221, 53–58. [Google Scholar] [CrossRef]
- Rao, A.M.; Richter, E.; Bandow, S.; Chase, B.; Eklund, P.C.; Williams, K.A.; Fang, S.; Subbaswamy, K.R.; Menon, M.; Thess, A.; et al. Diameter-selective Raman scattering from vibrational modes in carbon nanotubes. Science 1997, 275, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.S.; White, W.B. Characterization of diamond films by Raman-spectroscopy. J. Mater. Res. 1989, 4, 385–393. [Google Scholar] [CrossRef]
- Tan, P.H.; Deng, Y.M.; Zhao, Q. Temperature-dependent Raman spectra and anomalous Raman phenomenon of highly oriented pyrolytic graphite. Phys. Rev. B 1998, 58, 5435–5439. [Google Scholar] [CrossRef]
- Pocsik, I.; Hundhausen, M.; Koos, M.; Ley, L. Origin of the D peak in the Raman spectrum of microcrystalline graphite. J. Non Cryst. Solids 1998, 227, 1083–1086. [Google Scholar] [CrossRef]
- Matthews, M.J.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Endo, M. Origin of dispersive effects of the Raman D band in carbon materials. Phys. Rev. B 1999, 59, R6585–R6588. [Google Scholar] [CrossRef]
- Thomsen, C.; Reich, S. Double resonant Raman scattering in graphite. Phys. Rev. Lett. 2000, 85, 5214–5217. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Jorio, A.; Souza, A.G.; Dresselhaus, G.; Dresselhaus, M.S.; Pimenta, M.A. Probing phonon dispersion relations of graphite by double resonance Raman scattering. Phys. Rev. Lett. 2002, 88, 027401. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cancado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; da Silva, M.G.; Ferreira, E.H.M.; Hof, F.; Kampioti, K.; Huang, K.; Penicaud, A.; Achete, C.A.; Capaz, R.B.; Jorio, A. Disentangling contributions of point and line defects in the Raman spectra of graphene-related materials. 2D Mater. 2017, 4, 015039. [Google Scholar]
- Cançado, L.G.; Jorio, A.; Martins Ferreira, E.H.; Stavale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying defects in graphene via Raman spectroscopy at different excitation energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.H.M.; Moutinho, M.V.O.; Stavale, F.; Lucchese, M.M.; Capaz, R.B.; Achete, C.A.; Jorio, A. Evolution of the Raman spectra from single-, few-, and many-layer graphene with increasing disorder. Phys. Rev. B 2010, 82, 125429. [Google Scholar] [CrossRef]
- Giro, R.; Archanjo, B.S.; Martins Ferreira, E.H.; Capaz, R.B.; Jorio, A.; Achete, C.A. Quantifying defects in N-layer graphene via a phenomenological model of Raman spectroscopy. Nucl. Instrum. Methods Phys. Res. Sec. B Beam Interact. Mater. Atoms 2014, 319, 71–74. [Google Scholar] [CrossRef]
- Lucchese, M.M.; Stavale, F.; Ferreira, E.H.M.; Vilani, C.; Moutinho, M.V.O.; Capaz, R.B.; Achete, C.A.; Jorio, A. Quantifying ion-induced defects and Raman relaxation length in graphene. Carbon 2010, 48, 1592–1597. [Google Scholar] [CrossRef]
- Ribeiro-Soares, J.; Oliveros, M.E.; Garin, C.; David, M.V.; Martins, L.G.P.; Almeida, C.A.; Martins-Ferreira, E.H.; Takai, K.; Enoki, T.; Magalhaes-Paniago, R.; et al. Structural analysis of polycrystalline graphene systems by Raman spectroscopy. Carbon 2015, 95, 646–652. [Google Scholar] [CrossRef]
- Cong, C.X.; Yu, T.; Saito, R.; Dresselhaus, G.F.; Dresselhaus, M.S. Second-order overtone and combination Raman modes of graphene layers in the range of 1690–2150 cm−1. Acs Nano 2011, 5, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.N. Two-phonon Raman bands of bilayer graphene: Revisited. Carbon 2015, 91, 436–444. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K.S.; Casiraghi, C. Probing the nature of defects in graphene by Raman spectroscopy. Nano Lett. 2012, 12, 3925–3930. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, M.A.; del Corro, E.; Carvalho, B.R.; Fantini, C.; Malard, L.M. Comparative study of Raman spectroscopy in graphene and MoS2-type transition metal dichalcogenides. Accounts Chem. Res. 2015, 48, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.R.; Wang, Y.X.; Mignuzzi, S.; Roy, D.; Terrones, M.; Fantini, C.; Crespi, V.H.; Malard, L.M.; Pimenta, M.A. Intervalley scattering by acoustic phonons in two-dimensional MoS2 revealed by double-resonance Raman spectroscopy. Nat. Commun. 2017, 8, 14670. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.H.; Yang, T.; Yamamoto, M.; Zhou, L.; Ishikawa, R.; Ueno, K.; Tsukagoshi, K.; Zhang, Z.D.; Dresselhaus, M.S.; Saito, R. Double resonance Raman modes in monolayer and few-layer MoTe2. Phys. Rev. B 2015, 91, 205415. [Google Scholar] [CrossRef]
- Castiglioni, C.; Di Donato, E.; Tommasini, M.; Negri, F.; Zerbi, G. Multi-wavelength Raman response of disordered graphitic materials: Models and simulations. Synth. Met. 2003, 139, 885–888. [Google Scholar] [CrossRef]
- Castiglioni, C.; Negri, F.; Tommasini, M.; Di Donato, E.; Zerbi, G. Raman spectra and structure of sp2 carbon-based materials: Electron-phonon coupling, vibrational dynamics and Raman activity. Carbon 2006, 100, 381–402. [Google Scholar]
- Castiglioni, C.; Tommasini, M.; Zerbi, G. Raman spectroscopy of polyconjugated molecules and materials: Confinement effect in one and two dimensions. Philos. Trans. R. Soc. 2004, 362, 2425–2459. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, E.; Tommasini, M.; Fustella, G.; Brambilla, L.; Castiglioni, C.; Zerbi, G.; Simpson, C.D.; Mullen, K.; Negri, F. Wavelength-dependent Raman activity of D2h symmetry polycyclic aromatic hydrocarbons in the D-band and acoustic phonon regions. Chem. Phys. 2004, 301, 81–93. [Google Scholar] [CrossRef]
- Negri, F.; Castiglioni, C.; Tommasini, M.; Zerbi, G. A computational study of the Raman spectra of large polycyclic aromatic hydrocarbons: Toward molecularly defined subunits of graphite. J. Phys. Chem. A 2002, 106, 3306–3317. [Google Scholar] [CrossRef]
