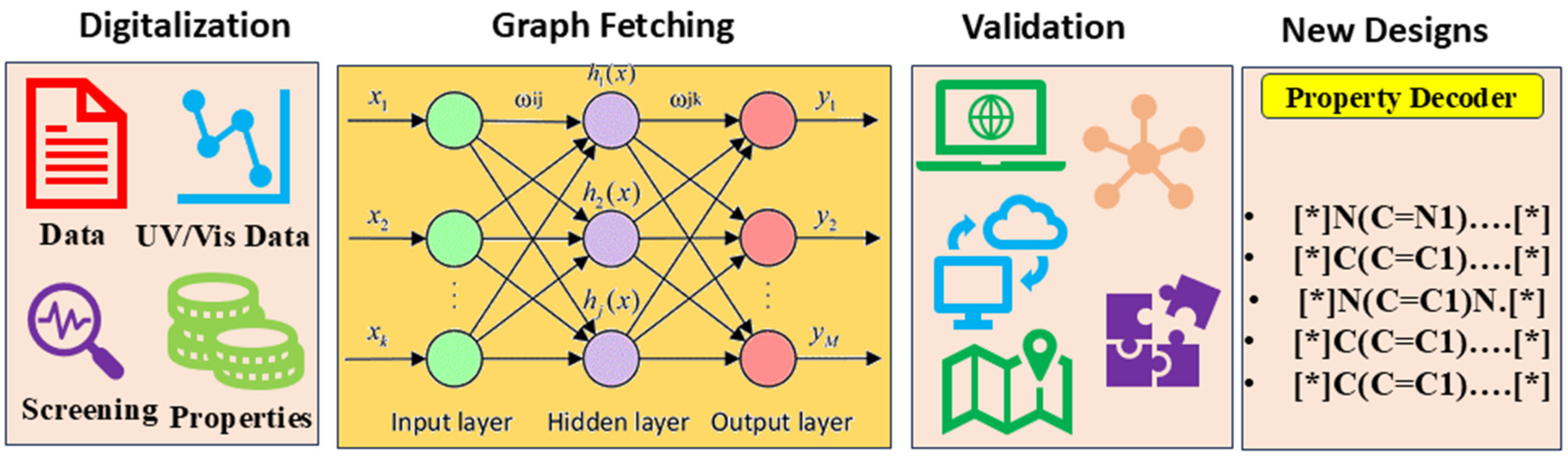
3.1. Descriptor Correlations
The integration of ML into polymer design has led to a significant transformation, drastically revolutionizing the discovery and optimization process [
19]. By leveraging vast repositories of polymer properties and performance data, ML algorithms can uncover hidden patterns and correlations that might have been overlooked by human researchers [
28]. This enables the precise prediction of polymer behavior based on chemical structure and composition, facilitating the rapid screening of potential candidates for specific applications, including sensors, photovoltaics, and biomaterials [
29]. Moreover, ML can facilitate the optimization of molecular structures to achieve desired properties, such as improved strength, thermal stability, and electrical conductivity, thus streamlining the design process and reducing the time and cost involved in trial-and-error experimentation. The top-performing tokens underwent regression analysis across various graph neural network models, yielding promising outcomes. Specifically, the Aromatic Carbonyls token attained an impressive R
2 value of 0.91 and a low RMSE value of 0.0021 when assessed using a Random Forest model (
Table 1).
The Aromatic_Heterocycles token exhibited even superior performance, achieving an R2 value of 0.92 and an RMSE value of 0.0032 when evaluated using a Decision Tree model. The Aromatic_Rings token also showed robust correlations, yielding an R2 value of 0.89 and an RMSE of 0.0029 when analyzed using the Random Forest model. Meanwhile, the HAcceptors token demonstrated a strong correlation, with an R2 value of 0.87 and an RMSE of 0.0018 when evaluated using the Decision Tree model. The HeteroAtoms token, however, exhibited the best fit with the xGBoost model, attaining an R2 value of 0.79 and an RMSE of 0.0009. The Rotatable Bonds token displayed moderate predictive performance, achieving an R2 value of 0.82 and an RMSE of 0.0031 when implemented using the Gradient-Boosting model. The Ring Count token showed comparable results, with an R2 value of 0.83 and an RMSE of 0.0021 when analyzed using the Random Forest model. The Fr_Benzene token exhibited strong performance, with an R2 value of 0.86 and an RMSE of 0.016 with the Random Forest model. In contrast, the Fr_Bicyclic token achieved an R2 value of 0.83 and an RMSE of 0.015 when utilizing the Decision Tree model. Finally, the Fr-ether and Fr_thiophene tokens exhibited moderate to strong predictive performance, attaining R2 values of 0.79 and 0.85, respectively, when applying Random Forest models.
