Abstract
A modular strategy for the molecular design of silicone-based antifoaming agents was developed by precisely controlling the architecture of poly (methylhydrosiloxane) (PMHS). Sixteen PMHS variants were synthesized by systematically varying the siloxane chain length (L1–L4), backbone composition (D3T1 vs. D30T1), and terminal group chemistry (H- vs. M-type). These structural modifications resulted in a broad range of Si-H functionalities, which were quantitatively analyzed and correlated with defoaming performance. The PMHS matrices were integrated with high-viscosity PDMS, a nonionic surfactant, and covalently grafted fumed silica—which was chemically matched to each PMHS backbone—to construct formulation-specific defoaming systems with enhanced interfacial compatibility and colloidal stability. Comprehensive physicochemical characterization via FT-IR, 1H NMR, GPC, TGA, and surface tension analysis revealed a nonmonotonic relationship between Si-H content and defoaming efficiency. Formulations containing 0.1–0.3 wt% Si-H achieved peak performance, with suppression efficiencies up to 96.6% and surface tensions as low as 18.9 mN/m. Deviations from this optimal range impaired performance due to interfacial over-reactivity or reduced mobility. Furthermore, thermal stability and molecular weight distribution were found to be governed by repeat unit architecture and terminal group selection. Compared with conventional EO/PO-modified commercial defoamers, the PMHS-based systems exhibited markedly improved suppression durability and formulation stability in high-viscosity environments. These results establish a predictive structure–property framework for tailoring antifoaming agents and highlight PMHS-based formulations as advanced foam suppressors with improved functionality. This study provides actionable design criteria for high-performance silicone materials with strong potential for application in thermally and mechanically demanding environments such as coating, bioprocessing, and polymer manufacturing.
1. Introduction
Foaming presents a persistent challenge across a range of industrial operations such as polymerization, distillation, fermentation, and the processing of viscous liquids. Excessive foam generation compromises heat and mass transfer, reduces operational throughput, and increases maintenance demands.
To mitigate these issues, organomodified siloxane-based antifoaming agents—particularly those incorporating ethylene oxide/propylene oxide (EO/PO) segments—are widely employed due to their high interfacial activity and favorable dispersion characteristics under low-viscosity or ambient conditions [1,2,3,4]. However, their effectiveness is diminished in high-temperature and high-viscosity environments. The EO/PO segments are susceptible to dehydration and restricted mobility, resulting in reduced performance [5,6,7]. Furthermore, their synthesis requires multistep grafting of EO/PO chains onto PDMS backbones, limiting both structural tunability and scalability [6].
In advanced applications such as precision polymerization, high-viscosity coatings, and continuous fermentation, long-term foam suppression stability under dynamic thermal and rheological stresses is essential [8,9,10]. However, the rigid architecture of conventional agents constrains adaptability, resulting in less predictable performance. While empirical studies have explored the influence of EO/PO block length and chain conformation, no established framework quantitatively links structural factors—such as Si-H content, end-group identity, or chain architecture—to functional performance [6,11,12].
Poly (methylhydrosiloxane) (PMHS) offers a structurally versatile alternative, featuring adjustable Si-H group densities and chain lengths that enable predictable interfacial behavior [13,14,15]. PMHS-based agents exhibit excellent thermal resistance and chemical reactivity, making them promising candidates for challenging conditions [8,16,17,18]. Nonetheless, comprehensive studies that correlate Si-H content with antifoaming efficiency or interfacial tension remain limited. Additionally, a multivariable design framework integrating backbone structure, terminal group effects, and filler compatibility has yet to be fully developed.
Fumed silica is frequently introduced to enhance formulation stability, but conventional surface treatments—such as random silanization or basic hydrophobization—often lack thermal durability and structural consistency [16,17,18,19,20,21], thereby limiting long-term foam suppression efficiency.
To address these limitations, a structure–function-guided formulation strategy was developed based on PMHS as the core siloxane matrix. Sixteen formulations were synthesized, incorporating controlled variations in Si-H content, chain length, terminal group chemistry, high-viscosity PDMS addition, and surface-modified fumed silica. Each was evaluated for surface tension, thermal stability, storage durability, and foam suppression in high-viscosity acrylic systems that simulate demanding industrial conditions.
This study seeks to define quantitative correlations between molecular architecture and defoaming performance. The ultimate goal is to establish a predictive design framework for high-performance antifoaming agents capable of maintaining efficacy under severe thermal and mechanical stress.
In contrast to previous studies that relied on single-variable modifications or commercial additives, this work introduces a multivariable design framework that simultaneously tunes Si-H content, siloxane chain length, terminal group chemistry, and filler compatibility to systematically optimize antifoaming efficiency. To the best of our knowledge, this is the first study to define quantitative structure–performance correlations across such a broad parameter space for PMHS-based defoamers tailored to high-viscosity systems.
2. Experimental Methods
2.1. Synthesis of Hydrogen-Containing Polysiloxane-Based Antifoaming Agent Formulations
A total of sixteen PMHS-based antifoaming agent formulations were synthesized to quantitatively investigate structure–performance relationships. The siloxane backbone composition was adjusted by varying the ratio of non-reactive –Si(CH3)2–O (D) units to reactive –Si(H)(CH3)–O (T) units, resulting in two backbone series: D3T1 and D30T1.
Chain lengths were deliberately targeted at approximately 10, 40, 70, and 100 repeating units (designated L1 through L4) to span a broad molecular weight range—from short oligomers to long-chain polymers—and to enable systematic evaluation of the influence of siloxane chain length on defoaming performance, interfacial behavior, and structural stability. Terminal group structures were defined using tetramethyldisiloxane (TMDSO, H-type, CAS No. 3277-26-7, purity ≥ 97%, bp 71 °C, Macklin, Shanghai, China) or hexamethyldisiloxane (HMDS, M-type, CAS No. 107-46-0, purity ≥ 98%, bp 100 °C, Macklin) as chain stoppers.
