
Comparing Operational Approaches (Spectrophotometric, Electroanalytic and Chromatographic) to Quantify the Concentration of Emerging Contaminants: The Limit of Detection, the Uncertainty of Measurement, Applicability and Open Problems

 ,
,  ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Analytical Methods
2.2. Electro-Oxidation of ECs
2.3. Environmental Water Samples
3. Results
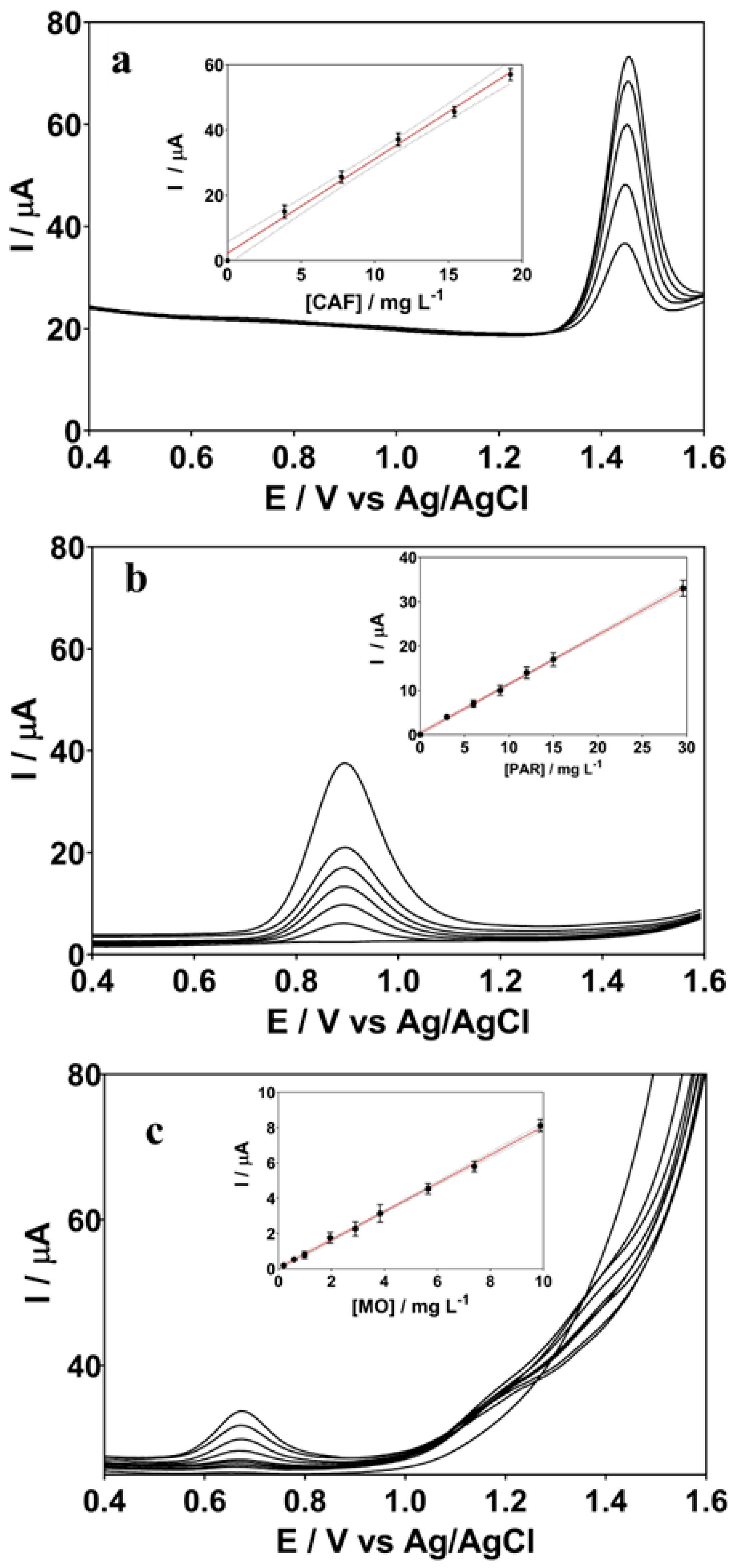
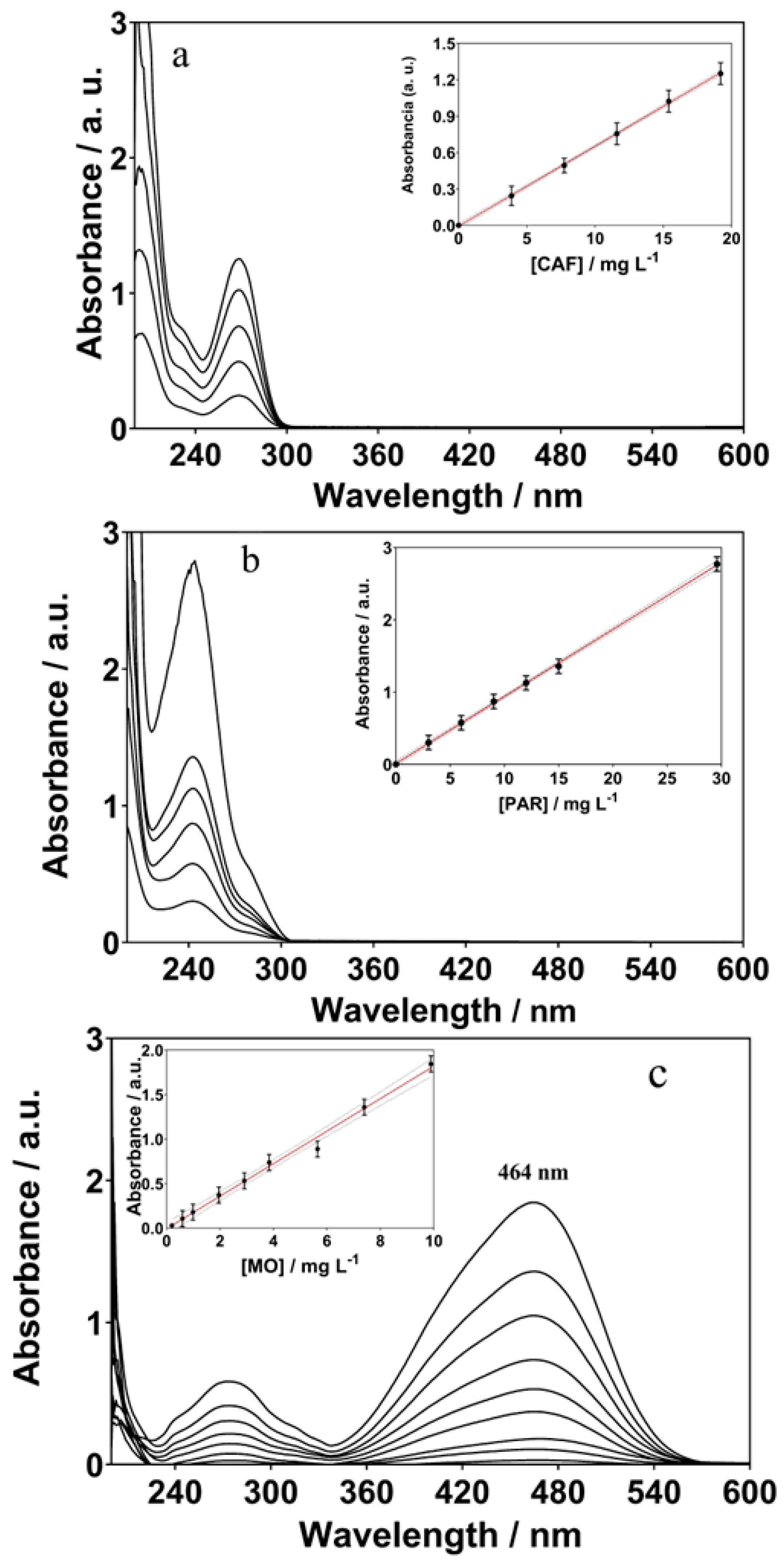
3.1. Electroanalysis of ECs by BDD Sensor and Comparison with Conventional Methods
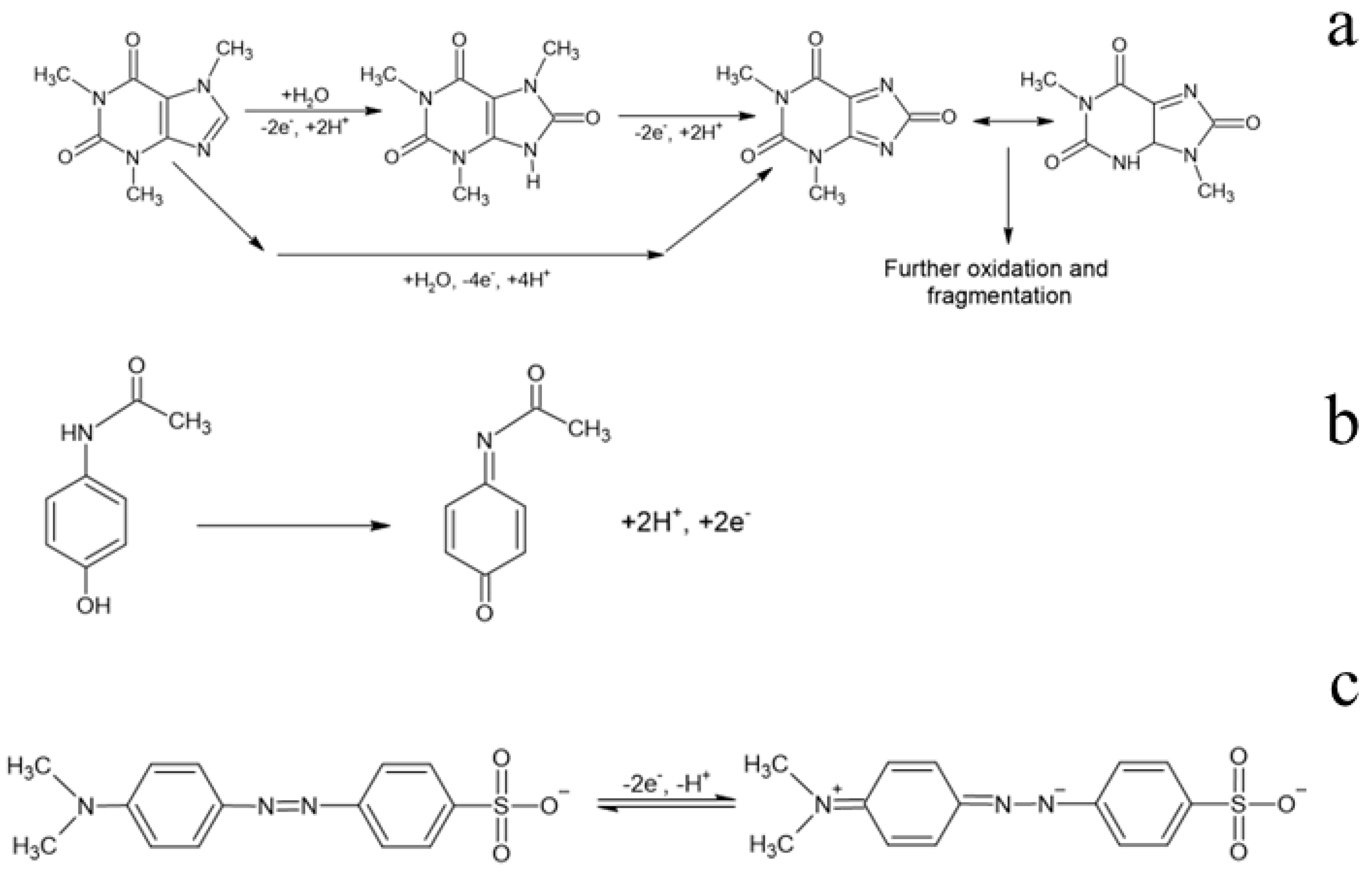
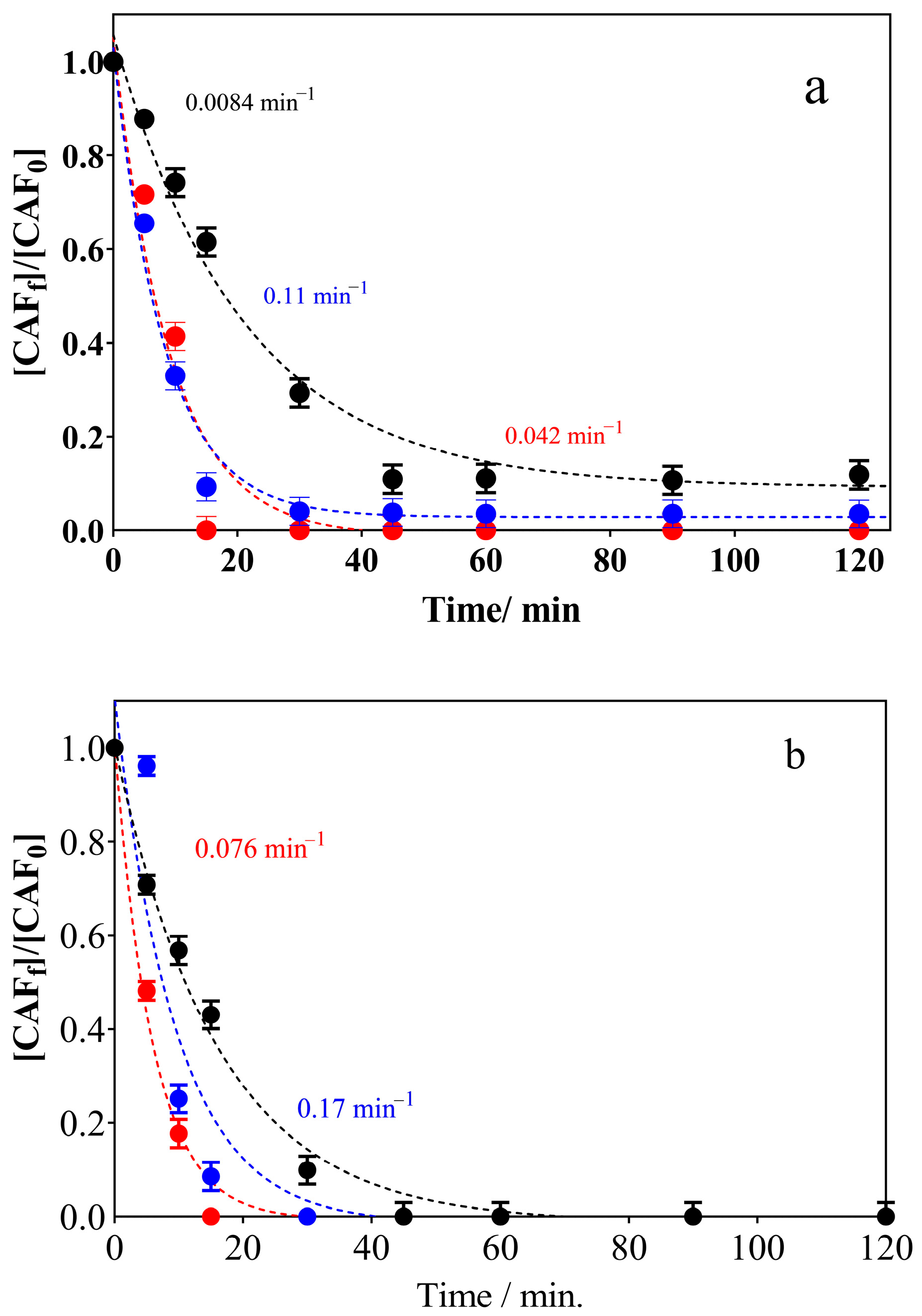
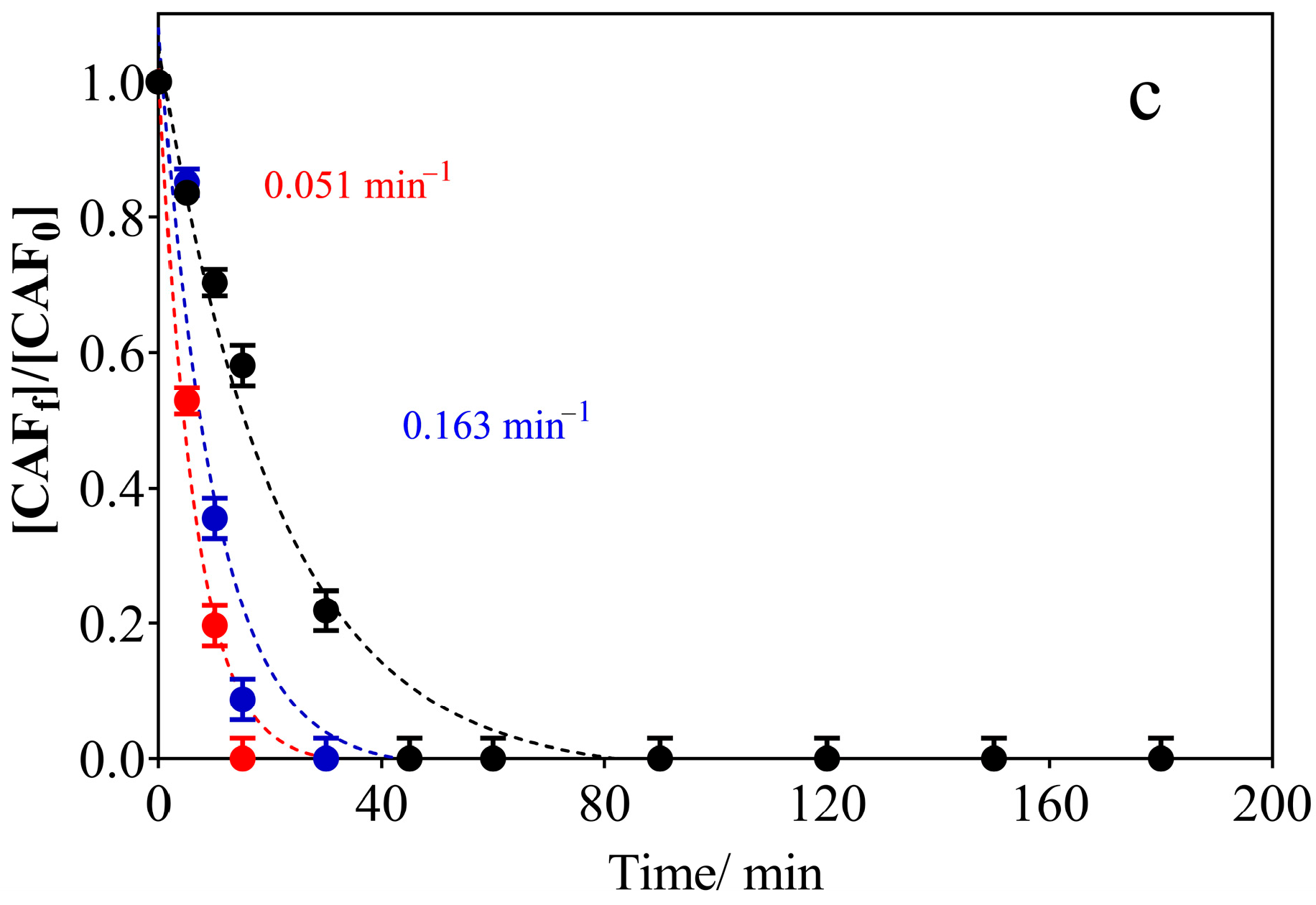
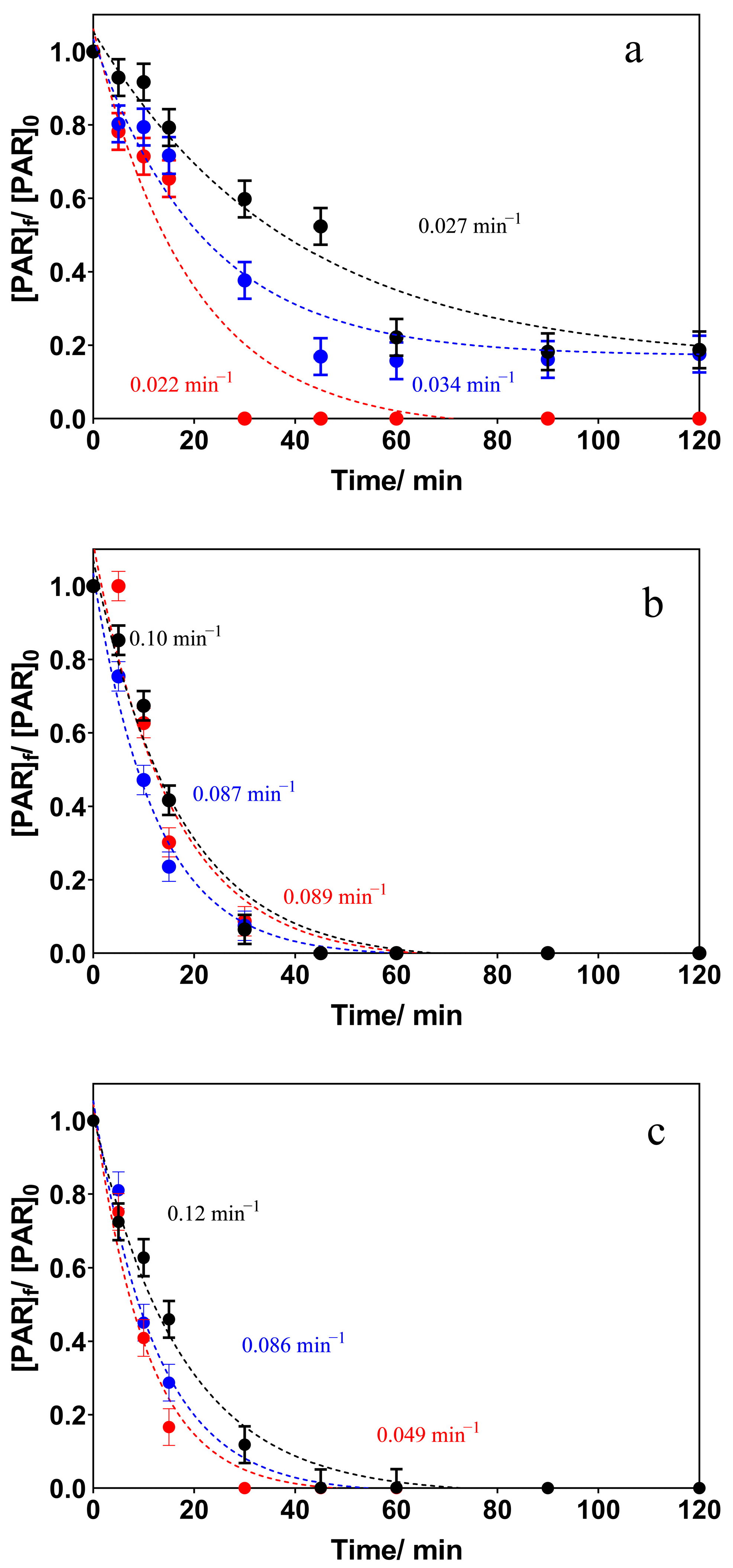
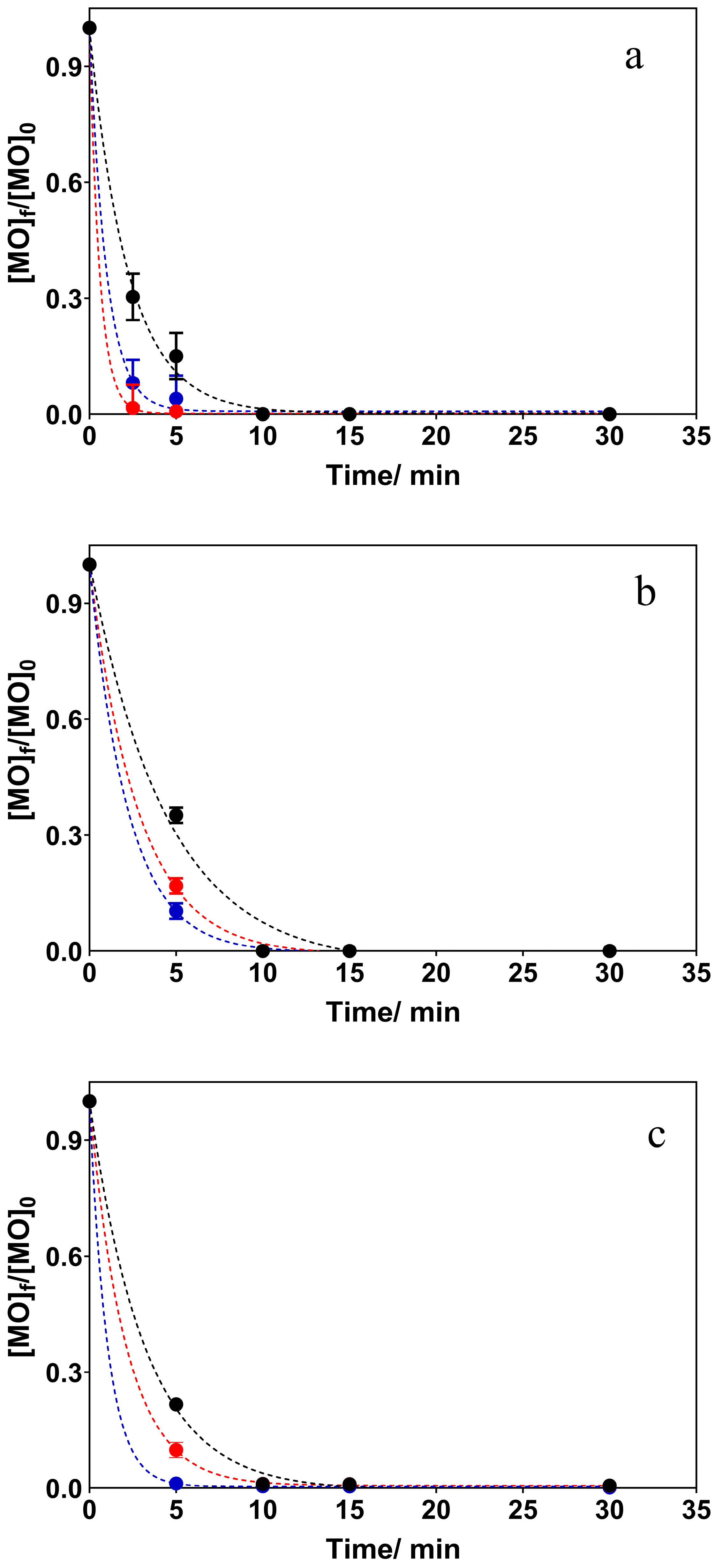
3.2. Electrochemical Oxidation Experiments
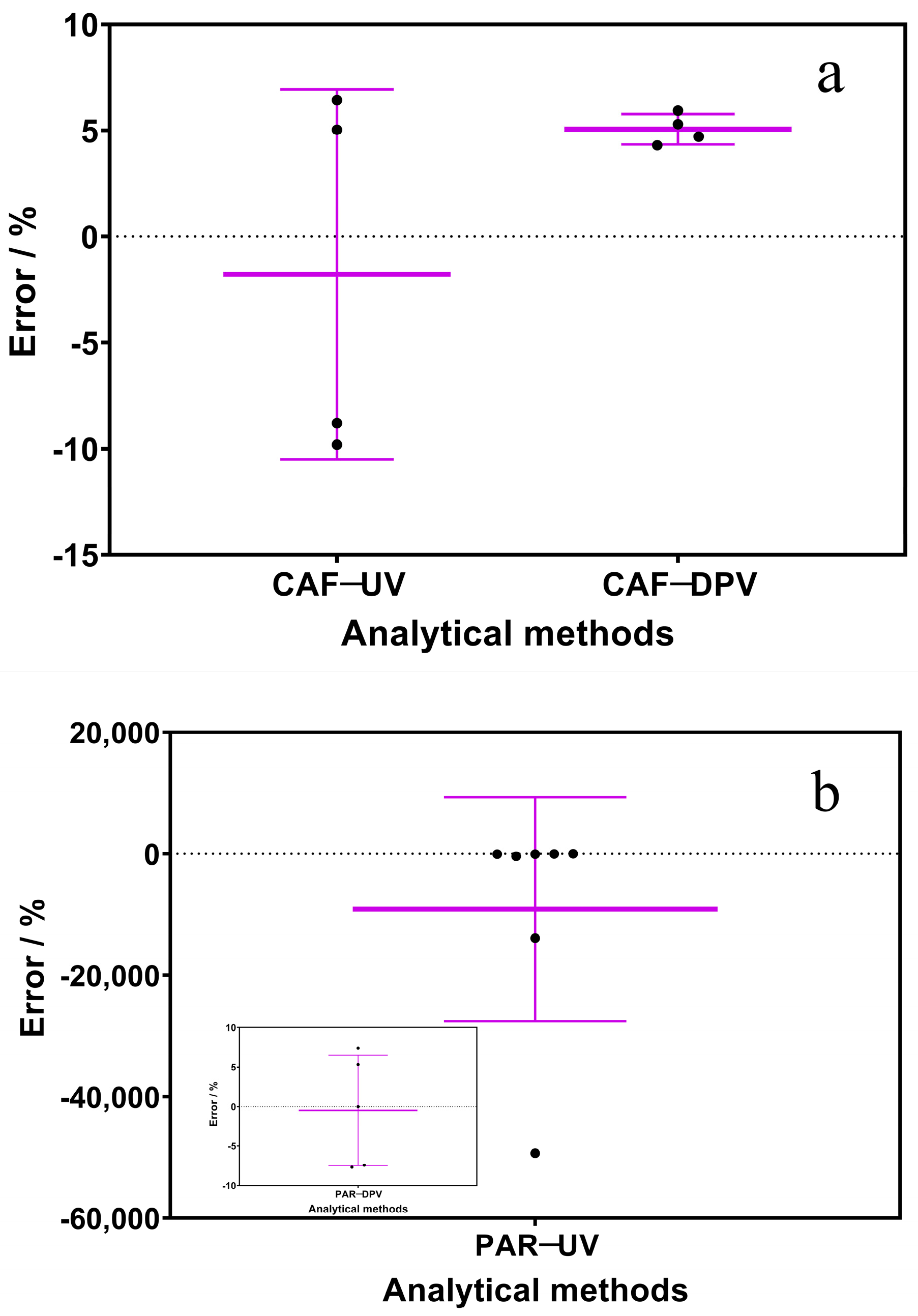
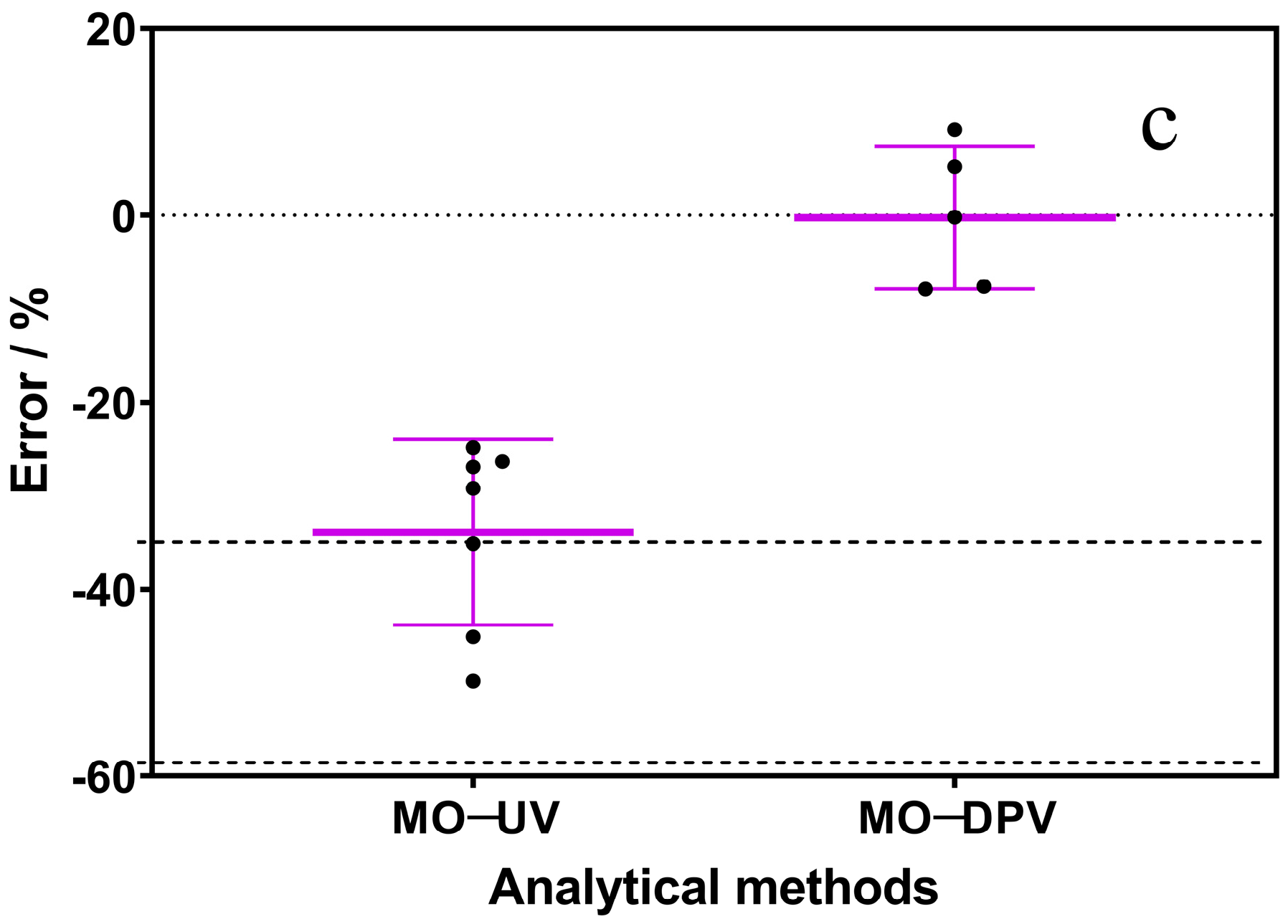
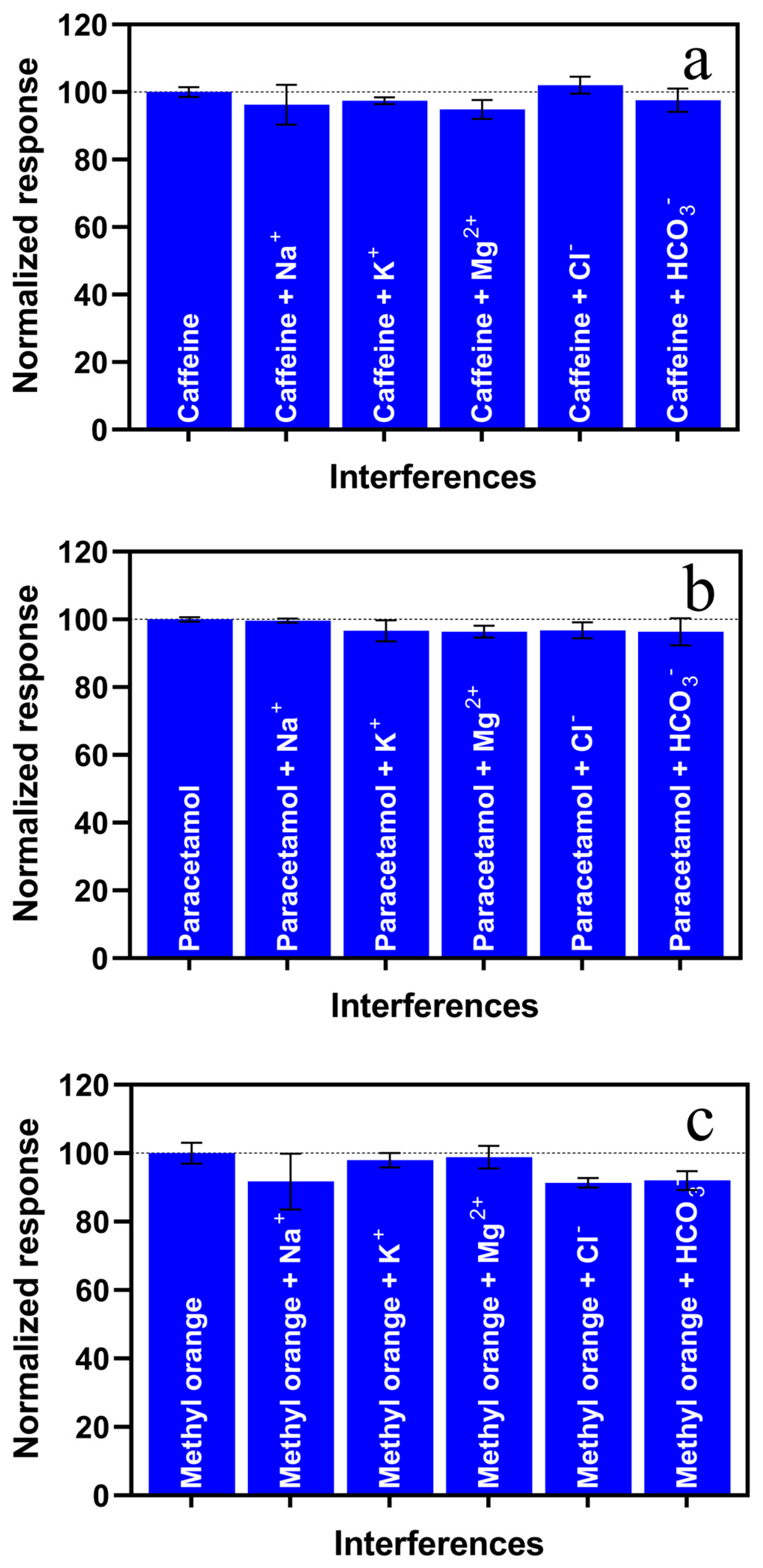
3.3. Interferences and Environmental Water Sample Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sundararaman, S.; Aravind Kumar, J.; Deivasigamani, P.; Devarajan, Y. Emerging Pharma Residue Contaminants: Occurrence, Monitoring, Risk and Fate Assessment—A Challenge to Water Resource Management. Sci. Total Environ. 2022, 825, 153897. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, L.; Li, Y.; Abuduwaili, J. Exploration of the Driving Factors and Distribution of Fecal Coliform in Rivers under a Traditional Agro-Pastoral Economy in Kyrgyzstan, Central Asia. Chemosphere 2022, 286, 131700. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.D.; Kimura, S.Y. Water Analysis: Emerging Contaminants and Current Issues. Anal. Chem. 2016, 88, 546–582. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, T.; Zhao, W.; Huang, J.; Wang, B.; Blaney, L.; Bu, Q.; Yu, G. Corrigendum to “Emerging Organic Contaminants in Chinese Surface Water: Identification of Priority Pollutants” [Engineering 11 (2022) 111–125]. Engineering 2022, 31, 139–140. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, J.H.; Lu, H.T.; Zhang, Q. Electrochemical Behavior and Voltammetric Determination of Paracetamol on Nafion/TiO2-Graphene Modified Glassy Carbon Electrode. Colloids Surf. B Biointerfaces 2011, 85, 289–292. [Google Scholar] [CrossRef]