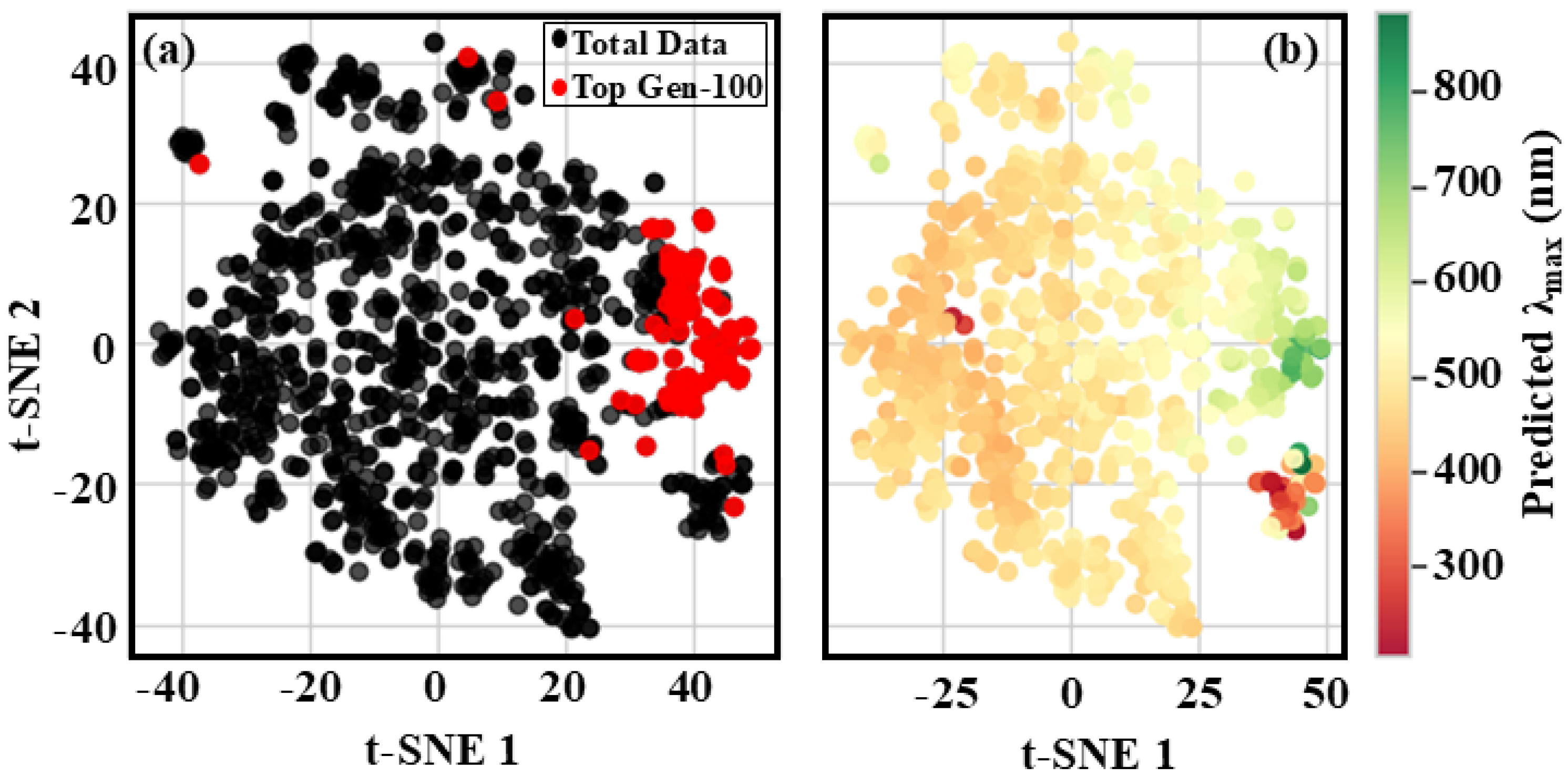
The t-SNE map constructed using these descriptors provided significant insights into the distribution patterns of the training and generation-1 datasets. The map illustrated a broad distribution of data points across both tSNE-1 and component 2, with tSNE-1 ranging from −50 to 50 and component 2 spanning from −80 to 80 (
Figure 3). Notably, the data points corresponding to both the actual and ground-predicted results were uniformly distributed across the map, highlighting a strong congruence between them. The uniform distribution of data points suggested that descriptors employed in this analysis effectively captured the intrinsic patterns and relationships within the dataset. This observation held particular significance, as it suggested that the model had effectively acquired meaningful representations of the data, exhibiting a strong correspondence with its inherent structural framework. The dispersion of data points across the entire map, rather than their concentration in a specific region, suggested that the model had successfully captured a comprehensive and diverse set of features, effectively representing the intrinsic properties of the polymer. Furthermore, the t-SNE map offered a visual depiction of the high-dimensional data, facilitating a more intuitive exploration and in-depth analysis of the relationships among various data points. The nearly uniform distribution of data points between the actual and ground-predicted results suggested that the model had effectively generalized to new, unseen data rather than merely memorizing the training instances.
This finding provided a compelling indication of the model with its capability to generate accurate predictions for new, unseen data, emphasizing the potential of this approach in expediting the design and discovery of new polymers with tailored properties.