Polymerization was conducted under nitrogen in a 500 mL four-neck round-bottom flask using octamethylcyclotetrasiloxane (CAS No. 556-67-2, purity ≥ 99%, bp 175 °C, Macklin), 2,4,6,8-tetramethylcyclotetrasiloxane (CAS No. 2370-88-9, purity ≥ 98%, bp 135 °C, Macklin), and chain stoppers as monomers. The reaction was carried out at 60 °C for 5 h, using 2.0 wt% sulfuric acid (H2SO4, CAS No. 7664-93-9, purity ≥ 98%, bp 330 °C, Tengyu New Materials, Huai’an, China) as the catalyst [22,23]. Upon completion, the reaction mixture was neutralized using solid sodium carbonate (CAS No. 497-19-8, purity ≥ 99.5%, mp 854 °C, Sinopharm, Beijing, China), and the pH was adjusted. Volatile components were removed through vacuum evaporation, yielding transparent liquid PMHS samples.
Each formulation was synthesized with controlled variations in backbone architecture and terminal group identity to create a foundational library for subsequent performance analysis.
2.2. Surface Treatment of Fumed Silica
To enhance the structural stability of the antifoaming agent formulations, surface treatment of fumed silica was conducted using PMHS. HDK H20 fumed silica (average particle size: 40 nm, Wacker Chemie AG, Suzhou, China) was pre-dried in an electric convection oven (DHG-9245A, Shanghai Yiheng Technology Instrument Co., Ltd., Shanghai, China) at 120 °C for 2 h prior to surface treatment. Approximately 20–30 g of fumed silica was used per drying batch. The drying temperature and time were selected based on established dry silanization protocols to ensure sufficient removal of physically adsorbed moisture while maintaining the integrity of the silica surface [14,24,25].
The dried fumed silica was combined with PMHS at a weight ratio of 10:1 and subjected to high-temperature dispersion. Dispersion was carried out in a 250 mL reactor under mechanical stirring at 170 °C for 7 h. Each PMHS sample used in the treatment was synthesized according to the structural variations described in Section 2.1.
Following dispersion, the reaction mixtures were cooled to ambient temperature and washed repeatedly with ethanol. Centrifugation was applied to remove unbound silicone oil, and the washed samples were dried at 80 °C to yield the final PMHS–fumed silica composites.
These treated powders were subsequently incorporated into antifoaming formulations to ensure structure-specific interfacial interaction.
2.3. Formulation and Preparation of Hydrogen-Containing Polysiloxane-Based Antifoaming Agents
Formulations of hydrogen-containing antifoaming agents were prepared by combining PMHS—containing 2 wt% of fumed silica that had been surface-modified using the identical PMHS structure—with high-viscosity polydimethylsiloxane (PDMS, viscosity ≈ 10,000 cSt, purity ≥ 99.9%, Shenzhen Jipeng Silicon Fluorine Material Co., Ltd., Shenzhen, China) and a nonionic surfactant, SPO (fatty alcohol polyoxypropylene ether, average PO ≈ 30, purity > 99%, Nantong Hai’an Petroleum Chemical Plant, Nantong, China), at a weight ratio of 4:3:3 (PMHS:PDMS:SPO).
Initially, the PMHS–fumed silica composite and PDMS were mixed at a 4:3 weight ratio and stirred at 1000 rpm for 1 h at 60 °C. Subsequently, SPO was added dropwise in a quantity equal to 3 parts by weight relative to the total formulation, and the mixture was stirred for an additional 1 h at the same temperature to ensure complete homogenization.
In this formulation, PMHS provided interfacial reactivity via Si-H functionalities and chemically anchored the fumed silica surface, which enhanced dispersion stability. PDMS contributed to bubble inhibition through viscosity-induced barrier effects, while SPO improved interfacial migration and accelerated foam collapse.
This 4:3:3 composition reflects a functionally optimized strategy aimed at achieving both long-term storage stability and effective foam suppression in high-viscosity environments.
2.4. Characterization Methods
The chemical structures and physicochemical properties of the synthesized PMHS-based antifoaming agents were characterized using the following analytical techniques.
Fourier transform infrared (FT-IR) spectroscopy was performed in attenuated total reflectance (ATR) mode using a Spectrum Two instrument (Model: L1600400, PerkinElmer, Waltham, MA, USA), with a spectral range of 400–4000 cm−1.
Gel permeation chromatography (GPC) was carried out using an Agilent 1260 Infinity system (Agilent Technologies, Santa Clara, CA, USA) equipped with a refractive index detector. A series of three Agilent PLgel columns—5 μm Mixed-C (300 × 7.5 mm), 10 μm Mixed-B (300 × 7.5 mm), and a 5 μm Guard column (50 × 7.5 mm)—were connected in series. Tetrahydrofuran (THF) was used as the mobile phase at a flow rate of 1.0 mL/min and a column temperature of 35 °C. Molecular weights were calibrated using polystyrene (PS) standards. The polydispersity index (PDI) was calculated as the ratio of weight-average molecular weight (Mw) to number-average molecular weight (Mn).
1H nuclear magnetic resonance (1H NMR) spectroscopy was conducted on a Bruker 600 MHz high-resolution NMR spectrometer (Magnet System 600′54 Ascend LH, Bruker, Karlsruhe, Germany), with deuterated chloroform (CDCl3) as the solvent. Tetramethylsilane (TMS) was not employed as an internal reference.
Thermogravimetric analysis (TGA) was performed using a TA Instruments SDT Q600 analyzer (TA Instruments, New Castle, DE, USA). Samples were loaded in alumina crucibles and heated to 506 °C at a rate of 10 °C/min under both nitrogen (120 mL/min) and ambient air atmospheres. Thermal degradation behavior was assessed based on weight loss up to 500 °C.
Surface tension measurements were obtained at 25 °C using a Q1000 automatic tensiometer (Shanghai Fangrui Instrument Co., Ltd., Shanghai, China), based on the du Noüy ring method.