- Negri, F.; di Donato, E.; Tommasini, M.; Castiglioni, C.; Zerbi, G.; Mullen, K. Resonance Raman contribution to the D band of carbon materials: Modeling defects with quantum chemistry. J. Chem. Phys. 2004, 120, 11889–11900. [Google Scholar] [CrossRef] [PubMed]
- Tommasini, M.; Di Donato, E.; Castiglioni, C.; Zerbi, G.; Severin, N.; Bohme, T.; Rabe, J.P. Resonant Raman spectroscopy of nanostructured carbon-based materials: The molecular approach. In Electronic Properties of Synthetic Nanostructures; Kuzmany, H., Fink, J., Mehring, M., Roth, S., Eds.; American Institute of Physics: College Park, MD, USA, 2004; Volume 723, pp. 334–338. [Google Scholar]
- Tommasini, M.; Castiglioni, C.; Zerbi, G. Raman scattering of molecular graphenes. Phys. Chem. Chem. Phys. 2009, 11, 10185–10194. [Google Scholar] [CrossRef] [PubMed]
- Maghsoumi, A.; Brambilla, L.; Castiglioni, C.; Mullen, K.; Tommasini, M. Overtone and combination features of G and D peaks in resonance Raman spectroscopy of the C78H26 polycyclic aromatic hydrocarbon. J. Raman Spectrosc. 2015, 46, 757–764. [Google Scholar] [CrossRef]
- Heller, E.J.; Yang, Y.; Kocia, L.; Chen, W.; Fang, S.A.; Borunda, M.; Kaxiras, E. Theory of graphene Raman scattering. Acs Nano 2016, 10, 2803–2818. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Lu, X.; Cong, C.X.; Yu, T.; Xiong, Q.H.; Quek, S.Y. Stacking sequence determines Raman intensities of observed interlayer shear modes in 2D layered materials—A general bond polarizability model. Sci. Rep. 2015, 5, 14565. [Google Scholar] [CrossRef] [PubMed]
- Benybassez, C.; Rouzaud, J.N. Characterization of carbonaceous materials by correlated electron and optical microscopy and raman microspectroscopy. Scanning Electron Microsc. 1985, 1, 119–132. [Google Scholar]
- Jawhari, T.; Roig, A.; Casado, J. Raman-spectroscopic characterization of some commercially available carbon-black materials. Carbon 1995, 33, 1561–1565. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Poschl, U. Raman micro spectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Vallerot, J.M.; Bourrat, X.; Mouchon, A.; Chollon, G. Quantitative structural and textural assessment of laminar pyrocarbons through Raman spectroscopy, electron diffraction and few other techniques. Carbon 2006, 44, 1833–1844. [Google Scholar] [CrossRef]
- Jacob, W.; Moller, W. On the structure of thin hydrocarbon films. Appl. Phys. Lett. 1993, 63, 1771–1773. [Google Scholar] [CrossRef]
- Hopf, C.; Angot, T.; Areou, E.; Duerbeck, T.; Jacob, W.; Martin, C.; Pardanaud, C.; Roubin, P.; Schwarz-Selinger, T. Characterization of temperature-induced changes in amorphous hydrogenated carbon thin films. Diam. Relat. Mater. 2013, 37, 97–103. [Google Scholar] [CrossRef]
- Schwarz-Selinger, T.; von Keudell, A.; Jacob, W. Plasma chemical vapor deposition of hydrocarbon films: The influence of hydrocarbon source gas on the film properties. J. Appl. Phys. 1999, 86, 3988–3996. [Google Scholar] [CrossRef]
- Wagner, J.; Ramsteiner, M.; Wild, C.; Koidl, P. Resonant raman-scattering of amorphous-carbon and polycrystalline diamond films. Phys. Rev. B 1989, 40, 1817–1824. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Li Bassi, A.; Tanner, B.K.; Stolojan, V.; Yuan, J.; Brown, L.M.; Rodil, S.E.; Kleinsorge, B.; Robertson, J. Density, sp3 fraction, and cross-sectional structure of amorphous carbon films determined by X-ray reflectivity and electron energy-loss spectroscopy. Phys. Rev. B 2000, 62, 11089–11103. [Google Scholar] [CrossRef]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Matus, M.; Kuzmany, H.; Kratschmer, W. Resonance raman-scattering and electronic-transitions in C60. Solid State Commun. 1991, 80, 839–842. [Google Scholar] [CrossRef]
- Sinha, K.; Menendez, J.; Hanson, R.C.; Adams, G.B.; Page, J.B.; Sankey, O.F.; Lamb, L.D.; Huffman, D.R. Evidence for solid-state effects in the electronic-structure of C60 films—A resonance-Raman study. Chem. Phys. Lett. 1991, 186, 287–290. [Google Scholar] [CrossRef]
- Vanloosdrecht, P.H.M.; Vanbentum, P.J.M.; Verheijen, M.A.; Meijer, G. Raman-scattering in single-crystal C60. Chem. Phys. Lett. 1992, 198, 587–595. [Google Scholar] [CrossRef]
- Monthioux, M.; Kuznetsov, V.L. Who should be given the credit for the discovery of carbon nanotubes? Carbon 2006, 44, 1621–1623. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Kataura, H.; Kumazawa, Y.; Maniwa, Y.; Umezu, I.; Suzuki, S.; Ohtsuka, Y.; Achiba, Y. Optical properties of single-wall carbon nanotubes. Synth. Met. 1999, 103, 2555–2558. [Google Scholar] [CrossRef]
- Barros, E.B.; Jorio, A.; Samsonidze, G.G.; Capaz, R.B.; Souza, A.G.; Mendes, J.; Dresselhaus, G.; Dresselhaus, M.S. Review on the symmetry-related properties of carbon nanotubes. Phys. Rep. Rev. Sec. Phys. Lett. 2006, 431, 261–302. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Jorio, A.; Souza, A.G.; Saito, R. Defect characterization in graphene and carbon nanotubes using Raman spectroscopy. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 5355–5377. [Google Scholar] [CrossRef] [PubMed]
- Malard, L.M.; Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectroscopy in graphene. Phys. Rep. Rev. Sec. Phys. Lett. 2009, 473, 51–87. [Google Scholar] [CrossRef]
- Mohr, M.; Maultzsch, J.; Thomsen, C. Splitting of the Raman 2D band of graphene subjected to strain. Phys. Rev. B 2010, 82, 201409. [Google Scholar] [CrossRef]
- Bonini, N.; Lazzeri, M.; Marzari, N.; Mauri, F. Phonon anharmonicities in graphite and graphene. Phys. Rev. Lett. 2007, 99, 176802. [Google Scholar] [CrossRef] [PubMed]
