
Non-Metal Doping as a First-Principles Study for Promoting the Hydrogen Evolution of Two-Dimensional Electride Y2C Electrocatalysts


Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
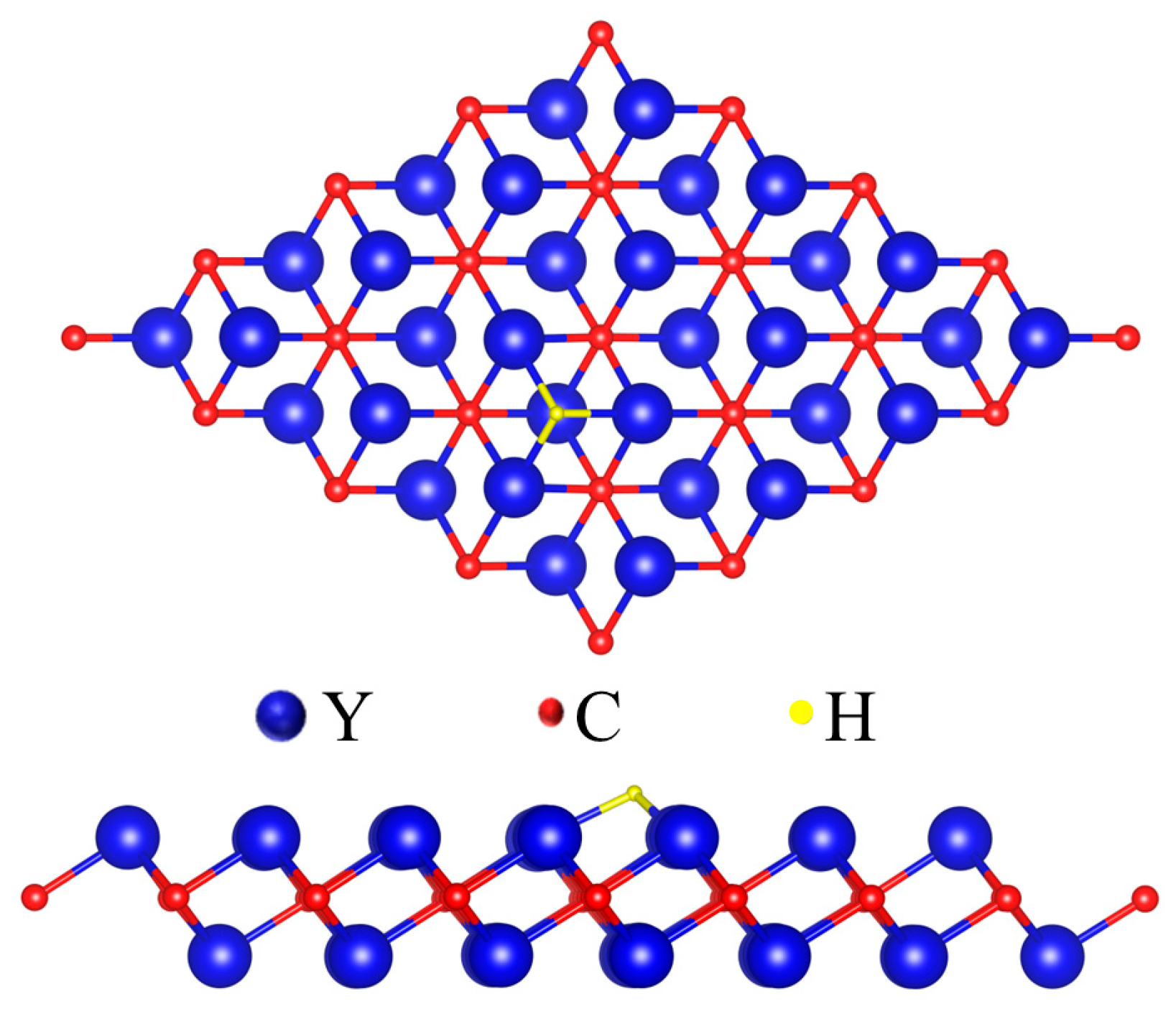
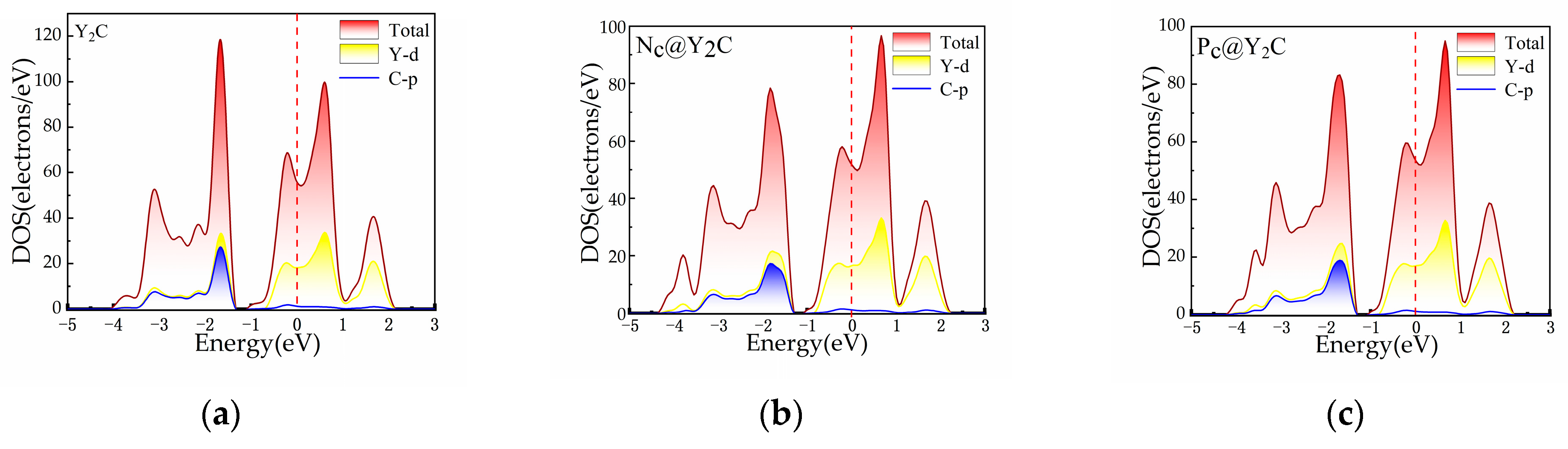
3.1. Catalytic Properties of Pristine Y2C
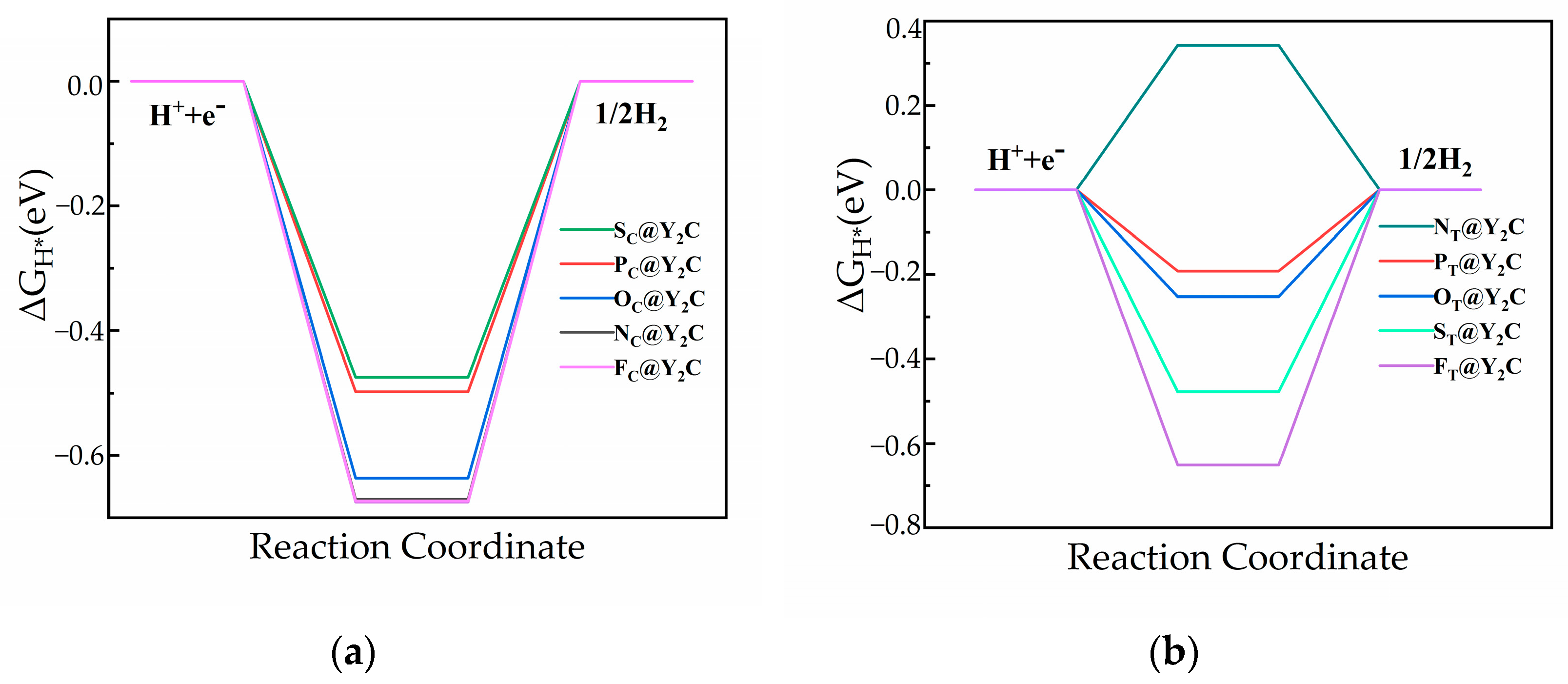
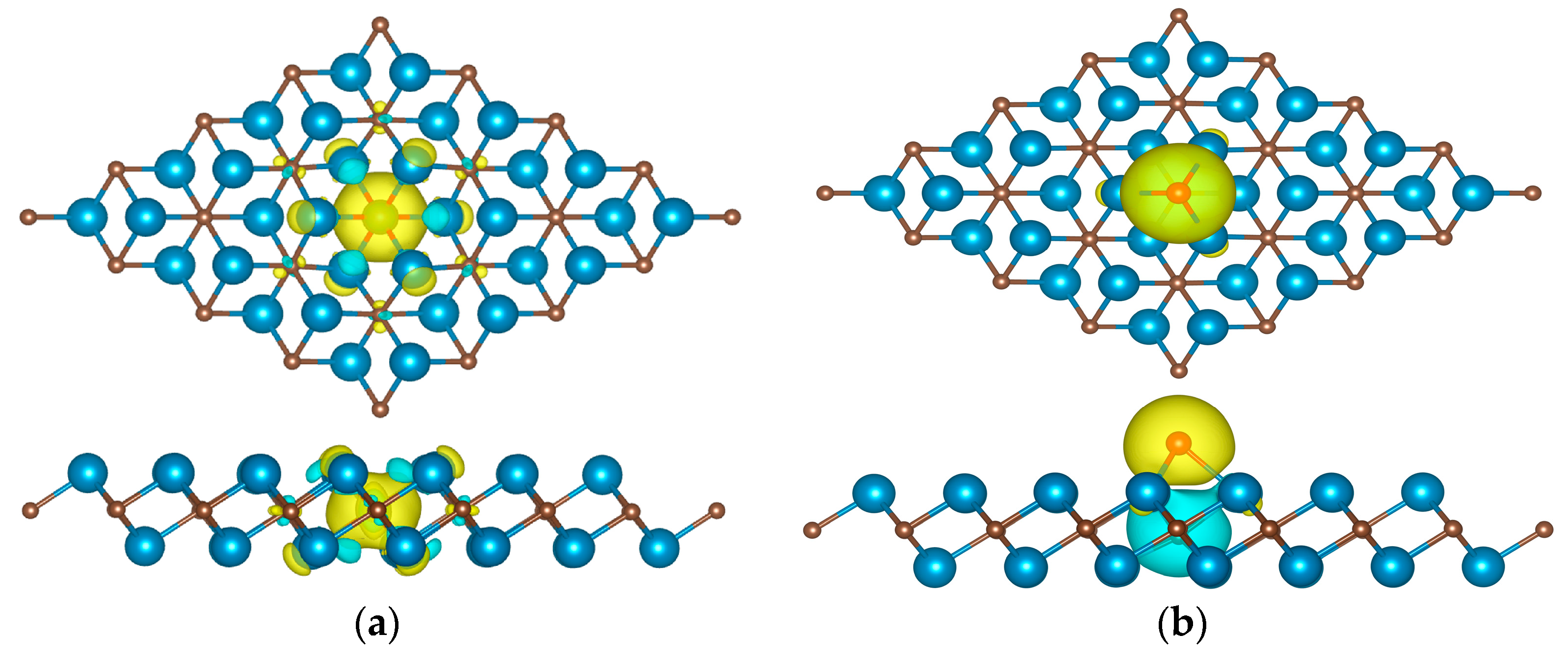
3.2. Catalytic Properties of Y2C Doped with Non-Metallic Atoms
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, T.; Zhang, D. A critical review of comparative global historical energy consumption and future demand: The story told so far. Energy Rep. 2020, 6, 1973–1991. [Google Scholar] [CrossRef]
- Ahmad, T.; Chen, H. A review on machine learning forecasting growth trends and their real-time applications in different energy systems. SCS 2020, 54, 102010. [Google Scholar] [CrossRef]
- Güney, T. Solar energy, governance and CO2 emissions. RENEY 2022, 184, 791–798. [Google Scholar] [CrossRef]