2.5. Evaluation of Antifoaming Performance
The antifoaming performance of the PMHS-based formulations was evaluated through comparative testing with three reference systems: Reference Sample 1 (RS1), Reference Sample 2 (RS2), and a commercial product (CP). These references were selected based on structural variation or commercial relevance and were included to benchmark performance in terms of foam suppression and storage stability.
Each test was conducted using a high-viscosity acrylic resin system provided by Zhejiang Fenghong New Material Co., Ltd. (Huzhou, China), which was specifically formulated to exhibit a high foaming tendency and serve as a representative industrial environment.
For each sample, a test formulation containing 1 wt% antifoaming agent and a control formulation without any additive were prepared. Both groups were subjected to high-speed stirring at 1000–1500 rpm for 5–10 min to induce foam generation.
Following agitation, the foamed samples were cast onto flat glass plates at a thickness of approximately 1–2 mm and allowed to rest for 2 min to stabilize the surface. Surface bubbles were counted using a magnifying glass or a low-magnification optical microscope (Model: SZ61, Olympus, Tokyo, Japan). All tests were conducted in triplicate under identical conditions, and average values were recorded.
Foam suppression efficiency was quantified based on the reduction in surface bubble count before and after additive introduction. This approach reflects the practical requirements of high-viscosity oil-based coating systems and provides a robust metric for evaluating the performance of silicone-based antifoaming agents under simulated industrial conditions.
Foam suppression efficiency (%) was calculated using the following equation:
where is the surface bubble count in the control formulation (without antifoaming agents), and is the surface bubble count in the test formulation containing the antifoaming agent. All bubble counts were obtained via optical observation 2 min after coating stabilization, and the values represent the average of three independent replicates.
2.6. Evaluation of Storage Stability
The storage stability of the antifoaming formulations was assessed through visual observation of phase separation over a period of 3 months at ambient temperature.
No external controls were applied to temperature or humidity during storage, and the samples were sealed and stored under conditions resembling practical storage environments. The stability of each formulation was categorized into three levels based on visual assessment. High stability referred to samples that exhibited no visible phase separation for more than three months, maintaining homogeneity throughout the storage period. Moderate stability indicated no phase separation during the initial three months, followed by a gradual onset of separation thereafter. Low stability was defined as the occurrence of clear and observable phase separation within the three-month storage duration.
Based on these criteria, a total of 19 formulations—including 16 synthesized PMHS-based antifoamers and 3 reference samples—were evaluated for storage stability.
3. Results and Discussion
3.1. Synthesis and Structural Characterization of PMHS-Based Antifoaming Agents
To elucidate the molecular architecture and terminal functionalities of the synthesized PMHS-based antifoaming agents, FT-IR spectroscopy was performed on four representative L2-series formulations. These formulations were systematically designed to vary in siloxane backbone structure—either D3T1 or D30T1—and terminal group identity (H-type or M-type), as illustrated in Figure 1a. This design enabled correlation between molecular structure and antifoaming performance. Characteristic absorption bands corresponding to Si–O–Si, Si–CH3, and Si-H were observed across all samples, confirming the molecular framework [26,27,28,29]. The intensity of Si-H-associated bands at 2156 cm−1 (stretching) and 912 cm−1 (bending) was found to increase proportionally with the SiH(CH3)–O unit content, particularly in the D3T1 series, indicating successful modulation of backbone reactivity through compositional adjustment [27]. Notably, no discernible spectral distinctions were observed between the different backbone types or terminal groups with respect to band positions or overall profiles.
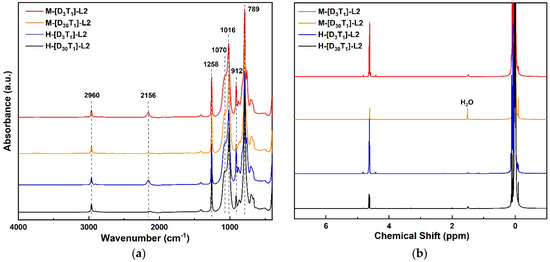
Figure 1.
(a) FT-IR and (b) 1H NMR spectra of four representative L2-series PMHS-based antifoaming agents, selected to illustrate key combinations of siloxane backbone compositions (D3T1 vs. D30T1) and terminal groups (M vs. H). Samples include M-[D3T1]-L2 (red), M-[D30T1]-L2 (yellow), H-[D3T1]-L2 (blue), and H-[D30T1]-L2 (black). All spectra were recorded in CDCl3. These four formulations were selected as representative structures for the structure–performance comparisons discussed throughout this study.
However, one notable exception was observed in the M-[D30T1]-L2 sample. In this case, the characteristic Si-H absorption bands at 2156 cm−1 (stretching) and 912 cm−1 (bending) were both significantly weakened in the FT-IR spectrum, consistent with the extremely low Si-H content (0.04 wt%) in this formulation, as confirmed by quantitative assessments summarized in Table S1 (Supplementary Materials). This reduction is structurally derived from the high D:T ratio (30:1) in the backbone and the absence of reactive Si-H terminals due to the use of HMDS (M-type) end groups.
Complementary insights were obtained through 1H NMR spectroscopy, which was conducted on seven PMHS formulations, including four mentioned above. In the NMR spectra (Figure 1b), a consistent singlet at 0.0–0.3 ppm—corresponding to methyl (Si–CH3) protons—was observed in all samples. In the D3T1-based samples, additional peaks between 4.7 and 5.0 ppm, which were attributable to Si-H protons, further supported the FT-IR observations.
In line with the FT-IR results, the corresponding Si-H resonance in the 1H NMR spectrum (typically observed around 4.7–5.0 ppm) exhibited markedly reduced intensity in the M-[D30T1]-L2 sample compared with other samples. These findings further confirm the compositional control over Si-H density and its spectroscopic detectability in ultra-low-concentration systems.
The integration of CH3/Si-H signals confirmed an increase in methyl content with longer siloxane chains (i.e., extended chain length via repeat units), especially in H-[D3T1]-L series, such as the H-[D3T1]-L2 sample. However, the signal intensity appeared to plateau, possibly due to rotational averaging or relaxation saturation effects.