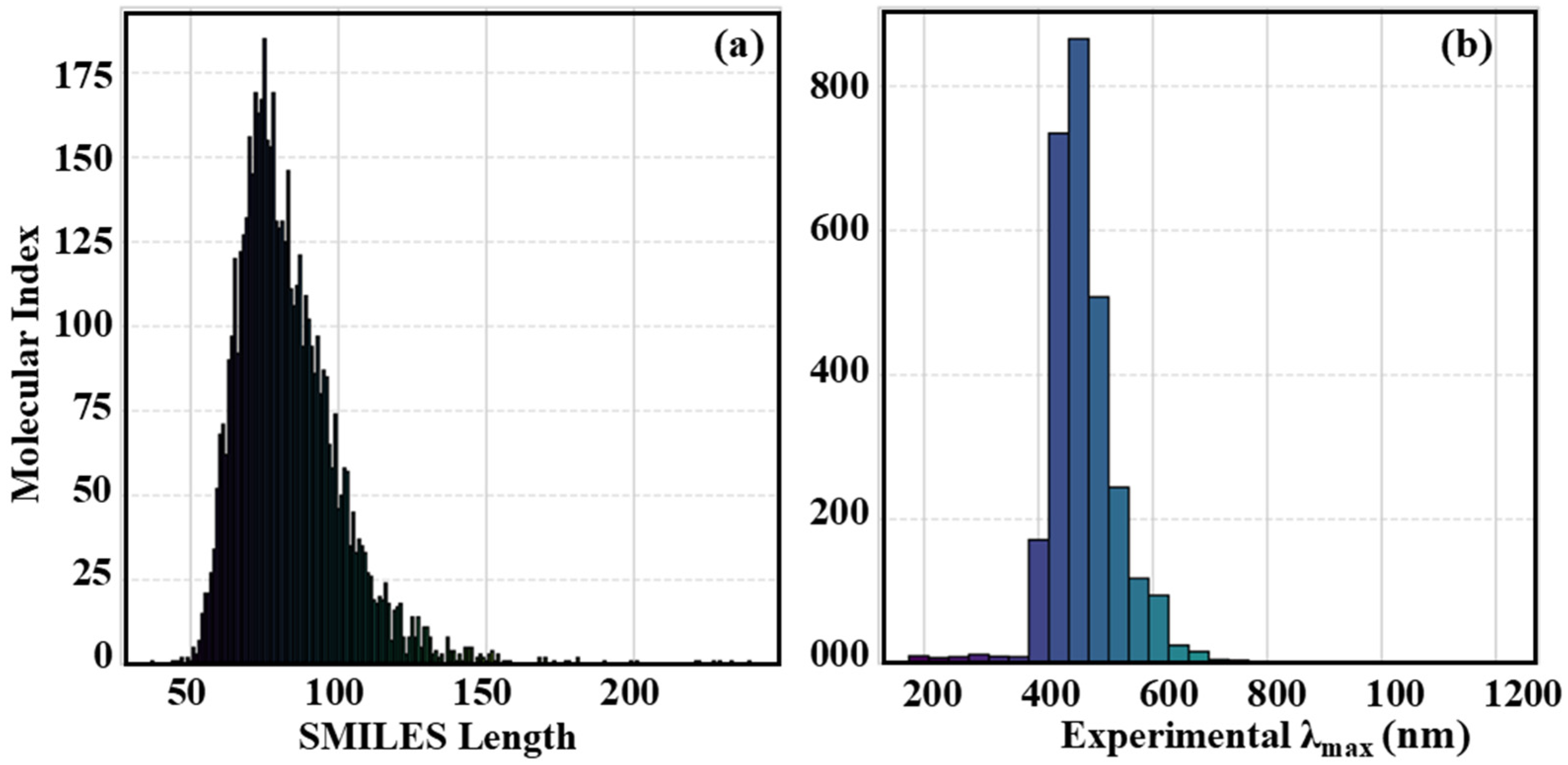
Beyond its impressive performance in predicting polymer properties, the synthetic feasibility of the generated polymers remained a critical factor for consideration. The synthetic accessibility metrics, ranging from 0.001–0.20, provided crucial insights into the feasibility of synthesizing these designed polymers within a laboratory environment. Notably, the majority of the data points displayed the highest density around a synthetic accessibility value of 0.05, suggesting that most of the designed polymers possessed moderate to high synthetic feasibility. This finding was particularly promising, as it suggested that a substantial proportion of the generated polymers were realistically synthesizable using contemporary laboratory methodologies and available resources. The high density of data points around 0.05 further suggested that the model had successfully generated polymers that were not only optimized for their predicted properties but also accounted for the practical considerations of their synthesis. This aspect was fundamental to any materials design endeavor, as accurately predicting a material with its properties was only one part of the challenge; ensuring its practical feasibility for synthesis and characterization was equally essential. The broad range of synthetic accessibility values, spanning from 0.001–0.20, further underscored the structural diversity of the generated polymers. While certain polymers may pose greater challenges in synthesis, others may be more readily obtainable. Its capacity to generate a diverse range of polymers with varying synthetic accessibility values could offer researchers a broader spectrum of potential opportunities to pursue.
3.2. Predicting λmax
In predicting the λ
max of a particular phenomenon, various regression models exhibited varying degrees of performance. An analysis of the results revealed that the Gradient-Boosting model stood out as the most effective, achieving a notable R
2 value of 0.86 and an exceptionally low RMSE of 0.0021. This showed that the model effectively captured the underlying patterns in the data with a high level of accuracy [
30]. In comparison, AdaBoost performed slightly less effectively, yielding an R
2 value of 0.81 and an RMSE of 0.0430 (
Table 2). Although still commendable, these metrics indicated that the model was comparatively less effective in predictive performance.
The K-Nearest Neighbor model exhibited an even lower performance, achieving an R2 value of 0.67 and an RMSE of 0.0051. This model struggled to capture the intricate patterns within the data, resulting in reduced predictive accuracy. The Decision Tree model demonstrated moderate performance, attaining an R2 value of 0.71 and an RMSE of 0.0009. Meanwhile, the xGBoost and LightGBM models exhibited moderate performance, yielding R2 values of 0.76 and 0.69, respectively, with corresponding RMSE values of 0.0022 and 0.0041. Notably, the differences in performance among these models could stem from various factors, including data complexity, the extent of hyperparameter tuning, and the specific nature of the phenomenon being predicted.