- Schwan, J.; Ulrich, S.; Batori, V.; Ehrhardt, H.; Silva, S.R.P. Raman spectroscopy on amorphous carbon films. J. Appl. Phys. 1996, 80, 440–447. [Google Scholar] [CrossRef]
- Chu, P.K.; Li, L.H. Characterization of amorphous and nanocrystalline carbon films. Mater. Chem. Phys. 2006, 96, 253–277. [Google Scholar] [CrossRef]
- Wang, Q.; Allred, D.D.; Knight, L.V. Deconvolution of the Raman spectrum of amorphous carbon. J. Raman Spectrosc. 1995, 26, 1039–1043. [Google Scholar] [CrossRef]
- Richter, H.; Wang, Z.P.; Ley, L. The one phonon raman-spectrum in microcrystalline silicon. Solid State Commun. 1981, 39, 625–629. [Google Scholar] [CrossRef]
- Puech, P.; Plewa, J.M.; Mallet-Ladeira, P.; Monthioux, M. Spatial confinement model applied to phonons in disordered graphene-based carbons. Carbon 2016, 105, 275–281. [Google Scholar] [CrossRef]
- Jorio, A.; Ferreira, E.H.M.; Moutinho, M.V.O.; Stavale, F.; Achete, C.A.; Capaz, R.B. Measuring disorder in graphene with the G and D bands. Phys. Status Solidi B 2010, 247, 2980–2982. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Saidi, W.A. Effects of topological defects and diatom vacancies on characteristic vibration modes and Raman intensities of zigzag single-walled carbon nanotubes. J. Phys. Chem. A 2014, 118, 7235–7241. [Google Scholar] [CrossRef] [PubMed]
- Saidi, W.A.; Norman, P. Probing single-walled carbon nanotube defect chemistry using resonance Raman spectroscopy. Carbon 2014, 67, 17–26. [Google Scholar] [CrossRef]
- Saidi, W.A.; Norman, P. Spectroscopic signatures of topological and diatom-vacancy defects in single-walled carbon nanotubes. Phys. Chem. Chem. Phys. 2014, 16, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Kudin, K.N.; Ozbas, B.; Schniepp, H.C.; Prud’homme, R.K.; Aksay, I.A.; Car, R. Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett. 2008, 8, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.L.; Farrar, L.W.; Saikin, S.K.; Andrade, X.; Aspuru-Guzik, A.; Polla, D.L. Measurement of the absolute Raman cross section of the optical phonons in type Ia natural diamond. Solid State Commun. 2012, 152, 204–209. [Google Scholar] [CrossRef]
- Skinner, J.G.; Nilsen, W.G. Absolute Raman scattering cross-section measurement of the 992 cm−1 line of benzene. J. Opt. Soc. Am. 1968, 58, 113–119. [Google Scholar] [CrossRef]
- Aggarwal, R.L.; Farrar, L.W.; Saikin, S.K.; Aspuru-Guzik, A.; Stopa, M.; Polla, D.L. Measurement of the absolute Raman cross section of the optical phonon in silicon. Solid State Commun. 2011, 151, 553–556. [Google Scholar] [CrossRef]
- Pettinger, B.; Picardi, G.; Schuster, R.; Ertl, G. Surface-enhanced and STM-tip-enhanced Raman spectroscopy at metal surfaces. Single Mol. 2002, 3, 285–294. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Ni, Z.H.; Shen, Z.X.; Wang, H.M.; Wu, Y.H. Interference enhancement of Raman signal of graphene. Appl. Phys. Lett. 2008, 92, 043121. [Google Scholar] [CrossRef]
- Peres, N.M.R. Colloquium: The transport properties of graphene: An introduction. Rev. Mod. Phys. 2010, 82, 2673–2700. [Google Scholar] [CrossRef]
- Beams, R.; Cancado, L.G.; Novotny, L. Raman characterization of defects and dopants in graphene. J. Phys. Condens. Matter 2015, 27, 083002. [Google Scholar] [CrossRef] [PubMed]
- Bayle, M.; Reckinger, N.; Huntzinger, J.R.; Felten, A.; Bakaraki, A.; Landois, P.; Colomer, J.F.; Henrard, L.; Zahab, A.A.; Sauvajol, J.L.; et al. Dependence of the Raman spectrum characteristics on the number of layers and stacking orientation in few-layer graphene. Phys. Status Solidi B 2015, 252, 2375–2379. [Google Scholar] [CrossRef]
- Poncharal, P.; Ayari, A.; Michel, T.; Sauvajol, J.L. Raman spectra of misoriented bilayer graphene. Phys. Rev. B 2008, 78, 113407. [Google Scholar] [CrossRef]
- Poncharal, P.; Ayari, A.; Michel, T.; Sauvajol, J.L. Effect of rotational stacking faults on the Raman spectra of folded graphene. Phys. Rev. B 2009, 79, 195417. [Google Scholar] [CrossRef]
- Das, A.; Chakraborty, B.; Sood, A.K. Raman spectroscopy of graphene on different substrates and influence of defects. Bull. Mater. Sci. 2008, 31, 579–584. [Google Scholar] [CrossRef]
- Tan, P.H.; Han, W.P.; Zhao, W.J.; Wu, Z.H.; Chang, K.; Wang, H.; Wang, Y.F.; Bonini, N.; Marzari, N.; Pugno, N.; et al. The shear mode of multilayer graphene. Nat. Mater. 2012, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Araujo, P.T.; Terrones, M.; Dresselhaus, M.S. Defects and impurities in graphene-like materials. Mater. Today 2012, 15, 98–109. [Google Scholar] [CrossRef]
- Casiraghi, C. Doping dependence of the Raman peaks intensity of graphene close to the Dirac point. Phys. Rev. B 2009, 80, 233407. [Google Scholar] [CrossRef]
- Casiraghi, C.; Pisana, S.; Novoselov, K.S.; Geim, A.K.; Ferrari, A.C. Raman fingerprint of charged impurities in graphene. Appl. Phys. Lett. 2007, 91, 233108. [Google Scholar] [CrossRef]
- Kalbac, M.; Reina-Cecco, A.; Farhat, H.; Kong, J.; Kavan, L.; Dresselhaus, M.S. The influence of strong electron and hole doping on the Raman intensity of chemical vapor-deposition graphene. ACS Nano 2010, 4, 6055–6063. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; Li, Q.Q.; Zou, Y.; Qian, Q.K.; Jin, Y.H.; Li, G.H.; Jiang, K.L.; Fan, S.S. The dependence of graphene Raman D-band on carrier density. Nano Lett. 2013, 13, 6170–6175. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.H.; Ponomarenko, L.A.; Nair, R.R.; Yang, R.; Anissimova, S.; Grigorieva, I.V.; Schedin, F.; Blake, P.; Shen, Z.X.; Hill, E.H.; et al. On resonant scatterers as a factor limiting carrier mobility in graphene. Nano Lett. 2010, 10, 3868–3872. [Google Scholar] [CrossRef] [PubMed]
- Calizo, I.; Ghosh, S.; Bao, W.Z.; Miao, F.; Lau, C.N.; Balandin, A.A. Raman nanometrology of graphene: Temperature and substrate effects. Solid State Commun. 2009, 149, 1132–1135. [Google Scholar] [CrossRef]