When comparing samples with identical D3T1 backbones but differing terminal groups, the CH3 integral for M-[D3T1]-L2 was approximately 10% higher than that of its H-type counterpart (24.80 vs. 22.52), reflecting the presence of six additional methyl groups in the HMDS (M-type) unit. More substantial differences were observed across backbone structures. For instance, the CH3 signal integrals for H-[D30T1]-L2 and M-[D30T1]-L2 were recorded at 220.62 and 97.66, respectively—several times greater than those of the D3T1 series—underscoring the dominant influence of repeat unit composition over terminal group identity.
Collectively, the FT-IR and NMR analyses validated the structural integrity and compositional control of the PMHS variants, establishing a robust foundation for subsequent structure–performance correlation in antifoaming applications.
3.2. Molecular Weight and Thermal Stability Characteristics According to PMHS Structure
The molecular weight characteristics of the synthesized PMHS-based antifoaming agents were evaluated using GPC. Both number-average (Mn) and weight-average molecular weights (Mw) increased with the number of dimethylsiloxane repeat units, as summarized in Table S2. For instance, the Mw of H-[D3T1]-L4 reached 44,335 g/mol—over tenfold higher than that of L1 (4649 g/mol) within the same backbone series—indicating that chain extension was facilitated by the higher incorporation of reactive –SiH(CH3)O– units.
Polydispersity index (PDI) analysis revealed that terminal group structure exerted a greater influence on molecular weight distribution than chain length. Within the D30T1-L4 series, the M-terminated variant (M-[D30T1]-L4) exhibited a narrower PDI (1.67) compared with its H-type counterpart (1.90). This suggests that the TMDSO-based H-terminal, which contained Si-H groups, may have induced side reactions such as uncontrolled chain extension or condensation. In contrast, the inert HMDS-based M-terminals provided more uniform termination, thereby reducing molecular weight dispersion.
Thermal stability was assessed via TGA under oxidative (O2) and inert (N2) conditions, with degradation behavior quantified at Td5, Td10, and Td20 (Figure 2, Table S2). In all cases, higher decomposition temperatures were observed under nitrogen, highlighting the role of oxidative initiation. Under inert (N2) conditions, thermal degradation predominantly proceeds through depolymerization via intramolecular back-biting reactions or random chain scission, requiring higher activation energies and, consequently, higher temperatures to initiate decomposition [14,30]. In contrast, under oxidative conditions (O2), the thermal stability significantly decreases due to radical-induced oxidation of reactive Si-H groups to silanol (Si–OH), facilitating subsequent crosslinking or decomposition at lower temperatures [14,30]. Under oxygen, H-type samples displayed reduced thermal stability attributable to terminal Si-H reactivity, which likely acted as an oxidation trigger. Conversely, under nitrogen, thermal behavior was governed more by chain length and Mn than by terminal chemistry.
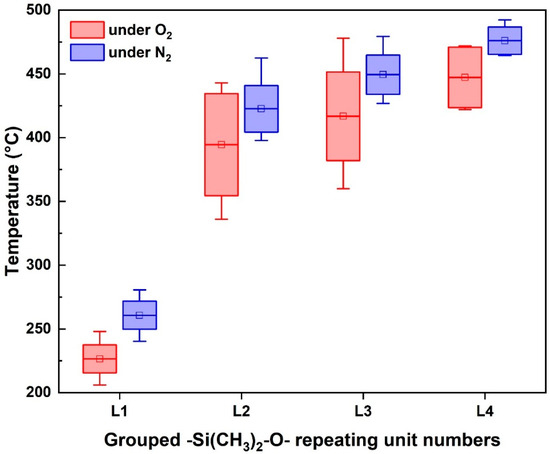
Figure 2.
Thermal degradation behavior of PMHS-based antifoaming agents evaluated under oxidative and inert conditions. Box plots show Td20 (temperature at 20% weight loss) for four series of PMHS samples (L1–L4), grouped by number of –Si(CH3)2–O– repeating units. All measurements were conducted by TGA under oxidative (O2, red) and inert (N2, blue) atmospheres. Each box represents the mean ± standard error (SE), and the whiskers denote the minimum and maximum values. Detailed values are summarized in Supplementary Materials Table S2.
A linear correlation between Mn and Td20 was observed, particularly in the L4 series, where Td20 exceeded 470 °C even in oxidative environments. Most PMHS samples showed Td20 values above 400 °C. However, the onset of degradation was likely influenced by internal Si-H oxidation. A notable separation between Td5 and Td20 under oxygen suggested thermal crosslinking through the Si-H-to-Si-OH transformation, followed by Si–O–Si condensation.
These mechanisms are closely tied to thermal decomposition and long-term material performance. To further elucidate the degradation pathways, follow-up studies employing FT-IR, GC–MS, or in situ TGA–FTIR may be warranted.
These results collectively demonstrate that thermal stability is modulated by multiple structural parameters. Mn, terminal group reactivity, and Si-H distribution interactively influenced the thermal decomposition profile. For example, in high-Si-H systems (D3T1), backbone reactivity dominated, and the H-type variants consistently exhibited superior thermal resistance. In contrast, for the low-Si-H D30T1 systems, terminal Si-H oxidation played a larger role in short-chain samples, with M-type variants showing improved stability under oxidative conditions. As chain length increased, this effect diminished, restoring H-type superiority.
Tables S2 and S3 support these observations. Increased thermal stability with higher repeat units was attributed to not only extended chain length but also to backbone stabilization, reduced terminal reactivity, and constrained chain mobility. These factors collectively enhanced oxidation resistance and thermal robustness. The D3T1–H series, in particular, exhibited a strong Mn–Td20 correlation.
Ultimately, precise control over backbone structure, repeat unit composition, and terminal group identity is essential for the development of thermally stable PMHS antifoaming agents. The insights gained provide design criteria for applications requiring extreme thermal durability, such as high-temperature fermentation, resin processing, and paint dispersion systems.