- El Bouabi, Y.; Farahi, A.; Labjar, N.; El Hajjaji, S.; Bakasse, M.; El Mhammedi, M.A. Square Wave Voltammetric Determination of Paracetamol at Chitosan Modified Carbon Paste Electrode: Application in Natural Water Samples, Commercial Tablets and Human Urines. Mater. Sci. Eng. C 2016, 58, 70–77. [Google Scholar] [CrossRef]
- Tyszczuk-Rotko, K.; Bȩczkowska, I.; Wójciak-Kosior, M.; Sowa, I. Simultaneous Voltammetric Determination of Paracetamol and Ascorbic Acid Using a Boron-Doped Diamond Electrode Modified with Nafion and Lead Films. Talanta 2014, 129, 384–391. [Google Scholar] [CrossRef]
- Mallah, M.A.; Sherazi, S.T.H.; Bhanger, M.I.; Mahesar, S.A.; Bajeer, M.A. A Rapid Fourier-Transform Infrared (FTIR) Spectroscopic Method for Direct Quantification of Paracetamol Content in Solid Pharmaceutical Formulations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 141, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Tefera, M.; Geto, A.; Tessema, M.; Admassie, S. Simultaneous Determination of Caffeine and Paracetamol by Square Wave Voltammetry at Poly(4-Amino-3-Hydroxynaphthalene Sulfonic Acid)-Modified Glassy Carbon Electrode. Food Chem. 2016, 210, 156–162. [Google Scholar] [CrossRef]
- Khaskheli, A.R.; Fischer, J.; Barek, J.; Vyskočil, V.; Sirajuddin; Bhanger, M.I. Differential Pulse Voltammetric Determination of Paracetamol in Tablet and Urine Samples at a Micro-Crystalline Natural Graphite-Polystyrene Composite Film Modified Electrode. Electrochim. Acta 2013, 101, 238–242. [Google Scholar] [CrossRef]
- Li, L.; Zou, D.; Xiao, Z.; Zeng, X.; Zhang, L.; Jiang, L.; Wang, A.; Ge, D.; Zhang, G.; Liu, F. Biochar as a Sorbent for Emerging Contaminants Enables Improvements in Waste Management and Sustainable Resource Use. J. Clean. Prod. 2019, 210, 1324–1342. [Google Scholar] [CrossRef]