- Salah, B.; Abdelgawad, A.; El-Demellawi, J.K.; Lu, Q.; Xia, Z.; Abdullah, A.M.; Eid, K. Scalable One-Pot Fabrication of Carbon-Nanofiber-Supported Noble-Metal-Free Nanocrystals for Synergetic-Dependent Green Hydrogen Production: Unraveling Electrolyte and Support Effects. ACS Appl. Mater. Interfaces 2024, 16, 18768–18781. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhu, M.; Li, Y. The Adsorption Behavior of Hydrogen on the PuO2(111) Surface: A DFT+U Study. Coatings 2024, 14, 195. [Google Scholar] [CrossRef]
- Wawrzyńczak, A.; Feliczak-Guzik, A. Hydrogen Production Using Modern Photocatalysts. Coatings 2024, 14, 366. [Google Scholar] [CrossRef]
- Jiao, S.; Yao, Z.; Xue, F.; Lu, Y.; Liu, M.; Deng, H.; Ma, X.; Liu, Z.; Ma, C.; Huang, H.; et al. Defect-rich one-dimensional MoS2 hierarchical architecture for efficient hydrogen evolution: Coupling of multiple advantages into one catalyst. Appl. Catal. B Environ. 2019, 258, 117964. [Google Scholar] [CrossRef]