3.3. Surface Modification of Fumed Silica Using PMHS
To improve compatibility between fumed silica and PMHS, a direct surface modification was performed, replacing commercially treated silica. This approach enabled structure-matched interactions, allowing for accurate correlation with antifoaming performance.
Surface grafting was confirmed via FT-IR spectroscopy (Figure 3a). Before chloroform washing, absorption bands were observed at 2160 cm−1 (Si-H stretching), 2960 cm−1 (Si–CH3 stretching), and 912 cm−1 (Si-H bending). After washing, the Si-H stretching band at 2160 cm−1 disappeared, indicating the removal of physically adsorbed PMHS. According to Zhou et al., the persistence of the 912 cm−1 peak after solvent washing typically indicates covalent bonding between PMHS and the silica surface [31]. In contrast, the near disappearance of this band in our samples suggests that physical adsorption predominated, and most of the PMHS was removed by chloroform washing. In contrast, the band at 2960 cm−1 remained, suggesting that Si–CH3 groups were covalently bonded to the silica surface. The 912 cm−1 band also persisted with reduced intensity, which may be attributed to mono- or di-substituted PMHS or physisorbed residues that remained after washing.
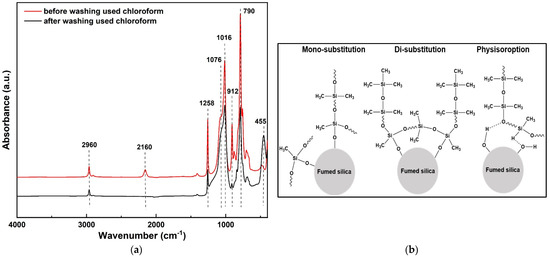
Figure 3.
(a) FT-IR spectra of fumed silica samples modified with PMHS before (red) and after (black) chloroform washing. (b) Proposed bonding modes of PMHS on silica: mono-substitution, di-substitution, and physisorption.
Although a single FT-IR band does not conclusively establish the immobilization mechanism, its persistence suggests the formation of Si–O–Si linkages between PMHS and the silica surface. Potential bonding configurations—including mono-substitution, di-substitution, and hydrogen bonding—are illustrated in Figure 3b.
The FT-IR results confirmed successful covalent grafting of PMHS onto the fumed silica surface. This immobilization is anticipated to play a pivotal role in enhancing dispersion stability, interfacial adsorption, and foam suppression behavior.
Each formulation incorporated fumed silica that was surface-modified using its corresponding PMHS—matched in backbone, chain length, and terminal group—ensuring that the performance reflected intrinsic PMHS–silica interfacial compatibility. This formulation-specific strategy offers higher structural fidelity than the use of a generic filler. The silica framework itself remained unchanged, as confirmed by the invariance of other FT-IR peaks. Given the tunability of the PMHS architecture, this strategy is expected to be applicable to various silica types and industrial systems [32].
3.4. Influence of Si-H Content on AntiFoaming Efficiency and Interfacial Properties
The Si-H content (wt%) was found to be the most decisive structural factor influencing both interfacial behavior and the foam suppression efficiency of the PMHS-based antifoaming agents. Based on 1H NMR quantification (Section 3.1), all formulations were categorized into three groups according to their Si-H content, <0.1%, 0.1%–0.3%, and >0.3%, as summarized in Table S3. Corresponding performance comparisons are illustrated in Figure 4.
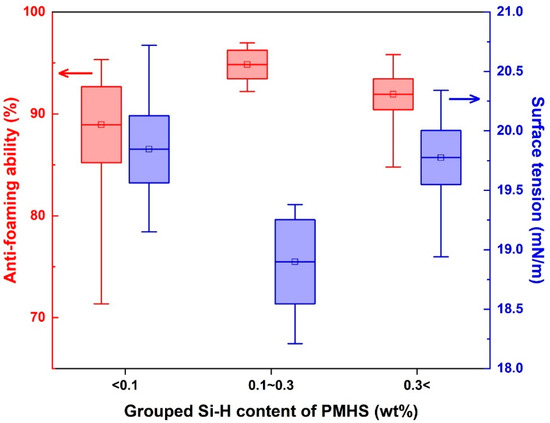
Figure 4.
Box plots comparing the antifoaming ability (red, left axis) and surface tension (blue, right axis) of PMHS-based formulations categorized by Si-H content: <0.1, 0.1–0.3, and >0.3 wt%. The middle group (0.1–0.3 wt%) exhibits superior antifoaming efficiency and lower surface tension with high reproducibility, identifying it as the optimal structural range. Each box represents the mean ± standard error (SE), and the whiskers denote the minimum and maximum values. The data represent 16 synthesized PMHS samples.
This structure–interfacial control–performance relationship is schematically summarized in Figure 5. The diagram highlights how structural parameters—including Si-H content, chain length, PDMS viscosity, and silica surface grafting—collectively regulate interfacial tension, grafting stability, and aggregation. These interfacial factors ultimately dictate foam suppression efficiency and storage stability in PMHS-based formulations.
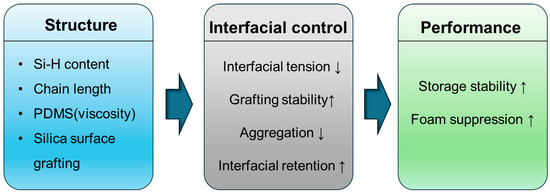
Figure 5.
Schematic representation of structure–interfacial control–performance relationships for PMHS-based antifoaming agents (In Figure 5, ‘PMDS’ was changed to ‘PDMS’).
The formulations with an Si-H content within the 0.1–0.3 wt% range demonstrated the highest defoaming efficiency (>95%) and the lowest surface tension (~18.9 mN/m). H-[D3T1]-L2 (Si-H = 0.26 wt%) yielded optimal performance, achieving 96.6% efficiency and 18.9 mN/m, along with low variability and high reproducibility. In contrast, both lower (<0.1%) and higher (>0.3%) Si-H concentrations resulted in decreased efficiency and greater performance fluctuation.