- Vieira, L.R.; Soares, A.M.V.M.; Freitas, R. Caffeine as a Contaminant of Concern: A Review on Concentrations and Impacts in Marine Coastal Systems. Chemosphere 2022, 286, 131675. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez del Rey, Z.; Granek, E.F.; Sylvester, S. Occurrence and Concentration of Caffeine in Oregon Coastal Waters. Mar. Pollut. Bull. 2012, 64, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Svorc, L. Determination of Caffeine: A Comprehensive Review on Electrochemical Methods. Int. J. Electrochem. Sci. 2013, 8, 5755–5773. [Google Scholar] [CrossRef]
- Indermuhle, C.; Martín de Vidales, M.J.; Sáez, C.; Robles, J.; Cañizares, P.; García-Reyes, J.F.; Molina-Díaz, A.; Comninellis, C.; Rodrigo, M.A. Degradation of Caffeine by Conductive Diamond Electrochemical Oxidation. Chemosphere 2013, 93, 1720–1725. [Google Scholar] [CrossRef]
- Pham, V.L.; Kim, D.G.; Ko, S.O. Advanced Oxidative Degradation of Acetaminophen by Carbon Catalysts: Radical vs Non-Radical Pathways. Environ. Res. 2020, 188, 109767. [Google Scholar] [CrossRef]
- Allaire, M.; Mackay, T.; Zheng, S.; Lall, U. Detecting Community Response to Water Quality Violations Using Bottled Water Sales. Proc. Natl. Acad. Sci. USA 2019, 116, 20917–20922. [Google Scholar] [CrossRef]
- da Costa Soares, I.C.; da Silva, Á.R.L.; de Moura Santos, E.C.M.; dos Santos, E.V.; da Silva, D.R.; Martínez-Huitle, C.A. Understanding the Electrochemical Oxidation of Dyes on Platinum and Boron–Doped Diamond Electrode Surfaces: Experimental and Computational Study. J. Solid State Electrochem. 2020, 24, 3245–3256. [Google Scholar] [CrossRef]
- Luo, Y.; Guo, W.; Ngo, H.H.; Nghiem, L.D.; Hai, F.I.; Zhang, J.; Liang, S.; Wang, X.C. A Review on the Occurrence of Micropollutants in the Aquatic Environment and Their Fate and Removal during Wastewater Treatment. Sci. Total Environ. 2014, 473–474, 619–641. [Google Scholar] [CrossRef]
- Bradley, I.; Straub, A.; Maraccini, P.; Markazi, S.; Nguyen, T.H. Iron Oxide Amended Biosand Filters for Virus Removal. Water Res. 2011, 45, 4501–4510. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Panizza, M. Electrochemical Oxidation of Organic Pollutants for Wastewater Treatment. Curr. Opin. Electrochem. 2018, 11, 62–71. [Google Scholar] [CrossRef]