- Song, D.; Wang, Y.; Lu, X.; Gao, Y.; Li, Y.; Gao, F. Ag nanoparticles-decorated nitrogen-fluorine co-doped monolayer MoS2 nanosheet for highly sensitive electrochemical sensing of organophosphorus pesticides. Sens. Actuators B Chem. 2018, 267, 5–13. [Google Scholar] [CrossRef]
- Li, C.; Zhu, D.; Cheng, S.; Zuo, Y.; Wang, Y.; Ma, C.; Dong, H. Recent research progress of bimetallic phosphides-based nanomaterials as cocatalyst for photocatalytic hydrogen evolution. Chin. Chem. Lett. 2022, 33, 1141–1153. [Google Scholar] [CrossRef]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Kolar, J. Alternative energy technologies. J. Environ. Qual. 2001, 10, 45–54. [Google Scholar] [CrossRef]
- Crabtree, G.W.; Dresselhaus, M.S.; Buchanan, M.V. The Hydrogen Economy. Phys. Today 2004, 57, 39–44. [Google Scholar] [CrossRef]
- Huang, T.; Shen, T.; Gong, M.; Deng, S.; Lai, C.; Liu, X.; Zhao, T.; Teng, L.; Wang, D. Ultrafine Ni-B nanoparticles for efficient hydrogen evolution reaction. Chin. J. Catal. 2019, 40, 1867–1873. [Google Scholar] [CrossRef]