- Calizo, I.; Bao, W.Z.; Miao, F.; Lau, C.N.; Balandin, A.A. The effect of substrates on the Raman spectrum of graphene: Graphene-on-sapphire and graphene-on-glass. Appl. Phys. Lett. 2007, 91, 201904. [Google Scholar] [CrossRef]
- Frank, O.; Mohr, M.; Maultzsch, J.; Thomsen, C.; Riaz, I.; Jalil, R.; Novoselov, K.S.; Tsoukleri, G.; Parthenios, J.; Papagelis, K.; et al. Raman 2D-band splitting in graphene: Theory and experiment. Acs Nano 2011, 5, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Huang, Q.S.; Chen, X.L.; Zhang, G.Y.; Gao, H.J. Substrate doping effects on Raman spectrum of epitaxial graphene on SiC. J. Appl. Phys. 2010, 107, 034305. [Google Scholar] [CrossRef]
- Das, A.; Pisana, S.; Chakraborty, B.; Piscanec, S.; Saha, S.K.; Waghmare, U.V.; Novoselov, K.S.; Krishnamurthy, H.R.; Geim, A.K.; Ferrari, A.C.; et al. Monitoring dopants by Raman scattering in an electrochemically top-gated graphene transistor. Nat. Nanotechnol. 2008, 3, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Novoselov, K.S.; Shin, H.S. Interaction between metal and graphene: Dependence on the Layer number of graphene. Acs Nano 2011, 5, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.G.; Mao, N.N.; Zhang, J. Graphene: A platform for surface-enhanced Raman spectroscopy. Small 2013, 9, 1206–1224. [Google Scholar] [CrossRef] [PubMed]
- Bronsgeest, M.S.; Bendiab, N.; Mathur, S.; Kimouche, A.; Johnson, H.T.; Coraux, J.; Pochet, P. Strain relaxation in CVD graphene: Wrinkling with shear lag. Nano Lett. 2015, 15, 5098–5104. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Siregar, S.; Hasdeo, E.H.; Kumamoto, Y.; Shen, C.C.; Cheng, C.C.; Li, L.J.; Saito, R.; Kawata, S. Deep-ultraviolet Raman scattering studies of monolayer graphene thin films. Carbon 2015, 81, 807–813. [Google Scholar] [CrossRef]
- Tyborski, C.; Herziger, F.; Gillen, R.; Maultzsch, J. Beyond double-resonant Raman scattering: Ultraviolet Raman spectroscopy on graphene, graphite, and carbon nanotubes. Phys. Rev. B 2015, 92, 041401. [Google Scholar] [CrossRef]
- Saito, R.; Nugraha, A.R.T.; Hasdeo, E.H.; Siregar, S.; Guo, H.H.; Yang, T. Ultraviolet Raman spectroscopy of graphene and transition-metal dichalcogenides. Phys. Status Solidi B 2015, 252, 2363–2374. [Google Scholar] [CrossRef]
- Zhou, W.; Zeng, J.W.; Li, X.F.; Xu, J.; Shi, Y.; Ren, W.; Miao, F.; Wang, B.G.; Xing, D.Y. Ultraviolet Raman spectra of double-resonant modes of graphene. Carbon 2016, 101, 235–238. [Google Scholar] [CrossRef]
- Herziger, F.; Calandra, M.; Gava, P.; May, P.; Lazzeri, M.; Mauri, F.; Maultzsch, J. Two-dimensional analysis of the double-resonant 2D Raman mode in bilayer graphene. Phys. Rev. Lett. 2014, 113, 187401. [Google Scholar] [CrossRef] [PubMed]
- Zandiatashbar, A.; Lee, G.-H.; An, S.J.; Lee, S.; Mathew, N.; Terrones, M.; Hayashi, T.; Picu, C.R.; Hone, J.; Koratkar, N. Effect of defects on the intrinsic strength and stiffness of graphene. Nat. Commun. 2014, 5, 3186. [Google Scholar] [CrossRef] [PubMed]
- Ferralis, N. Probing mechanical properties of graphene with Raman spectroscopy. J. Mater. Sci. 2010, 45, 5135–5149. [Google Scholar] [CrossRef]
- Yu, Q.K.; Jauregui, L.A.; Wu, W.; Colby, R.; Tian, J.F.; Su, Z.H.; Cao, H.L.; Liu, Z.H.; Pandey, D.; Wei, D.G.; et al. Control and characterization of individual grains and grain boundaries in graphene grown by chemical vapour deposition. Nat. Mater. 2011, 10, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, J.H.; Kim, M.J.; Dash, J.K.; Lee, C.H.; Joshi, R.; Lee, S.; Hone, J.; Soon, A.; Lee, G.H. Direct observation of grain boundaries in chemical vapor deposited graphene. Carbon 2017, 115, 147–153. [Google Scholar] [CrossRef]
- Chen, S.S.; Moore, A.L.; Cai, W.W.; Suk, J.W.; An, J.H.; Mishra, C.; Amos, C.; Magnuson, C.W.; Kang, J.Y.; Shi, L.; et al. Raman measurements of thermal transport in suspended monolayer graphene of variable sizes in vacuum and gaseous environments. Acs Nano 2011, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Metten, D.; Froehlicher, G.; Berciaud, S. Monitoring electrostatically-induced deflection, strain and doping in suspended graphene using Raman spectroscopy. 2D Mater. 2017, 4. [Google Scholar] [CrossRef]
- Suarez-Martinez, I.; Grobert, N.; Ewels, C.P. Nomenclature of sp2 carbon nanoforms. Carbon 2012, 50, 741–747. [Google Scholar] [CrossRef]
- Bianco, A.; Cheng, H.M.; Enoki, T.; Gogotsi, Y.; Hurt, R.H.; Koratkar, N.; Kyotani, T.; Monthioux, M.; Park, C.R.; Tascon, J.M.D.; et al. All in the graphene family—A recommended nomenclature for two-dimensional carbon materials. Carbon 2013, 65, 1–6. [Google Scholar] [CrossRef]
- Wick, P.; Louw-Gaume, A.E.; Kucki, M.; Krug, H.F.; Kostarelos, K.; Fadeel, B.; Dawson, K.A.; Salvati, A.; Vazquez, E.; Ballerini, L.; et al. Classification framework for graphene-based materials. Angew. Chem. Int. Ed. 2014, 53, 7714–7718. [Google Scholar] [CrossRef] [PubMed]
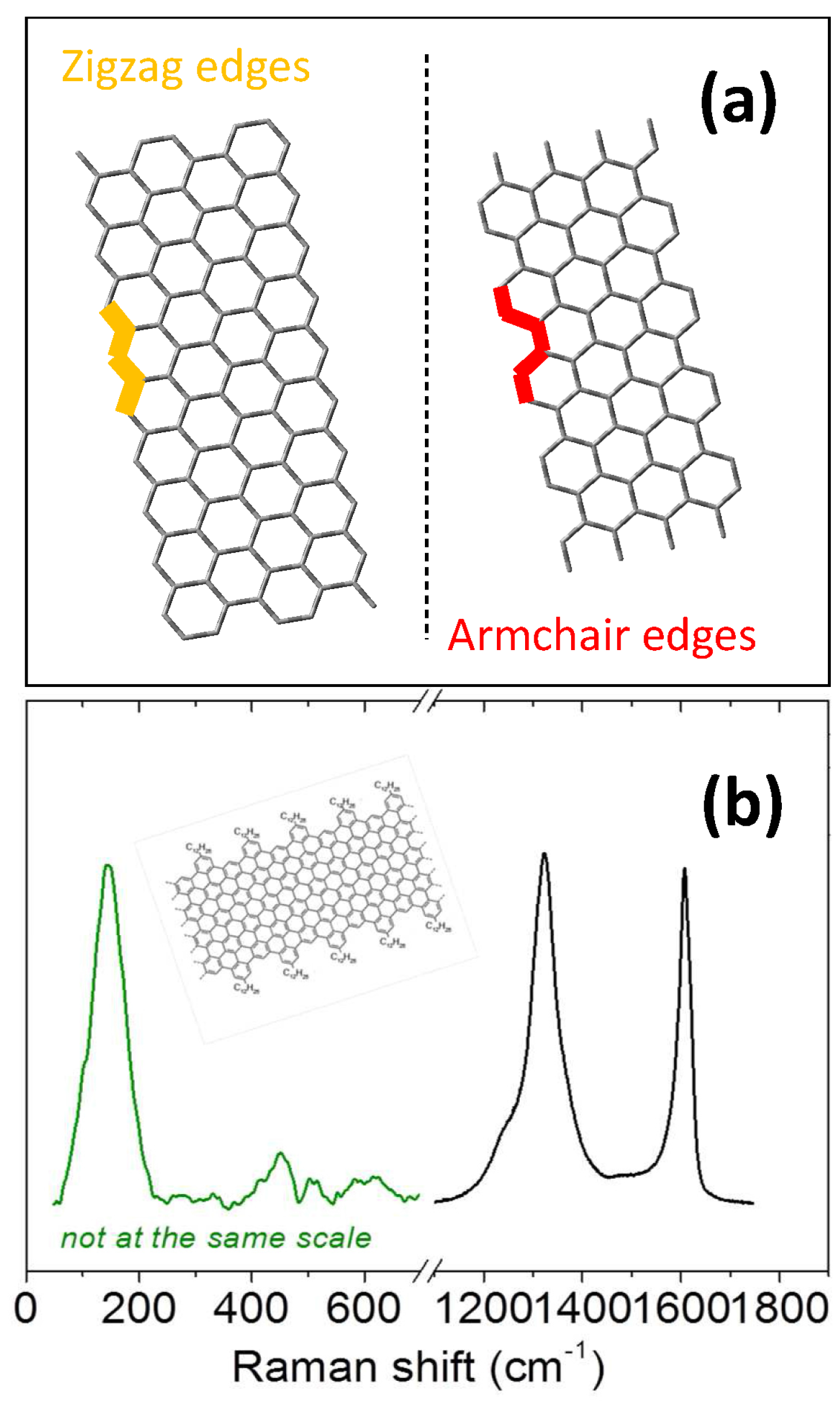
- Cancado, L.G.; Pimenta, M.A.; Neves, B.R.A.; Dantas, M.S.S.; Jorio, A. Influence of the atomic structure on the Raman spectra of graphite edges. Phys. Rev. Lett. 2004, 93, 247401. [Google Scholar] [CrossRef] [PubMed]
- You, Y.M.; Ni, Z.H.; Yu, T.; Shen, Z.X. Edge chirality determination of graphene by Raman spectroscopy. Appl. Phys. Lett. 2008, 93, 163112. [Google Scholar] [CrossRef]
- Casiraghi, C.; Hartschuh, A.; Qian, H.; Piscanec, S.; Georgi, C.; Fasoli, A.; Novoselov, K.S.; Basko, D.M.; Ferrari, A.C. Raman spectroscopy of graphene edges. Nano Lett. 2009, 9, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Tamakawa, D.; Tanaka, S.; Makino, T.; Hashimoto, A. Polarized microscopic laser Raman scattering spectroscopy for edge structure of epitaxial graphene and localized vibrational mode. Carbon 2014, 77, 1073–1081. [Google Scholar] [CrossRef]