- Bolong, N.; Ismail, A.F.; Salim, M.R.; Matsuura, T. A Review of the Effects of Emerging Contaminants in Wastewater and Options for Their Removal. Desalination 2009, 239, 229–246. [Google Scholar] [CrossRef]
- Rizzo, L.; Malato, S.; Antakyali, D.; Beretsou, V.G.; Đolić, M.B.; Gernjak, W.; Heath, E.; Ivancev-Tumbas, I.; Karaolia, P.; Lado Ribeiro, A.R.; et al. Consolidated vs New Advanced Treatment Methods for the Removal of Contaminants of Emerging Concern from Urban Wastewater. Sci. Total Environ. 2019, 655, 986–1008. [Google Scholar] [CrossRef] [PubMed]
- Krzeminski, P.; Tomei, M.C.; Karaolia, P.; Langenhoff, A.; Almeida, C.M.R.; Felis, E.; Gritten, F.; Andersen, H.R.; Fernandes, T.; Manaia, C.M.; et al. Performance of Secondary Wastewater Treatment Methods for the Removal of Contaminants of Emerging Concern Implicated in Crop Uptake and Antibiotic Resistance Spread: A Review. Sci. Total Environ. 2019, 648, 1052–1081. [Google Scholar] [CrossRef]
- Silva, T.A.; Zanin, H.; Corat, E.J.; Fatibello-Filho, O. Simultaneous Voltammetric Determination of Paracetamol, Codeine and Caffeine on Diamond-like Carbon Porous Electrodes. Electroanalysis 2017, 29, 907–916. [Google Scholar] [CrossRef]
- de Araújo, D.M.; Paiva, S.D.S.S.M.; Henrique, J.M.M.; Martínez-Huitle, C.A.; Dos Santos, E.V. Green Composite Sensor for Monitoring Hydroxychloroquine in Different Water Matrix. Materials 2021, 14, 4990. [Google Scholar] [CrossRef] [PubMed]
- Jlassi, K.; Oturan, M.A.; Fauzi Ismail, A.; Chehimi, M.M. Clean Water: Next Generation Technologies; Springer: Cham, Switzerland, 2024. [Google Scholar] [CrossRef]
- dos Santos, E.V.; Martínez-Huitle, C.A.; Rodrigo, M.A. The Electro-Refinery in Organics: A New Arising Concept for Valorization of Wastes. Curr. Opin. Electrochem. 2023, 39, 101267. [Google Scholar] [CrossRef]