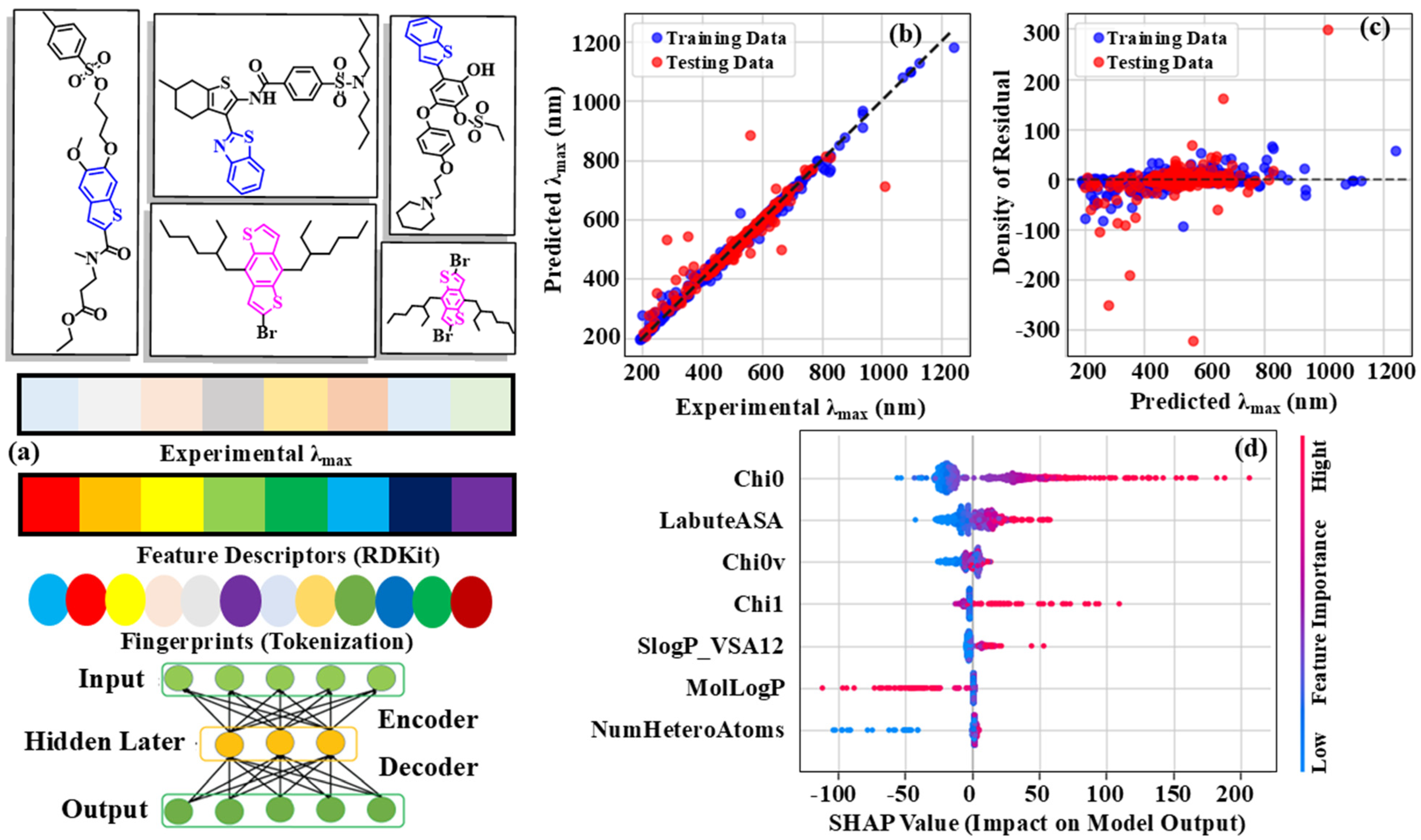
3.3. Regression Analysis
The Gradient-Boosting Regression analysis, which achieved an impressive R
2 value of 0.86 and an RMSE of 0.002, represented a noteworthy accomplishment. This algorithm integrated multiple weak models to construct a robust predictive model, which, in this case, was employed for regression analysis to estimate continuous values. The model performance was likely optimized through hyperparameter tuning, taking into account key factors such as the number of estimators, learning rate, maximum depth, and minimum sample size. Furthermore, feature engineering played a pivotal role in identifying and transforming the most relevant attributes from the dataset, ultimately improving its overall performance. This process likely encompassed handling missing values, feature scaling, and selecting relevant features through techniques such as mutual information, correlation analysis, or recursive feature elimination. The model performance was evaluated based on R
2 and RMSE metrics, offering valuable insight into its effectiveness in capturing the underlying patterns within the data (
Figure 4). An R
2 value of 0.86 signifies that the model accounted for approximately 86% of the variance in the target variable, whereas the RMSE value of 0.0021 indicated that the average difference between the actual and predicted values was around 0.0021 units. This underscored the model’s exceptional predictive accuracy. Furthermore, advanced interpretation techniques such as permutation importance and SHAP values were employed to analyze the factors influencing the model predictions and to identify the most significant features that contributed to their overall performance. The density of residuals scatter plot for the Gradient-Boosting Regression model provided a detailed representation of the model performance. The residuals, which indicated the deviations between observed and predicted values, were plotted against the predicted λ
max, reaching up to 1200 nm. The density of residuals ranged from −300 to 300, showing its overall accuracy, as the majority of residuals remained confined within a relatively narrow range. Under closer examination, the scatter plot revealed a predominantly uniform distribution of residuals across the predicted λ
max range.