- Ren, W.C.; Saito, R.; Gao, L.B.; Zheng, F.W.; Wu, Z.S.; Liu, B.L.; Furukawa, M.; Zhao, J.P.; Chen, Z.P.; Cheng, H.M. Edge phonon state of mono- and few-layer graphene nanoribbons observed by surface and interference co-enhanced Raman spectroscopy. Phys. Rev. B 2010, 81, 035412. [Google Scholar] [CrossRef]
- Mazzamuto, F.; Saint-Martin, J.; Valentin, A.; Chassat, C.; Dollfus, P. Edge shape effect on vibrational modes in graphene nanoribbons: A numerical study. J. Appl. Phys. 2011, 109, 064516. [Google Scholar] [CrossRef]
- Saito, R.; Furukawa, M.; Dresselhaus, G.; Dresselhaus, M.S. Raman spectra of graphene ribbons. J. Phys. Condens. Matter 2010, 22, 334203. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhou, H.Q.; Zhang, Z.X.; Tang, D.S.; Chen, M.J.; Yang, H.C.; Wang, G.; Yang, H.F.; Gu, C.Z.; Sun, L.F. Experimental observation of radial breathing-like mode of graphene nanoribbons. Appl. Phys. Lett. 2012, 100, 101904. [Google Scholar] [CrossRef]
- Zhou, J.; Dong, J. Vibrational property and Raman spectrum of carbon nanoribbon. Appl. Phys. Lett. 2007, 91, 173108. [Google Scholar] [CrossRef]
- Verzhbitskiy, I.A.; De Corato, M.; Ruini, A.; Molinari, E.; Narita, A.; Hu, Y.; Schwab, M.G.; Bruna, M.; Yoon, D.; Milana, S.; et al. Raman fingerprints of atomically precise graphene nanoribbons. Nano Lett. 2016, 16, 3442–3447. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, C.; Prezzi, D. Raman spectroscopy of graphene nanoribbons: A review. In GraphITA: Selected papers from the Workshop on Synthesis, Characterization and Technological Exploitation of Graphene and 2D Materials Beyond Graphene; Carbon Nanostructures; Morandi, V., Ottaviano, L., Eds.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Reich, S.; Thomsen, C.; Maultzsch, J. Carbon Nanotubes: Basic Concepts and Physical Properties; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Saito, R.; Dresselhaus, G.; Dresselhaus, M.S. Physical Properties of Carbon Nanotubes; Imperial College Press: London, UK, 1998. [Google Scholar]
- Bohn, J.E.; Etchegoin, P.G.; le Ru, E.C.; Xiang, R.; Chiashi, S.; Maruyama, S. Estimating the Raman cross sections of single carbon nanotubes. Acs Nano 2010, 4, 3466–3470. [Google Scholar] [CrossRef] [PubMed]
- Sauvajol, J.-L.; Anglaret, E.; Rols, S.; Stephan, O. Spectroscopies on carbon nanotubes. In Understanding Carbon Nanotubes: From Basics to Applications; Loiseau, A., Launois, P., Petit, P., Roche, S., Salvetat, J.-P., Eds.; Springer: Berlin, Germany, 2006; Volume 677, pp. 277–334. [Google Scholar]
- Ghavanloo, E.; Fazelzadeh, S.A.; Rafii-Tabar, H. Analysis of radial breathing-mode of nanostructures with various morphologies: A critical review. Int. Mater. Rev. 2015, 60, 312–329. [Google Scholar] [CrossRef]
- Jorio, A.; Pimenta, M.A.; Souza, A.G.; Saito, R.; Dresselhaus, G.; Dresselhaus, M.S. Characterizing carbon nanotube samples with resonance Raman scattering. New J. Phys. 2003, 5, 139. [Google Scholar] [CrossRef]
- Brown, S.D.M.; Jorio, A.; Corio, P.; Dresselhaus, M.S.; Dresselhaus, G.; Saito, R.; Kneipp, K. Origin of the Breit-Wigner-Fano lineshape of the tangential G-band feature of metallic carbon nanotubes. Phys. Rev. B 2001, 63, 155414. [Google Scholar] [CrossRef]
- Song, L.; Ci, L.J.; Sun, L.F.; Jin, C.H.; Liu, L.F.; Ma, W.J.; Liu, D.F.; Zhao, X.W.; Luo, S.D.; Zhang, Z.X.; et al. Large-scale synthesis of rings of bundled single-walled carbon nanotubes by floating chemical vapor deposition. Adv. Mater. 2006, 18, 1817–1821. [Google Scholar] [CrossRef]
- Ren, Y.; Song, L.; Ma, W.J.; Zhao, Y.C.; Sun, L.F.; Gu, C.Z.; Zhou, W.Y.; Xie, S.S. Additional curvature-induced Raman splitting in carbon nanotube ring structures. Phys. Rev. B 2009, 80, 113412. [Google Scholar] [CrossRef]
- Dunk, P.W.; Niwa, H.; Shinohara, H.; Marshall, A.G.; Kroto, H.W. Large fullerenes in mass spectra. Mol. Phys. 2015, 113, 2359–2361. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Eklund, P.C. Raman scattering in fullerenes. J. Raman Spectrosc. 1996, 27, 351–371. [Google Scholar] [CrossRef]
- Kuzmany, H.; Pfeiffer, R.; Hulman, M.; Kramberger, C. Raman spectroscopy of fullerenes and fullerene-nanotube composites. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 2375–2406. [Google Scholar] [CrossRef] [PubMed]
- Bardelang, D.; Giorgi, M.; Pardanaud, C.; Hornebecq, V.; Rizzato, E.; Tordo, P.; Ouari, O. Organic multishell isostructural host-guest crystals: Fullerenes C60 inside a nitroxide open framework. Chem. Commun. 2013, 49, 3519–3521. [Google Scholar] [CrossRef] [PubMed]
- Jishi, R.A.; Dresselhaus, M.S.; Dresselhaus, G.; Wang, K.A.; Zhou, P.; Rao, A.M.; Eklund, P.C. Vibrational-mode frequencies in C70. Chem. Phys. Lett. 1993, 206, 187–192. [Google Scholar] [CrossRef]
- Eklund, P.C.; Rao, A.M.; Zhou, P.; Wang, Y. Photochemical transformation of C60 and C70 films. Thin Solid Films 1995, 257, 185–203. [Google Scholar] [CrossRef]
- Brazhkin, V.V.; Lyapin, A.G.; Popova, S.V.; Voloshin, R.N.; Antonov, Y.V.; Lyapin, S.G.; Kluev, Y.A.; Naletov, A.M.; Melnik, N.N. Metastable crystalline and amorphous carbon phases obtained from fullerite C60 by high-pressuse-high-temperature treatment. Phys. Rev. B 1997, 56, 11465–11472. [Google Scholar] [CrossRef]
- Karousis, N.; Suarez-Martinez, I.; Ewels, C.P.; Tagmatarchis, N. Structure, properties, functionalization, and applications of carbon nanohorns. Chem. Rev. 2016, 116, 4850–4883. [Google Scholar] [CrossRef] [PubMed]