- de Oliveira Silva, K.N.; Rodrigo, M.A.; dos Santos, E.V. Electrochemical Treatment of Soil-Washing Effluent with Boron-Doped Diamond Electrodes: A Review. Curr. Opin. Solid State Mater. Sci. 2021, 25, 100962. [Google Scholar] [CrossRef]
- Zazou, H.; Oturan, N.; Sönmez Çelebi, M.; Hamdani, M.; Oturan, M.A. Cold Incineration of 1,2-Dichlorobenzene in Aqueous Solution by Electrochemical Advanced Oxidation Using DSA/Carbon Felt, Pt/Carbon Felt and BDD/Carbon Felt Cells. Sep. Purif. Technol. 2019, 208, 184–193. [Google Scholar] [CrossRef]
- Clematis, D.; Panizza, M. Application of Boron-Doped Diamond Electrodes for Electrochemical Oxidation of Real Wastewaters. Curr. Opin. Electrochem. 2021, 30, 100844. [Google Scholar] [CrossRef]
- Lotfi, S.; Veisi, H.; Karmakar, B. A Convenient Strategy for the Electrochemical Evaluation of Acetaminophen and Caffeine in Combined Drugs and Biological Samples over TiO2@polymethyldopa/Pd Nanocomposite Functionalized Glassy Carbon Electrodes. Measurement 2021, 186, 110156. [Google Scholar] [CrossRef]
- Monteiro, M.K.S.; Santos, E.C.M.M.; Silva, D.R.; Martínez-Huitle, C.A.; dos Santos, E.V. Simultaneous Determination of Paracetamol and Caffeine in Pharmaceutical Formulations and Synthetic Urine Using Cork-Modified Graphite Electrodes. J. Solid State Electrochem. 2020, 24, 1789–1800. [Google Scholar] [CrossRef]
- Martin, K.K.; Ouattara, L. Electroanalytical Investigation on Paracetamol on Boron-Doped Diamond Electrode by Voltammetry. Am. J. Anal. Chem. 2019, 10, 562–578. [Google Scholar] [CrossRef]
- Kang, N.; Ji, L.; Zhao, J.; Zhou, X.; Weng, X.; Li, H.; Zhang, X.; Yang, F. Uniform Growth of Fe3O4 Nanocubes on the Single-Walled Carbon Nanotubes as an Electrosensor of Organic Dyes and the Study on Its Catalytic Mechanism. J. Electroanal. Chem. 2019, 833, 70–78. [Google Scholar] [CrossRef]
- Miller, J.N. Basic Statistical Methods for Analytical Chemistry. Part 2. Calibration and Regression Methods. A Review. Analyst 1991, 116, 3–14. [Google Scholar] [CrossRef]