This suggested that the model consistently captured the underlying patterns in the data without demonstrating notable biases or clustering around specific regions. The lack of distinct patterns or structures in the residual distribution suggested that the model neither overfitted nor underfitted the data, thereby further strengthening its reliability. Notably, the residuals were primarily concentrated around the zero line, indicating that the model predictions were generally accurate and closely aligned with the observed values. The residuals exhibited a relatively uniform spread, with no discernible patterns of increasing or decreasing variance as the predicted λmax increased. This suggested that the model performance remained robust and consistent across the range of predicted values, further reinforcing its reliability.
3.4. Feature Importance
The SHapley Additive exPlanations (SHAP) [
31] value beeswarm plot provided a comprehensive analysis of feature importance in the Gradient-Boosting Regression model, identifying the most influential factors contributing to the model predictions. As expected,
emerged as the most influential feature, prominently dominating the plot with its high SHAP values. This outcome was anticipated, given
recognized role as a key molecular structure descriptor and its effectiveness in capturing fundamental aspects of molecular topology. LabuteASA closely followed
, serving as another prominent molecular descriptor that encodes crucial information about molecular surface area and shape. Its high SHAP values indicated that the model was strongly dependent on this feature for making accurate predictions. The inclusion of
as the third most important feature further reinforced the model’s focus on molecular structure, as
is a related descriptor that captures subtle variations in molecular topology. The
, a descriptor associated with molecular flexibility, also emerged as a key contributor to the model predictions, emphasizing the role of molecular dynamics in determining λ
max. The relatively high SHAP values of SlogP-VSA12, a descriptor related to molecular hydrophobicity, suggested that the model also utilizes information about molecular lipophilicity for making predictions. This was not surprising, considering the established significance of hydrophobic interactions in molecular interactions. Although less influential, MolLogP and NumHeteroAtoms still contributed to the model predictions, offering
Supplementary Information about molecular properties such as logP and the number of heteroatoms. The ranking of these features in the SHAP value plot offered valuable insights into the underlying mechanisms governing λ
max, underscoring the significance of molecular structure, shape, and properties in determining this crucial parameter.
3.5. Cross Validation
The Gradient-Boosting model, combined with K-Fold cross-validation analysis [
32], produced a series of Mean Squared Errors (MSEs) across five folds. The MSEs for each fold were 1.7, 2.2, 2.05, 1.6, and 1.4, respectively. An examination of the MSEs revealed that the model performance remained relatively consistent across the five folds, with no notable outliers or anomalies. The average MSE across all folds was approximately 1.83, indicating that the model was able to predict the target variable with a moderate level of precision. The lowest MSE of 1.4 was recorded in fold 5, suggesting that the model performed marginally better on this particular subset of the data (
Figure 5). Conversely, the highest MSE of 2.2 was observed in fold 2, suggesting that the model encountered slightly more difficulty with this subset of the data. However, the differences in MSEs across the folds were relatively small, implying that the model was robust and demonstrated good generalization to new, unseen data. Overall, the K-Fold cross-validation analysis revealed that the Gradient-Boosting model was a reliable and consistent performer, demonstrating a moderate level of accuracy in predicting the target variable. The model performance was not highly sensitive to the specific subset of data used for training, which is a desirable trait in ML models.
The overfitting analysis of the Gradient-Boosting model provided the Root Mean Squared Errors (RMSEs) for both the training and validation sets across five folds. A comprehensive analysis of the RMSE values revealed that the model exhibited a moderate level of overfitting rather than experiencing severe overfitting. The RMSE values for the validation set spanned from 1.2 to 1.5, with an average value of approximately 1.33. In contrast, the RMSE values for the training sets varied between 1.0 and 1.25, with an average of approximately 1.12. The difference between the RMSE values for the validation and training sets was relatively small, suggesting that the model did not exhibit severe overfitting to the training data. However, the RMSE values for the validation sets showed a slight increase compared to those of the training sets, indicating that the model was not generalizing perfectly to new, unseen data. This phenomenon is commonly observed in ML models, particularly when dealing with complex datasets. It is important to highlight that the RMSE values remained consistent across the five folds, without any significant outliers or anomalies. This analysis suggested that despite moderate overfitting, the model was robust and generalized effectively to new data. Overall, the overfitting analysis concluded that the Gradient-Boosting model performed reasonably well, exhibiting a moderate level of overfitting. The model performance could be enhanced by tuning the hyperparameters, applying regularization techniques, or collecting additional data to mitigate the risk of overfitting.