- Iijima, S.; Yudasaka, M.; Yamada, R.; Bandow, S.; Suenaga, K.; Kokai, F.; Takahashi, K. Nano-aggregates of single-walled graphitic carbon nano-horns. Chem. Phys. Lett. 1999, 309, 165–170. [Google Scholar] [CrossRef]
- Pena-Alvarez, M.; del Corro, E.; Langa, F.; Baonza, V.G.; Taravillo, M. Morphological changes in carbon nanohorns under stress: A combined Raman spectroscopy and TEM study. RSC Adv. 2016, 6, 49543–49550. [Google Scholar] [CrossRef]
- Sasaki, K.; Sekine, Y.; Tateno, K.; Gotoh, H. Topological Raman band in the carbon nanohorn. Phys. Rev. Lett. 2013, 111, 116801. [Google Scholar] [CrossRef] [PubMed]
- Pardanaud, C.; Martin, C.; Giacometti, G.; Mellet, N.; Pegourie, B.; Roubin, P. Thermal stability and long term hydrogen/deuterium release from soft to hard amorphous carbon layers analyzed using in-situ Raman spectroscopy. Comparison with Tore Supra deposits. Thin Solid Films 2015, 581, 92–98. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Giacometti, G.; Roubin, P.; Pegourie, B.; Hopf, C.; Schwarz-Selinger, T.; Jacob, W.; Buijnsters, J.G. Long-term H-release of hard and intermediate between hard and soft amorphous carbon evidenced by in situ Raman microscopy under isothermal heating. Diam. Relat. Mater. 2013, 37, 92–96. [Google Scholar] [CrossRef]
- Niwase, K.; Tanabe, T.; Sugimoto, M.; Fujita, F.E. Modification of graphite structure by D+ and He+ bombardment. J. Nucl. Mater. 1989, 162, 856–860. [Google Scholar] [CrossRef]
- Oschatz, M.; Pre, P.; Dorfler, S.; Nickel, W.; Beaunier, P.; Rouzaud, J.N.; Fischer, C.; Brunner, E.; Kaskel, S. Nanostructure characterization of carbide-derived carbons by morphological analysis of transmission electron microscopy images combined with physisorption and Raman spectroscopy. Carbon 2016, 105, 314–322. [Google Scholar] [CrossRef]
- Da Costa, J.P.; Weisbecker, P.; Farbos, B.; Leyssale, J.M.; Vignoles, G.L.; Germain, C. Investigating carbon materials nanostructure using image orientation statistics. Carbon 2015, 84, 160–173. [Google Scholar] [CrossRef]
- Niwase, K. Irradiation-induced amorphization of graphite. Phys. Rev. B 1995, 52, 15785–15798. [Google Scholar] [CrossRef]
- Pardanaud, C.; Martin, C.; Cartry, G.; Ahmad, A.; Schiesko, L.; Giacometti, G.; Carrere, M.; Roubin, P. In-plane and out-of-plane defects of graphite bombarded by H, D and He investigated by atomic force and Raman microscopies. J. Raman Spectrosc. 2015, 46, 256–265. [Google Scholar] [CrossRef]
- Banhart, F.; Kotakoski, J.; Krasheninnikov, A.V. Structural defects in graphene. Acs Nano 2011, 5, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Kotakoski, J.; Krasheninnikov, A.V.; Kaiser, U.; Meyer, J.C. From point defects in graphene to two-dimensional amorphous carbon. Phys. Rev. Lett. 2011, 106, 105505. [Google Scholar] [CrossRef] [PubMed]
- Elman, B.S.; Dresselhaus, M.S.; Dresselhaus, G.; Maby, E.W.; Mazurek, H. Raman-scattering from ion-implanted graphite. Phys. Rev. B 1981, 24, 1027–1034. [Google Scholar] [CrossRef]
- Compagnini, G.; Puglisi, O.; Foti, G. Raman spectra of virgin and damaged graphite edge planes. Carbon 1997, 35, 1793–1797. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujitsuka, M.; Kitajima, M. Finite size effect on raman-scattering of graphite microcrystals. Chem. Phys. Lett. 1990, 172, 205–208. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitajima, M. Real-time raman measurements of graphite under Ar+ irradiation. Appl. Phys. Lett. 1991, 59, 1550–1552. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitajima, M. Ion-irradiation effects on the phonon correlation length of graphite studies by raman-spectroscopy. Phys. Rev. B 1992, 45, 78–82. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Verzhbitskiy, I.; Davey, R.; Casiraghi, C. Raman study on defective graphene: Effect of the excitation energy, type, and amount of defects. Phys. Rev. B 2013, 88, 035426. [Google Scholar] [CrossRef]
- Sato, K.; Saito, R.; Oyama, Y.; Jiang, J.; Cancado, L.G.; Pimenta, M.A.; Jorio, A.; Samsonidze, G.G.; Dresselhaus, G.; Dresselhaus, M.S. D-band Raman intensity of graphitic materials as a function of laser energy and crystallite size. Chem. Phys. Lett. 2006, 427, 117–121. [Google Scholar] [CrossRef]
- Meyer, J.C.; Kisielowski, C.; Erni, R.; Rossell, M.D.; Crommie, M.F.; Zettl, A. Direct imaging of lattice atoms and topological defects in graphene membranes. Nano Lett. 2008, 8, 3582–3586. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.Y.; Yeo, J.J.; Liu, Z.S. A molecular dynamics study of the thermal conductivity of graphene nanoribbons containing dispersed Stone-Thrower-Wales defects. Carbon 2012, 50, 4887–4893. [Google Scholar] [CrossRef]
- Thrower, P.A. The study of defects in graphite by transmission electron microscopy. In Chemistry and Physics of Carbon; Walker, P.L., Jr., Ed.; Marcel Dekker: New York, NY, USA, 1969. [Google Scholar]
- Wu, G.; Dong, J.M. Raman characteristic peaks induced by the topological defects of carbon nanotube intramolecular junctions. Phys. Rev. B 2006, 73, 245414. [Google Scholar] [CrossRef]
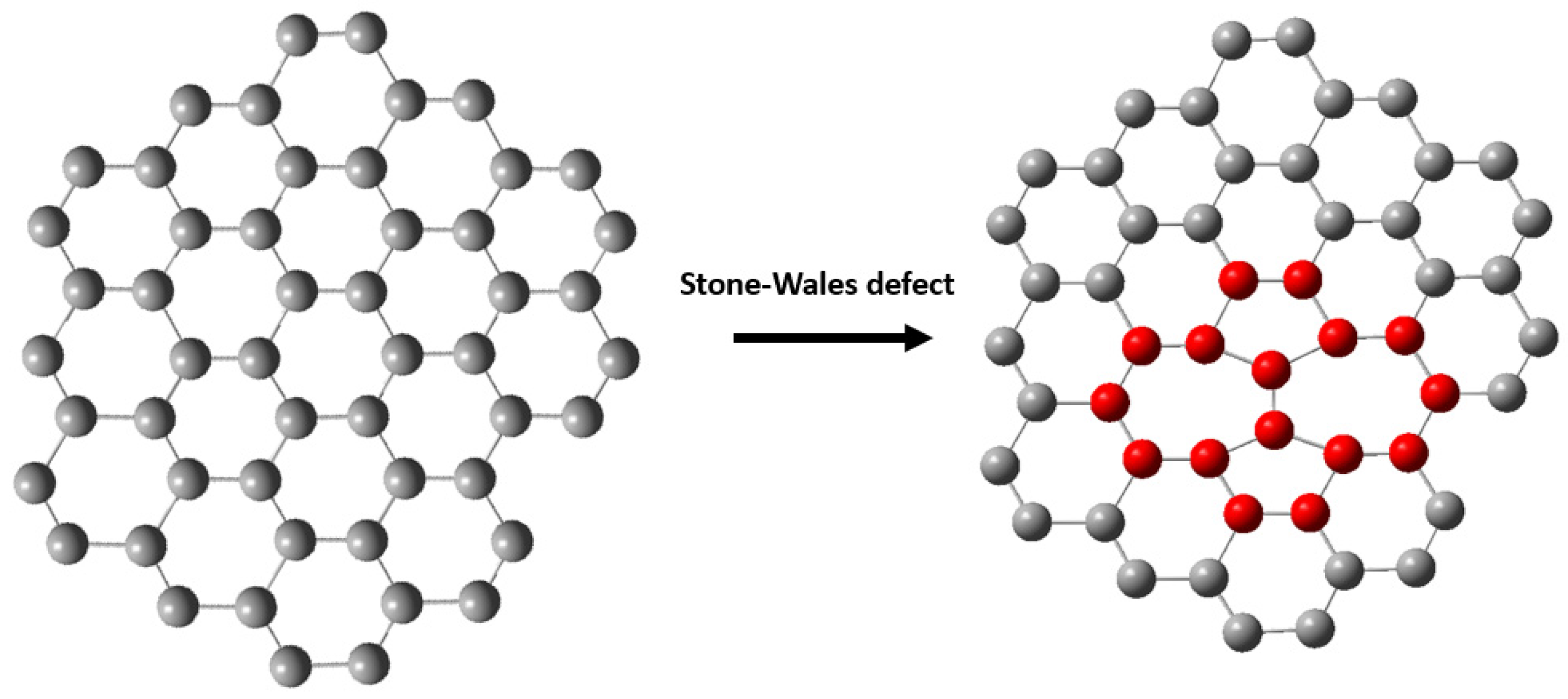
- Fujimori, T.; Radovic, L.R.; Silva-Tapia, A.B.; Endo, M.; Kaneko, K. Structural importance of Stone-Thrower-Wales defects in rolled and flat graphenes from surface-enhanced Raman scattering. Carbon 2012, 50, 3274–3279. [Google Scholar] [CrossRef]
- Shirodkar, S.N.; Waghmare, U.V. Electronic and vibrational signatures of Stone-Wales defects in graphene: First-principles analysis. Phys. Rev. B 2012, 86, 165401. [Google Scholar] [CrossRef]
- Itoh, T.; Yamamoto, Y.S.; Biju, V.; Tamaru, H.; Wakida, S. Fluctuating single sp2 carbon clusters at single hotspots of silver nanoparticle dimers investigated by surface-enhanced resonance Raman scattering. AIP Adv. 2015, 5, 127113. [Google Scholar] [CrossRef]