- Desimoni, E.; Brunetti, B. About Estimating the Limit of Detection of Heteroscedastic Analytical Systems. Anal. Chim. Acta 2009, 655, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Whitaker Sena, J.D.; Lowe, P.A.; Lee, S.W. Significant Predictors of Test Anxiety among Students with and without Learning Disabilities. J. Learn. Disabil. 2007, 40, 360–376. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, H. The Impact of COVID-19 on Anxiety in Chinese University Students. Front. Psychol. 2020, 11, 1168. [Google Scholar] [CrossRef]
- Belay, A.; Ture, K.; Redi, M.; Asfaw, A. Measurement of Caffeine in Coffee Beans with UV/Vis Spectrometer. Food Chem. 2008, 108, 310–315. [Google Scholar] [CrossRef]
- Monteiro, M.K.S.; Paiva, S.S.M.; da Silva, D.R.; Vilar, V.J.P.; Martínez-Huitle, C.A.; dos Santos, E.V. Novel Cork-Graphite Electrochemical Sensor for Voltammetric Determination of Caffeine. J. Electroanal. Chem. 2019, 839, 283–289. [Google Scholar] [CrossRef]
- Freitas de Araújo, K.C.; de Oliveira Silva, K.N.; Monteiro, M.K.; Ribeiro Da Silva, D.; Santos, E.V.d.; Quiroz, M.A.; Martínez-Huitle, C.A. Using Persulfate Electrogenerated at Boron-Doped Diamond Electrodes as an Ex -Situ Oxidation Approach: Storage and Service-Life Solution Parameters. J. Electrochem. Soc. 2022, 169, 033506. [Google Scholar] [CrossRef]
- Molina, J.; Cases, F.; Moretto, L.M. Graphene-Based Materials for the Electrochemical Determination of Hazardous Ions. Anal. Chim. Acta 2016, 946, 9–39. [Google Scholar] [CrossRef] [PubMed]
- Panizza, M.; Cerisola, G. Application of Diamond Electrodes to Electrochemical Processes. Electrochim. Acta 2005, 51, 191–199. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Direct and Mediated Anodic Oxidation of Organic Pollutants. Chem. Rev. 2009, 109, 6541–6569. [Google Scholar] [CrossRef]
- Oliveira, K.S.G.C.; dos Santos, E.V.; Loor-Urgilés, L.D.; Shabanloo, A.; Martínez-Huitle, C.A. The World Impact of Boron Doped Diamond Electrodes and Low-Cost Strategies for Novel Production Systems for Sustainable Wastewater Treatment. Curr. Opin. Electrochem. 2025, 50, 101648. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Parameters | DPV | UV-Vis | HPLC |