This nonmonotonic trend indicates that not only the presence but also the precise concentration of Si-H groups is crucial. Insufficient Si-H reduces grafting potential and interfacial reactivity, whereas excess Si-H increases aggregation tendency or restricts interfacial diffusion due to excessive reactivity. Thus, the balance between reactive site density and molecular mobility plays a central role in optimizing performance.
Importantly, the same optimal Si-H range coincided with efficient fumed silica surface modification, as detailed in Section 3.3. Most samples that retained Si–CH3 peaks after chloroform washing—indicative of successful chemical grafting—belonged to this range.
A consistent inverse relationship between surface tension and foam suppression was observed across all samples (Figure 4), reinforcing the accepted mechanism whereby surface-active siloxanes disrupt interfacial film stability. This suggests that surface tension may serve as a practical quality control parameter in industrial defoamer development. Nevertheless, these evaluations were conducted under ambient conditions. Although thermal performance was not directly assessed in this study, reports in the literature indicate that under industrial conditions (T > 120 °C, high shear rates, and elevated viscosity), interfacial viscoelasticity and Si-H group reactivity play vital roles in sustaining antifoaming efficiency [33,34,35,36].
The 0.1–0.3 wt% range was thus experimentally validated as the optimal Si-H concentration for achieving high suppression efficiency, interfacial activity, and stable grafting. Although this study focused on experimental optimization, the identified threshold offers a promising basis for future computational analysis. Approaches such as DFT-based activation energy profiling or MD simulations of interfacial aggregation may elucidate the molecular-level mechanisms underlying Si-H-mediated defoaming.
Overall, the defoaming performance and interfacial behavior of PMHS-based antifoaming agents exhibited a strong correlation with the Si-H content and siloxane chain architecture. In particular, formulations with Si-H content around 0.26 wt% consistently demonstrated the most favorable balance between interfacial activity and foam suppression efficiency. This is in agreement with previous reports indicating that moderate Si-H content enhances surface reactivity and interfacial compatibility while minimizing excessive crosslinking or hydrophobicity-related aggregation tendencies [31,37].
3.5. Comparison of Performance for PMHS-Based Antifoaming Agents
The performance of the developed PMHS-based antifoaming agent was comparatively evaluated against three reference formulations—RS1, RS2, and CP—with respect to foam suppression efficiency and storage stability (Table 1). These reference formulations were designed to reflect systematic variations in key structural features, including the use of PMHS, a fumed silica surface modification strategy, and the inclusion of high-viscosity PDMS.

Table 1.
Antifoaming performance and storage stability of 16 PMHS-based formulations categorized by siloxane backbone composition (D3T1 vs. D30T1), terminal group (Si-H vs. Si–CH3), and chain length (L1–L4). Three reference samples are included: RS1 (PMHS-based but without PDMS), RS2 (PDMS-based without PMHS), and CP (commercially available organomodified silicone-based antifoaming agent). Storage stability is categorized as ◎ (stable over 3 months), ○ (partial phase separation), and ✕ (visible phase separation). All samples were evaluated in high-viscosity acrylic resin systems.
RS1 was prepared by modifying hydrophilic silica with hydrogen-containing PMHS (H-[D30T1]-L4), identical to the formulation employed herein. The treated silica was dispersed in the same PMHS matrix and blended with a nonionic surfactant (SPO) at a 1:1 weight ratio, intentionally excluding high-viscosity PDMS (10,000 cSt) to isolate the contribution of the PMHS-functionalized silica. This formulation achieved a foam suppression efficiency of 92.06% but exhibited phase separation within three months, indicating low storage stability. These observations emphasize the essential role of high-viscosity PDMS in maintaining dispersion and interfacial homogeneity.
RS2 excluded PMHS entirely and employed thermally treated high-viscosity PDMS for silica surface modification. The modified silica was then dispersed in a PDMS matrix and combined with SPO in a 1:1 ratio. This system, lacking defined Si-H functionalities and end-group architecture, relies on weak physical interactions or partial thermal bonding. Consequently, RS2 achieved a lower foam suppression efficiency of 71.36% and demonstrated poor storage stability. The absence of reactive groups limited interfacial activity and hydrophobic retention.
CP, a commercially available organomodified silicone defoamer, was used as a market-standard comparator. Although its precise composition remains proprietary, such formulations typically include EO/PO-modified silicones and hydrophobic fillers. CP demonstrated a suppression efficiency of 87.03% and showed excellent storage stability.
In contrast, the PMHS-based antifoaming agent developed in this study exhibited superior defoaming performance and robust storage stability across all metrics. This outcome was attributed to the synergistic contribution of several structural design factors: optimized Si-H content, tailored chain length, high-viscosity PDMS dispersion stabilization, and chemically grafted fumed silica surfaces. These features collectively enhanced both short-term foam elimination and long-term formulation integrity. The formulation’s resilience under demanding conditions suggests its potential reclassification as a high-performance foam suppressor rather than a conventional antifoaming agent. The comparative analysis confirms that strategic structural integration is critical in engineering advanced silicone-based defoamers capable of sustaining performance under high-viscosity and long-term storage conditions, with potential applicability to systems under a thermal load.
4. Conclusions
A systematic molecular-level design strategy was established for silicone-based antifoaming agents, and its validity was quantitatively demonstrated through structural parameter optimization. Key variables—including repeat unit composition (Mn), Si-H content (wt%), terminal group structure (M- or H-type), the incorporation of high-viscosity PDMS, and backbone-matched fumed silica modification—were precisely controlled to elucidate structure–performance correlations.
Maximum defoaming efficiency (96.6%) and minimum surface tension (18.9 mN/m) were achieved at an Si-H content of 0.26 wt%, confirming this condition as the optimal formulation. The integration of chemically grafted fumed silica and high-viscosity PDMS contributed synergistically to interfacial stability and long-term dispersion retention.
The simultaneous control of repeat unit length and Si-H density marked a significant advancement over conventional single-variable optimization. This multidimensional tuning strategy provided reproducible performance across diverse conditions and enhanced formulation robustness.