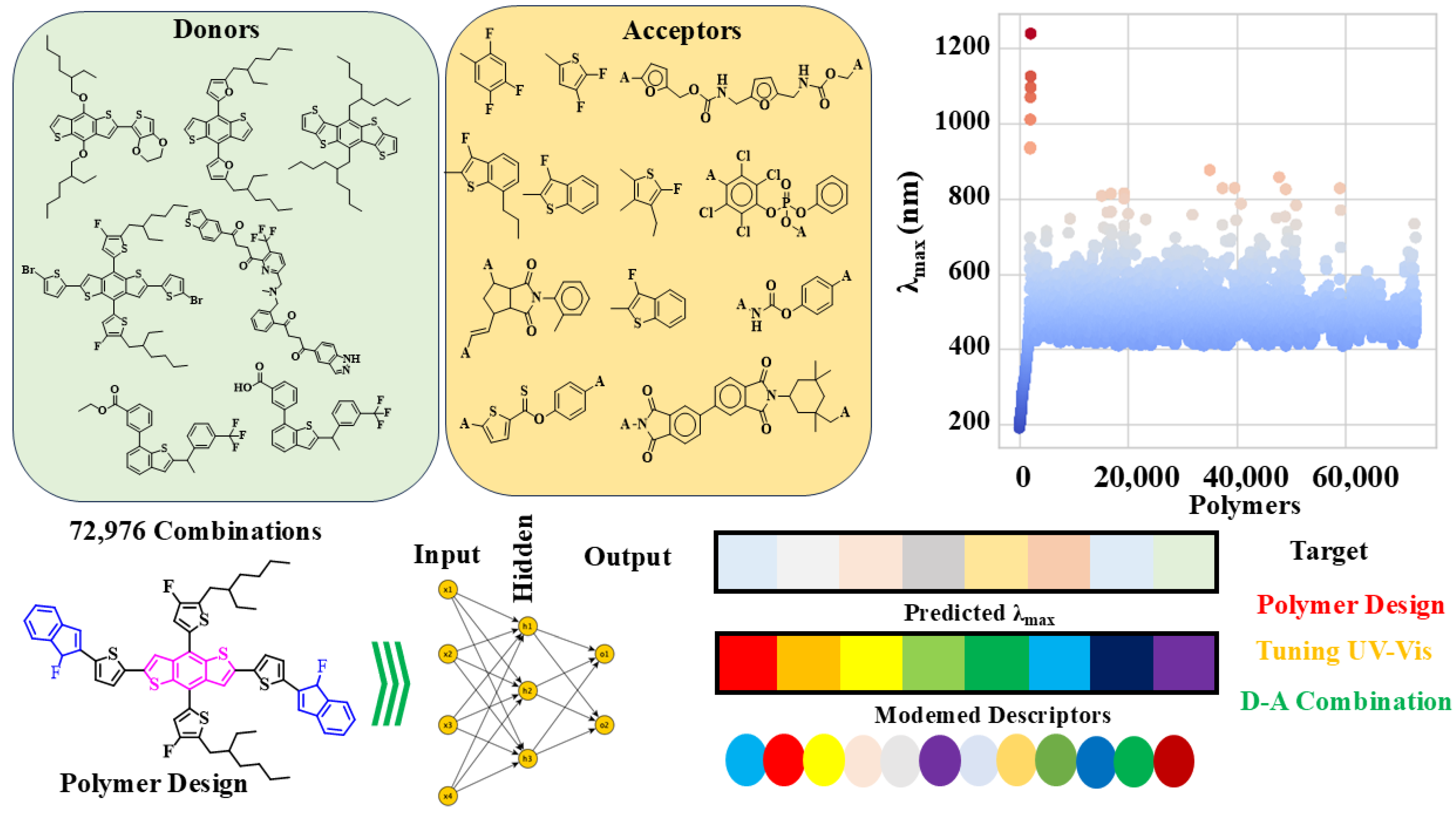
3.8. Throughput Screening for New Combinations
The collected data and trained model facilitated the design of 72,996 new combinations, each associated with their respective λ
max. This achievement was significant, as it enabled the exploration of a vast chemical space and the identification of novel compounds with favorable properties. The new combinations were generated by combining different molecular structures and functional groups, considering both the principles of chemistry and the limitations of the experimental design. The resulting compounds exhibited considerable diversity, encompassing a wide range of chemical properties, such as logP, molecular weight, and topological polar surface area. This diversity was crucial for investigating the structure–activity relationships and identifying the key factors that drive the λ
max of the compounds (
Figure 8).
Different descriptors were developed for new combinations to aid in the accessibility studies. These descriptors included molecular fingerprints, such as MACCS and ECFP, which represented the structural characteristics of the compounds. In addition, physicochemical properties, including logP, molecular weight, and topological polar surface area, were computed for each compound. These descriptors were subsequently employed to analyze the correlations between the chemical structures and their λmax. The design of the descriptors played a crucial role in the success of the accessibility studies. By integrating molecular fingerprints and physicochemical properties, the researchers were able to successfully capture the fundamental chemical and physical factors that influence the λmax of the compounds. This approach facilitated the identification of key structural features and functional groups responsible for the λmax, offering valuable insights into the underlying mechanisms that govern the absorption process. Furthermore, the descriptors were utilized to develop ML models capable of predicting the λmax of new compounds. This enabled the rapid screening of extensive compound libraries, facilitating the identification of those likely to exhibit desirable absorption properties. The ML models were trained on an extensive dataset of experimental λmax and validated with a selected subset of the data.