- Podila, R.; Rao, R.; Tsuchikawa, R.; Ishigami, M.; Rao, A.M. Raman spectroscopy of folded and scrolled graphene. Acs Nano 2012, 6, 5784–5790. [Google Scholar] [CrossRef] [PubMed]
- Larouche, N.; Stansfield, B.L. Classifying nanostructured carbons using graphitic indices derived from Raman spectra. Carbon 2010, 48, 620–629. [Google Scholar] [CrossRef]
- Yehliu, K.; Vander Wal, R.L.; Boehman, A.L. Development of an HRTEM image analysis method to quantify carbon nanostructure. Combust. Flame 2011, 158, 1837–1851. [Google Scholar] [CrossRef]
- Pre, P.; Huchet, G.; Jeulin, D.; Rouzaud, J.N.; Sennour, M.; Thorel, A. A new approach to characterize the nanostructure of activated carbons from mathematical morphology applied to high resolution transmission electron microscopy images. Carbon 2013, 52, 239–258. [Google Scholar] [CrossRef]
- Bourrat, X.; Langlais, F.; Chollon, G.; Vignoles, G.L. Low temperature pyrocarbons: A review. J. Braz. Chem. Soc. 2006, 17, 1090–1095. [Google Scholar] [CrossRef]
- Rouzaud, J.N.; Oberlin, A.; Benybassez, C. Carbon-films—Structure and microtexture (optical and electron-microscopy, raman-spectroscopy). Thin Solid Films 1983, 105, 75–96. [Google Scholar] [CrossRef]
- Brunetto, R.; Pino, T.; Dartois, E.; Cao, A.T.; d’Hendecourt, L.; Strazzulla, G.; Brechignac, P. Comparison of the Raman spectra of ion irradiated soot and collected extraterrestrial carbon. Icarus 2009, 200, 323–337. [Google Scholar] [CrossRef]
- Schmid, J.; Grob, B.; Niessner, R.; Ivleva, N.P. Multiwavelength Raman microspectroscopy for rapid prediction of soot oxidation reactivity. Anal. Chem. 2011, 83, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Ciajolo, A. Effect of the flame environment on soot nanostructure inferred by Raman spectroscopy at different excitation wavelengths. Combust. Flame 2015, 162, 2431–2441. [Google Scholar] [CrossRef]
- Ess, M.N.; Ferry, D.; Kireeva, E.D.; Niessner, R.; Ouf, F.X.; Ivleva, N.P. In situ Raman microspectroscopic analysis of soot samples with different organic carbon content: Structural changes during heating. Carbon 2016, 105, 572–585. [Google Scholar] [CrossRef]
- Bourrat, X.; Fillion, A.; Naslain, R.; Chollon, G.; Brendle, M. Regenerative laminar pyrocarbon. Carbon 2002, 40, 2931–2945. [Google Scholar] [CrossRef]
- Bourrat, X.; Lavenac, J.; Langlais, F.; Naslain, R. The role of pentagons in the growth of laminar pyrocarbon. Carbon 2001, 39, 2376–2380. [Google Scholar] [CrossRef]
- Vallerot, J.M. Matrice de Pyrocarbone: Propriétés, Structure et Anisotropie Optique. Ph.D. Thesis, Université de Bordeaux 1, Bordeaux, France, 2004. (In French). [Google Scholar]
- Prawer, S.; Nemanich, R.J. Raman spectroscopy of diamond and doped diamond. Philos. Trans. R. Soc. 2004, 362, 2537–2565. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Sedghi, S.; Silvestre-Albero, A.; Andersson, G.G.; Sharma, A.; Pendleton, P.; Rodriguez-Reinoso, F.; Kaneko, K.; Biggs, M.J. Raman spectroscopy study of the transformation of the carbonaceous skeleton of a polymer-based nanoporous carbon along the thermal annealing pathway. Carbon 2015, 85, 147–158. [Google Scholar] [CrossRef]
- May, P.; Lazzeri, M.; Venezuela, P.; Herziger, F.; Callsen, G.; Reparaz, J.S.; Hoffmann, A.; Mauri, F.; Maultzsch, J. Signature of the two-dimensional phonon dispersion in graphene probed by double-resonant Raman scattering. Phys. Rev. B 2013, 87, 075402. [Google Scholar] [CrossRef]
- Herziger, F.; Tyborski, C.; Ochedowski, O.; Schleberger, M.; Maultzsch, J. Double-resonant LA phonon scattering in defective graphene and carbon nanotubes. Phys. Rev. B 2014, 90, 45431. [Google Scholar] [CrossRef]
- Chernyak, S.A.; Ivanov, A.S.; Maslakov, K.I.; Egorov, A.V.; Shen, Z.X.; Savilov, S.S.; Lunin, V.V. Oxidation, defunctionalization and catalyst life cycle of carbon nanotubes: A Raman spectroscopy view. Phys. Chem. Chem. Phys. 2017, 19, 2276–2285. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Torres, J.C.; Wirtz, L.; Pichler, T. Raman spectroscopy of graphite intercalation compounds: Charge transfer, strain, and electron-phonon coupling in graphene layers. Phys. Status Solidi B 2014, 251, 2337–2355. [Google Scholar] [CrossRef]
- Allen, M.J.; Tung, V.C.; Kaner, R.B. Honeycomb carbon: A review of graphene. Chem. Rev. 2010, 110, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, M.S.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 2002, 51, 1–186. [Google Scholar] [CrossRef]
- Abdelkader, A.M.; Cooper, A.J.; Dryfe, R.A.W.; Kinloch, I.A. How to get between the sheets: A review of recent works on the electrochemical exfoliation of graphene materials from bulk graphite. Nanoscale 2015, 7, 6944–6956. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Scharff, P.; Risch, K.; Romanus, H.; Muller, R. Synthesis of C60 intercalated graphite. Solid State Commun. 2004, 131, 153–155. [Google Scholar] [CrossRef]
- Zhao, W.J.; Tan, P.H.; Liu, J.; Ferrari, A.C. Intercalation of few-layer graphite flakes with FeCl3: Raman determination of fermi level, layer by layer decoupling, and stability. J. Am. Chem. Soc. 2011, 133, 5941–5946. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Carotenuto, G.; de Nicola, S.; Camerlingo, C.; Ambrogi, V.; Carfagna, C. Synthesis and characterization of highly intercalated graphite bisulfate. Nanoscale Res. Lett. 2017, 12, 167. [Google Scholar] [CrossRef] [PubMed]
- Eklund, P.C.; Falardeau, E.R.; Fischer, J.E. Raman-scattering in low stage compounds of graphite intercalated with AsF5, HNO3 and SbCl5. Solid State Commun. 1979, 32, 631–634. [Google Scholar] [CrossRef]