Compared with EO/PO-modified commercial products, the developed formulation exhibited superior foam suppression, improved structural tunability, and greater storage stability. These findings highlight its applicability to industrial processes such as fermentation, coating, and lubrication, where mechanical and thermal stresses are critical. By validating a functional group–directed interfacial control mechanism within a polymer–inorganic hybrid system, this study offers not only mechanistic insight but also a practical framework for the development of advanced silicone-based antifoaming agents.
Importantly, among the synthesized formulations, those with Si-H content in the 0.1–0.3 wt% range demonstrated optimal defoaming performance and structural stability, making them promising candidates for large-scale production. These compositions achieved high synthetic reproducibility, excellent thermal durability (Td20 > 400 °C), and long-term storage stability. However, several practical limitations should be considered: the surface modification of fumed silica requires elevated temperatures (≥170 °C), which may pose challenges for continuous manufacturing processes; additionally, the incorporation of high-viscosity PDMS (~10,000 cSt) can lead to increased formulation viscosity, potentially affecting processability. Despite these considerations, the demonstrated robustness under industrially relevant conditions underscores the scalability and applicability of these formulations in high-viscosity systems.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/coatings15080894/s1. Figure S1. Generalized molecular structure of poly(methylhydrosiloxane) (PMHS) used in this study. Table S1. Summary of synthetic yields, 1H NMR integration values, Si-H content, density, and surface tension for 16 synthesized PMHS samples. Table S2. Summary of molecular weight characteristics (GPC) and thermal degradation behavior (TGA) of 16 PMHS-based antifoaming agents under oxidative (O2) and inert (N2) atmospheres. Table S3. Si-H content, density, and surface tension of 16 PMHS-based antifoaming agents grouped by backbone composition (D3T1 or D30T1) and terminal group (H-type or M-type).
Author Contributions
Conceptualization, S.K. and C.L.; methodology, S.K. and H.S.; software, J.H.; validation, S.K., C.L. and X.F.; formal analysis, X.Z.; investigation, M.S.; resources, G.T.; data curation, S.K.; writing—original draft preparation, S.K.; writing—review and editing, C.L., H.B. and G.T.; visualization, J.H.; supervision, H.B. and G.T.; project administration, H.B.; funding acquisition, C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the 2024 Jiangsu Province Industry University Research Cooperation Project (BY20240470), the 2024 Jiangsu Province Science and Technology Deputy General Project (FZ20240998), and Research Projects for Horizontal Cooperation in Universities (11130200125029).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
Authors Soyeon Kim, Junyao Huang, Xiang Feng, Hong Sun, Xiaoli Zhan, Mingkui Shi were employed by the company Zhejiang Fenghong New Material Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Kekevi, B.; Berber, H.; Yıldırım, H. Synthesis and characterization of silicone-based surfactants as anti-foaming agents. J. Surfact. Deterg. 2012, 15, 73–81. [Google Scholar] [CrossRef]
- Owen, M.J.; Dvornic, P.R. (Eds.) Surface Applications of Silicones. In Silicone Surface Science; Springer: Dordrecht, The Netherlands, 2012; pp. 355–374. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Xu, D.; Lin, Y.; Song, W.; Wang, Y.; Xu, F.; Chen, G. Preparation and performance evaluation of CNF-enhanced polyether-modified polysiloxane defoamer. J. Surfact. Deterg. 2025, 28, 363–372. [Google Scholar] [CrossRef]
- Han, S.; Xu, J.; Zheng, G.; Sun, Y. A dissertation on polysiloxane-antifoaming-agents: Antifoaming principles, synthesis and compound. Adv. Mater. Res. 2011, 301–303, 26–30. [Google Scholar] [CrossRef]
- Floyd, D.T. Organo-modified silicone copolymers for cosmetic use. In Cosmetic and Pharmaceutical Applications of Polymers; Gebelein, C.G., Carraher, C.E., Eds.; Springer: New York, NY, USA, 1991; pp. 49–72. [Google Scholar]
- Hill, R.M. (Ed.) Siloxane surfactants. In Silicone Surfactants, 2nd ed.; Routledge: London, UK, 2019; pp. 1–48. ISBN 978-1-138-32908-6. [Google Scholar]
- Shukla, A.K.; Roy, A.K.D.; Chauhan, P.S. Evaluate surfactants as antifoam substitutes in amine systems. Hydrocarb. Process. 2013, 92, 67–72. [Google Scholar]
- Ng, E.L.S.; Lau, K.K.; Partoon, B.; Lim, S.F.; Chin, S.Y. Selection Criteria for Antifoams Used in the Acid Gas Sweetening Process. Ind. Eng. Chem. Res. 2021, 60, 13438–13462. [Google Scholar] [CrossRef]
- Garrett, P.R. Mechanisms of Antifoam Action. In Foam Films and Foams; Gochev, G., Miller, R., Eds.; CRC Press: Boca Raton, FL, USA, 2018; pp. 331–367. [Google Scholar]
- Junker, B. Foam and its mitigation in fermentation systems. Biotechnol. Prog. 2007, 23, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Christiano, S.P. Dispersion Processes. In Silicone Dispersions; Springer: Cham, Switzerland, 2017; pp. 217–238. [Google Scholar] [CrossRef]
- Petroff, L.J.; Snow, S.A. Silicone Surfactants. In Silicone Surface Science; Owen, M.J., Dvornic, P.R., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 243–280. [Google Scholar] [CrossRef]