3.9. Computational Studies
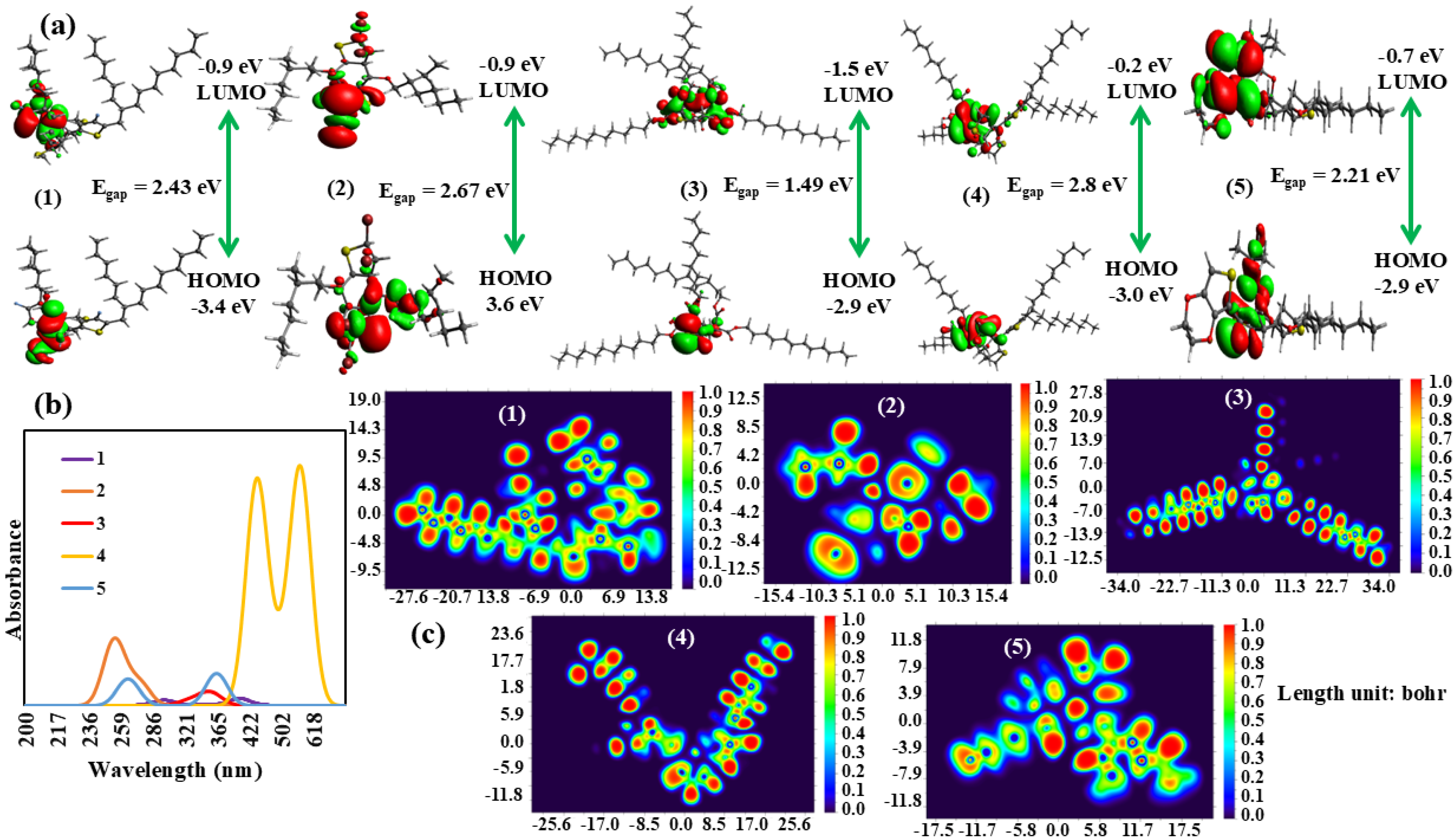
A computational study of selected polymers, utilizing the Density Functional Theory (DFT), was successfully validated to provide insights into their electronic properties. Their molecular geometries were optimized to their ground state energy minima using the CAM-B3LYP functional with the 6-31+G(d,p) basis set. Additionally, the computed TD-DFT spectra were obtained at the same level as the theory and basis set. Notably, the analysis revealed that the charges on the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were primarily localized within the donor regions, exhibiting opposite charges [
34]. This intriguing phenomenon suggested that the donor regions were crucial in governing the electronic behavior of polymers. In sharp contrast, the alkyl chains incorporated into these polymers were found to lack significant charges, implying that they do not substantially influence the electronic properties of the materials. These computational findings offered valuable insights for the design and development of novel polymers with tailored electronic properties, laying the foundation for potential applications in emerging fields such as optoelectronics and organic electronics. The computed UV-Vis parameters for the five polymers revealed distinct features. Polymer 1, exhibiting an energy of 3.10 eV and a wavelength of 401 nm, displayed a comparatively low oscillator strength (
f) of 0.0012, reflecting a moderate absorption intensity (
Figure 9). The predominant contribution to this polymer arose from the HOMO→LUMO transition, which accounted for the entirety (100%) of the transition. In contrast, polymer 2 exhibited a higher energy of 4.89 eV and a shorter wavelength of 257 nm, accompanied by a slightly increased oscillator strength (
f) of 0.0016. The primary contribution to this polymer originated from the HOMO − 1→LUMO + 1 transition, which was responsible for 70% of the overall transition (
Table 3). Polymers 3 and 5 displayed similar characteristics, exhibiting energies of 3.46 and 3.43 eV, along with wavelengths of 358 and 363 nm, respectively. Both polymers displayed relatively low
f of 0.0014 and 0.0055, respectively, with the HOMO→LUMO transition being the dominant contributor, accounting for 94 and 99% of the transition, respectively.