- Chacon-Torres, J.C.; Wirtz, L.; Pichler, T. Manifestation of charged and strained graphene layers in the Raman response of graphite intercalation compounds. ACS Nano 2013, 7, 9249–9259. [Google Scholar] [CrossRef] [PubMed]
- Solin, S.A. Raman and IR studies of graphite intercalates. Phys. B+C 1980, 99, 443–452. [Google Scholar] [CrossRef]
- Calandra, M.; Mauri, F. Theoretical explanation of superconductivity in C6Ca. Phys. Rev. Lett. 2005, 95, 237002. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R Rep. 2002, 37, 129–281. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, X.; Lin, Y.; Wang, F. A ternary phase diagram for amorphous carbon. Carbon 2015, 94, 202–213. [Google Scholar] [CrossRef]
- Dillon, R.O.; Woollam, J.A.; Katkanant, V. Use of raman-scattering to investigate disorder and crystallite formation in as-deposited and annealed carbon-films. Phys. Rev. B 1984, 29, 3482–3489. [Google Scholar] [CrossRef]
- Peter, S.; Guenther, M.; Gordan, O.; Berg, S.; Zahn, D.R.T.; Seyller, T. Experimental analysis of the thermal annealing of hard a-C:H films. Diam. Relat. Mater. 2014, 45, 43–57. [Google Scholar] [CrossRef]
- Mangolini, F.; Rose, F.; Hilbert, J.; Carpick, R.W. Thermally induced evolution of hydrogenated amorphous carbon. Appl. Phys. Lett. 2013, 103, 161605. [Google Scholar] [CrossRef]
- Rose, F.; Wang, N.; Smith, R.; Xiao, Q.-F.; Inaba, H.; Matsumura, T.; Saito, Y.; Matsumoto, H.; Dai, Q.; Marchon, B.; et al. Complete characterization by Raman spectroscopy of the structural properties of thin hydrogenated diamond-like carbon films exposed to rapid thermal annealing. J. Appl. Phys. 2014, 116, 123516. [Google Scholar] [CrossRef]
- Casiraghi, C. Effect of hydrogen on the UV Raman intensities of diamond-like carbon. Diam. Relat. Mater. 2011, 20, 120–122. [Google Scholar] [CrossRef]
- Wagner, J.; Wild, C.; Koidl, P. Resonance effects in raman-scattering from polycrystalline diamond films. Appl. Phys. Lett. 1991, 59, 779–781. [Google Scholar] [CrossRef]
- Buijnsters, J.G.; Gago, R.; Jimenez, I.; Camero, M.; Agullo-Rueda, F.; Gomez-Aleixandre, C. Hydrogen quantification in hydrogenated amorphous carbon films by infrared, Raman, and X-ray absorption near edge spectroscopies. J. Appl. Phys. 2009, 105, 093510. [Google Scholar] [CrossRef]
- Cui, W.G.; Lai, Q.B.; Zhang, L.; Wang, F.M. Quantitative measurements of sp3 content in DLC films with Raman spectroscopy. Surf. Coat. Technol. 2010, 205, 1995–1999. [Google Scholar] [CrossRef]
- Wu, A.; Cremer, D. Correlation of the vibrational spectra of isotopomers: Theory and application. J. Phys. Chem. A 2003, 107, 10272–10279. [Google Scholar] [CrossRef]
- Mallet-Ladeira, P.; Puech, P.; Toulouse, C.; Cazayous, M.; Ratel-Ramond, N.; Weisbecker, P.; Vignoles, G.L.; Monthioux, M. A Raman study to obtain crystallite size of carbon materials: A better alternative to the Tuinstra-Koenig law. Carbon 2014, 80, 629–639. [Google Scholar] [CrossRef]
- Martin, C.; Pegourie, B.; Ruffe, R.; Marandet, Y.; Giacometti, G.; Pardanaud, C.; Languille, P.; Panayotis, S.; Tsitrone, E.; Roubin, P. Structural analysis of eroded carbon fiber composite tiles of Tore Supra: Insights on ion transport and erosion parameters. Phys. Scr. 2011, 2011, 01024. [Google Scholar] [CrossRef]
- Ruiz, M.P.; de Villoria, R.G.; Millera, A.; Alzueta, M.U.; Bilbao, R. Influence of the temperature on the properties of the soot formed from C2H2 pyrolysis. Chem. Eng. J. 2007, 127, 1–9. [Google Scholar] [CrossRef]
- Chen, P.W.; Huang, F.L.; Yun, S.R. Optical characterization of nanocarbon phases in detonation soot and shocked graphite. Diam. Relat. Mater. 2006, 15, 1400–1404. [Google Scholar] [CrossRef]
- Beyssac, O.; Goffe, B.; Petitet, J.P.; Froigneux, E.; Moreau, M.; Rouzaud, J.N. On the characterization of disordered and heterogeneous carbonaceous materials by Raman spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 2267–2276. [Google Scholar] [CrossRef]
- Farbos, B.; Weisbecker, P.; Fischer, H.E.; da Costa, J.P.; Lalanne, M.; Chollon, G.; Germain, C.; Vignoles, G.L.; Leyssale, J.M. Nanoscale structure and texture of highly anisotropic pyrocarbons revisited with transmission electron microscopy, image processing, neutron diffraction and atomistic modeling. Carbon 2014, 80, 472–489. [Google Scholar] [CrossRef]
- Shin, Y.Y.; Lozada-Hidalgo, M.; Sambricio, J.L.; Grigorieva, I.V.; Geim, A.K.; Casiraghi, C. Raman spectroscopy of highly pressurized graphene membranes. Appl. Phys. Lett. 2016, 108, 221907. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | sp3 (at.%) | H (at.%) | Eg (eV) | Hardness (GPa) | Density (g/cm3) |
|---|---|---|---|---|---|
| ta-C:H | 70 | 25–30 | 2–2.5 | >20 | 2.4 |
| PLCH | 70 | 40–60 | 2–4 | soft | <1.2 |
| DLCH | 40–60 | 20–40 | 1–2 | <20 | 2.0 |
| GLCH | <30 | <20 | <1 | soft | 1.6 |
| GLCHH | 30 | 30–40 | >1 | soft | 1.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merlen, A.; Buijnsters, J.G.; Pardanaud, C. A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons. Coatings 2017, 7, 153. https://doi.org/10.3390/coatings7100153
Merlen A, Buijnsters JG, Pardanaud C. A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons. Coatings. 2017; 7(10):153. https://doi.org/10.3390/coatings7100153
Chicago/Turabian StyleMerlen, Alexandre, Josephus Gerardus Buijnsters, and Cedric Pardanaud. 2017. "A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons" Coatings 7, no. 10: 153. https://doi.org/10.3390/coatings7100153
APA StyleMerlen, A., Buijnsters, J. G., & Pardanaud, C. (2017). A Guide to and Review of the Use of Multiwavelength Raman Spectroscopy for Characterizing Defective Aromatic Carbon Solids: from Graphene to Amorphous Carbons. Coatings, 7(10), 153. https://doi.org/10.3390/coatings7100153