- Chrusciel, J. Progress in chemistry of polymethylhydrosiloxanes. Polimery 1999, 44, 462–471. [Google Scholar] [CrossRef]
- Chruściel, J.J. Hydrosilyl-Functional Polysiloxanes: Synthesis, Reactions and Applications. In Reactive and Functional Polymers Volume One: Biopolymers, Polyesters, Polyurethanes, Resins and Silicones; Springer: Cham, Switzerland, 2020; pp. 329–414. [Google Scholar] [CrossRef]
- Chruściel, J.J. Modifications of Textile Materials with Functional Silanes, Liquid Silicone Softeners, and Silicone Rubbers—A Review. Polymers 2022, 14, 4382. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, V.; Giermanska-Kahn, C.F.; Langevin, D.; Pouchelon, A. Polydimethylsiloxane (PDMS)-Based Antifoams. Colloids Surf. A Physicochem. Eng. Asp. 1997, 122, 103–120. [Google Scholar] [CrossRef]
- Jha, B.K.; Christiano, S.P.; Shah, D.O. Silicone antifoam performance: Correlation with spreading and surfactant monolayer packing. Langmuir 2000, 16, 9947–9954. [Google Scholar] [CrossRef]
- Owen, M.J. (Ed.) Foam control. In Foam Control in the Process Industries; American Chemical Society: Washington, DC, USA, 2010; pp. 269–283. [Google Scholar] [CrossRef]
- Murray, B.S.; Ettelaie, R. Foam stability: Proteins and nanoparticles. Curr. Opin. Colloid Interface Sci. 2004, 9, 314–320. [Google Scholar] [CrossRef]
- Hunter, T.N.; Pugh, R.J.; Franks, G.V.; Jameson, G.J. The role of particles in stabilising foams and emulsions. Adv. Colloid Interface Sci. 2008, 137, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Delahaije, R.J.B.M.; Wierenga, P.A. Hydrophobicity Enhances the Formation of Protein-Stabilized Foams. Molecules 2022, 27, 2358. [Google Scholar] [CrossRef] [PubMed]
- LeGrow, G.E.; Petroff, L.J. Silicone polyether copolymers: Synthetic methods and chemical compositions. In Silicone Surfactants; Routledge: London, UK, 2019; pp. 49–64. [Google Scholar]
- Fu, K.; Xu, M.; Zhong, R.; Yang, Z.; Zhou, A.A. Preparation, Characterization and Application of Polyether and Long-Chain Alkyl Co-Modified Polydimethylsiloxane. J. Polym. Res. 2019, 26, 261. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Walls, H.J.; Khan, S.A. Rheology of Silica Dispersions in Organic Liquids: New Evidence for Solvation Forces Dictated by Hydrogen Bonding. Langmuir 2000, 16, 7920–7930. [Google Scholar] [CrossRef]
- Ramakrishna, S.N.; Caruso, R.A. Surface Modification of Fumed Silica by Dry Silanization. Langmuir 2020, 36, 10813–10821. [Google Scholar] [CrossRef]
- Kim, S.; Zhu, H.; Demirci, A.; Yamamoto, S.; Miyashia, T.; Mitsuishi, M. Cyclosiloxane polymer bearing dynamic boronic acid: Synthesis and bottom-up nanocoating. Polym. Chem. 2019, 10, 5228–5235. [Google Scholar] [CrossRef]
- Li, Y.F.S.; Zang, C.G.; Zhang, Y.L. Effect of the structure of hydrogen-containing silicone oil on the properties of silicone rubber. Mater. Chem. Phys. 2020, 248, 122734. [Google Scholar] [CrossRef]
- Wang, C.; Sun, C.; Ding, F.; Yi, Y. Study on the synthesis of fluoroalkyl and polyether co-modified polysiloxane and appraisal of its foam-breaking and -inhibiting performance. J. Chin. Chem. Soc. 2017, 64, 674–682. [Google Scholar] [CrossRef]
- Lin, W.; Sun, Y.; Zheng, J.; Zheng, Y.; Yan, L.; Jiang, B.; Yang, W.; Chen, H.; Zhang, X. Surface modification of sol-gel silica antireflective coatings by F-PMHS: A simple method for improvement of amphiphobicity. Coatings 2018, 8, 57. [Google Scholar] [CrossRef]
- Tian, M.; Zhao, C.; Jiang, Y.; Yu, J.; Wang, L. Kinetic study of thermal degradation of polydimethylsiloxane: The effect of molecular weight on thermal stability in inert atmosphere. Polym. Degrad. Stab. 2020, 179, 109259. [Google Scholar] [CrossRef]
- Lau, W.W.Y.; Cade, D.; Lu, J.R.; Thomas, R.K. Interactions of hydrophobically modified polymers with surfactant assemblies. Langmuir 1999, 15, 4092–4098. [Google Scholar]
- O’lenick, A.J.; Siltech, L.L.C.; Dacula, G. Basic silicone chemistry–A review. Silicone Spect. 2009, 1. [Google Scholar]
- Liu, J.; Liu, P.; Du, J.; Wang, Q.; Chen, X.; Zhao, L. Review on High-Temperature-Resistant Viscoelastic Surfactant Fracturing Fluids: State-of-the-Art and Perspectives. Energy Fuels 2023, 37, 9790–9821. [Google Scholar] [CrossRef]
- Cun, M.; Mao, J.; Sun, H.; Wei, G.; Tang, F.; Zhang, W.; Tian, J.; Yang, X.; Lin, C. Development of a novel temperature-resistant and salt-resistant double-cationic surfactant with “super thick hydration layer” for clean fracturing fluid. Colloids Surf. A Physicochem. Eng. Asp. 2021, 617, 126389. [Google Scholar] [CrossRef]
- Raghavan, S.R.; Kaler, E.W. Highly viscoelastic wormlike micellar solutions formed by cationic surfactants with long unsaturated tails. Langmuir 2001, 17, 300–306. [Google Scholar] [CrossRef]
- Huang, Z.; Mao, J.; Cun, M.; Yang, X.; Lin, C.; Zhang, Y. Polyhydroxy cationic viscoelastic surfactant for clean fracturing fluids: Study on the salt tolerance and the effect of salt on the high temperature stability of wormlike micelles. J. Mol. Liq. 2022, 366, 120301. [Google Scholar] [CrossRef]
- Gao, X.; Huang, H.; Zhang, X.; Zhang, W.; Zhu, Y.; Wang, B. Preparation and performance evaluation of CNF-enhanced polyether-modified silicone antifoam emulsions. J. Surfactants Deterg. 2024, 27, 329–339. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).