|---|---|---|---|---|
| CAF | Limit range (mg L−1) | 3.88–19.20 | 3.88–19.20 | 3.88–19.20 |
| Correlation coefficient (r2) | 0.995 | 0.994 | 0.981 | |
| Slope (b) | 2.88 | 0.066 | 1.33 | |
| Intercept (a) | 2.20 | 0.0067 | 1.02 | |
| LOD (mg L−1) | 0.69 | 3.68 | 0.52 | |
| LQ (mg L−1) | 1.69 | 4.39 | 1.40 | |
| PAR | Limit range (mg L−1) | 3.02–29.6 | 3.02–29.6 | 3.02–29.6 |
| Correlation coefficient (r2) | 0.999 | 0.992 | 0.998 | |
| Slope (b) | 1.09 | 0.092 | 2.156 | |
| Intercept (a) | 4.96 | 0.015 | 1.359 | |
| LOD (mg L−1) | 0.84 | 9.82 | 0.95 | |
| LQ (mg L−1) | 1.31 | 11.38 | 2.01 | |
| MO | Limit range (mg L−1) | 0.19–9.89 | 0.19–9.89 | 0.19–9.89 |
| Correlation coefficient (r2) | 0.998 | 0.992 | 0.995 | |
| Slope (b) | 8.04 | 0.182 | 1.593 | |
| Intercept (a) | 2.65 | 0.0063 | 0.0014 | |
| LOD (mg L−1) | 0.46 | 1.39 | 0.68 | |
| LQ (mg L−1) | 1.26 | 4.02 | 2.38 |
| Analyte | DPV | UV-vis | HPLC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Added/mg L−1 | Found/mg L−1 | Recovery/% | Added/mg L−1 | Found/mg L−1 | Recovery/% | Added/mg L−1 | Found/mg L−1 | Recovery/% | ||
| Tap water | CAF | 5.0 | 4.66 ± 0.05 | 93.1 ± 1.1 | 5.0 | 1.44 ± 0.30 | 28.8 ± 21.0 | 5.0 | 5.96 ± 0.01 | 119.2 ± 0.1 |
| PAR | 5.0 | 4.64 ± 0.13 | 92.8 ± 2.8 | 5.0 | 7.88 ± 0.21 | 157.5 ± 2.7 | 5.0 | 5.47 ± 0.01 | 109.4 ± 0.1 | |
| MO | 5.0 | 4.36 ± 0.08 | 87.3 ± 1.9 | 5.0 | 4.26 ± 0.02 | 85.2 ± 0.5 | 5.0 | 5.57 ± 0.28 | 111.4 ± 4.9 | |
| Well water | CAF | 5.0 | 4.85 ± 0.18 | 97.0 ± 3.8 | 5.0 | 6.51 ± 0.04 | 130.3 ± 0.6 | 5.0 | 5.81 ± 0.01 | 116.2 ± 0.11 |
| PAR | 5.0 | 4.34 ± 0.13 | 86.8 ± 2.9 | 5.0 | 2.90 ± 0.01 | 58.0 ± 0.1 | 5.0 | 5.90 ± 0.01 | 118.1 ± 0.1 | |
| MO | 5.0 | 5.41 ± 0.35 | 108.2 ± 6.5 | 5.0 | 4.95 ± 0.01 | 99.0 ± 0.1 | 5.0 | 5.35 ± 0.13 | 107.0 ± 2.5 | |
| Lagoon water | CAF | 5.0 | 5.83 ± 0.27 | 116.5 ± 4.6 | 5.0 | 3.16 ± 0.06 | 63.2 ± 1.9 | 5.0 | 5.66 ± 0.01 | 113.3 ± 0.04 |
| PAR | 5.0 | 4.43 ± 0.15 | 88.6 ± 3.5 | 5.0 | 3.94 ± 0.14 | 78.8 ± 3.6 | 5.0 | 4.82 ± 0.01 | 96.3 ± 0.2 | |