Polymer 4 was characterized by a significantly lower energy of 2.21 eV and a longer wavelength of 563 nm, coupled with a relatively high
f of 0.0414. The HOMO→LUMO transition was the predominant contributor to this polymer, responsible for 81% of the total transition. In addition to the electronic transitions, the electron localization functions (ELFs) [
35] of these polymers yielded valuable insights into the spatial distribution of electrons. The red spots observed at the terminal regions of the polymers indicated regions of high electron density, which were likely engaged in electronic transitions. These regions may have been critical in determining the electronic and optical properties of polymers. For instance, in polymer 1, the ELF demonstrated a high electron density at the terminal regions, which corresponded to the HOMO→LUMO transition. This suggested that the electrons were localized in these regions, resulting in a moderate absorption intensity. In contrast, the ELF of polymer 2 exhibited a more dispersed electron density, which could be attributed to the involvement of HOMO − 1 and LUMO + 1 orbitals in the electronic transition. The similar electronic transitions observed in polymers 3 and 5 were also mirrored in their ELFs, which exhibited comparable electron density patterns.
The high electron density at the terminal regions in these polymers suggested a strong localization of electrons, which resulted in the observed low oscillator strengths. Polymer 4, characterized by its distinct electronic transition, displayed a more intricate ELF pattern featuring multiple regions of high electron density. This could be related to the longer wavelength and higher oscillator strength observed in this polymer.
The transition density matrix (TDM) analysis [
36] provided comprehensive insights into the electronic transitions of the five polymers. The TDM plots, representing the probability density of the electronic transition, provided insights into the spatial distribution of the transition dipole moment. For polymer 1, the TDM plot exhibited a significant contribution from the ends of the polymer chain, signifying a strong dipole moment along the chain axis. This was consistent with the HOMO→LUMO transition, which predominantly governed this polymer. However, the TDM plot of polymer 2 exhibited a more intricate pattern, with contributions from both the terminal and the central regions of the chain, reflecting the involvement of HOMO − 1 and LUMO + 1. The TDM plots of polymers 3 and 5 showed comparable patterns, with predominant contributions originating from the ends of the chain, which corresponded to their HOMO→LUMO transitions. However, the TDM plot of polymer 4 displayed a unique pattern, with contributions from various regions along the chain, including both the ends and the middle, indicating a more delocalized transition dipole moment. This was consistent with the lower energy and longer wavelength observed in this polymer, which could be attributed to a more extended conjugation throughout the polymer chain. The TDM analysis also yielded insights regarding the directionality of the transition dipole moment, which could significantly impact the optical properties of the polymers. For instance, polymers 1 and 3 displayed a strong dipole moment along the chain axis, which might lead to a high absorption coefficient for light polarized in this direction. In contrast, polymer 4 exhibited a more isotropic transition dipole moment, which could lead to a lower absorption coefficient but may potentially enhance fluorescence efficiency.
The charge density difference (CDD) [
37] analysis offered a comprehensive understanding of the electronic rearrangement that occurred during the electronic transitions of the five polymers. The CDD plots, which depicted the variation in electron density between the ground and excited states, revealed the areas of the polymer chain where electron transfer or redistribution took place during the transition. For polymer 1, the CDD plot indicated a notable transfer of electron density from the ends of the polymer chain towards the central region, which was consistent with the HOMO→LUMO transition. This suggested that the electrons were excited from HOMO to LUMO, leading to a redistribution of electron density along the chain. In contrast, the CDD plot of polymer 2 exhibited a more intricate pattern, showing both increases and decreases in electron density along the chain, which reflected the involvement of HOMO − 1 and LUMO + 1 orbitals. The CDD plots of polymers 3 and 5 exhibited comparable patterns, showing electron density transfer from the ends to the central region, which was consistent with their HOMO→LUMO transitions (
Figure 10). However, the CDD plot of polymer 4 revealed a distinct pattern characterized by a substantial increase in electron density along the entire chain, suggesting electron delocalization during the transition. This showed consistency with the lower energy and longer wavelength of this polymer, which could be attributed to a more extended conjugation throughout the chain. Additionally, the CDD analysis revealed variations in electron density distribution between the ground and excited states, which could impact the optical properties of the polymers.
For instance, the CDD plot of polymer 1 revealed a notable increase in electron density near the ends of the chain, potentially leading to a higher absorption coefficient for light polarized in this direction. In comparison, the CDD plot of polymer 4 exhibited a more uniform increase in electron density across the chain, which might result in a lower absorption coefficient but could potentially improve fluorescence efficiency.
The global chemical reactivity parameters [
38] of the five polymers offered crucial insights into their reactivity and chemical behavior. The ionization potential (IP) and electron affinity (EA) values demonstrated the energy required to remove or add an electron to the system, respectively. The electronegativity (
χ) values, spanning from 1.63–2.28 eV, suggested that those polymers could exhibit moderate electronegativities, reflecting a balance between their electron-withdrawing and electron-donating abilities. The hardness (
η) and softness (
σ) values revealed information about the system with its resistance to electron transfer and its tendency to donate or accept electrons, respectively. The hardness values, ranging between 0.74–1.39 eV, suggested that these polymers exhibited moderate hardness, implying an intermediate resistance to electron transfer (
Table 4).
The σ values, spanning from 0.36–0.67 eV, revealed that these polymers exhibited moderate σ, suggesting their ability to moderately donate or accept electrons. The electrophilicity index (ω) values, varying from 0.95–3.34 eV, explained their ability to accept electrons and form bonds. The elevated electrophilicity index values suggested that some of these polymers exhibited a stronger inclination to form bonds and react with nucleophiles.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}