| MO | 5.0 | 5.21 ± 0.10 | 104.3 ± 2.0 | 5.0 | 4.87 ± 0.01 | 97.4 ± 0.07 | 5.0 | 5.46 ± 0.15 | 109.1 ± 2.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco de Oliveira, M.S.; de Queiroz, J.L.A.; Medeiros de Araújo, D.; Monteiro, M.K.S.; Duarte Magalhaes, K.G.; Martínez-Huitle, C.A.; dos Santos, E.V. Comparing Operational Approaches (Spectrophotometric, Electroanalytic and Chromatographic) to Quantify the Concentration of Emerging Contaminants: The Limit of Detection, the Uncertainty of Measurement, Applicability and Open Problems. Coatings 2025, 15, 719. https://doi.org/10.3390/coatings15060719
Franco de Oliveira MS, de Queiroz JLA, Medeiros de Araújo D, Monteiro MKS, Duarte Magalhaes KG, Martínez-Huitle CA, dos Santos EV. Comparing Operational Approaches (Spectrophotometric, Electroanalytic and Chromatographic) to Quantify the Concentration of Emerging Contaminants: The Limit of Detection, the Uncertainty of Measurement, Applicability and Open Problems. Coatings. 2025; 15(6):719. https://doi.org/10.3390/coatings15060719
Chicago/Turabian StyleFranco de Oliveira, Marconi Sandro, Jorge Leandro Aquino de Queiroz, Danyelle Medeiros de Araújo, Mayra Kerolly Sales Monteiro, Karen Giovanna Duarte Magalhaes, Carlos Alberto Martínez-Huitle, and Elisama Vieira dos Santos. 2025. "Comparing Operational Approaches (Spectrophotometric, Electroanalytic and Chromatographic) to Quantify the Concentration of Emerging Contaminants: The Limit of Detection, the Uncertainty of Measurement, Applicability and Open Problems" Coatings 15, no. 6: 719. https://doi.org/10.3390/coatings15060719
APA StyleFranco de Oliveira, M. S., de Queiroz, J. L. A., Medeiros de Araújo, D., Monteiro, M. K. S., Duarte Magalhaes, K. G., Martínez-Huitle, C. A., & dos Santos, E. V. (2025). Comparing Operational Approaches (Spectrophotometric, Electroanalytic and Chromatographic) to Quantify the Concentration of Emerging Contaminants: The Limit of Detection, the Uncertainty of Measurement, Applicability and Open Problems. Coatings, 15(6), 719. https://doi.org/10.3390/coatings15060719