- Benck, J.D.; Hellstern, T.R.; Kibsgaard, J.; Chakthranont, P.; Jaramillo, T.F. Catalyzing the Hydrogen Evolution Reaction (HER) with Molybdenum Sulfide Nanomaterials. ACS Catal. 2014, 4, 3957–3971. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef]
- Amiinu, I.S.; Pu, Z.; Liu, X.; Owusu, K.A.; Monestel, H.G.R.; Boakye, F.O.; Zhang, H.; Mu, S. Multifunctional Mo–N/C@MoS2 Electrocatalysts for HER, OER, ORR, and Zn–Air Batteries. Adv. Funct. Mater. 2017, 27, 1702300. [Google Scholar] [CrossRef]
- Kang, M.; Lin, C.; Yang, H.; Guo, Y.; Liu, L.; Xue, T.; Liu, Y.; Gong, Y.; Zhao, Z.; Zhai, T.; et al. Proximity Enhanced Hydrogen Evolution Reactivity of Substitutional Doped Monolayer WS2. ACS Appl. Mater. Interfaces 2021, 13, 19406–19413. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Luo, W.; Ma, L.; Yu, M.; Ren, X.; He, M.; Polen, S.; Click, K.; Garrett, B.; Lu, J.; et al. Dimeric [Mo2S12]2− Cluster: A Molecular Analogue of MoS2 Edges for Superior Hydrogen-Evolution Electrocatalysis. Angew. Chem. Int. Ed. 2015, 54, 15181–15185. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Huang, Y.; Fu, J. Recent advances in electrocatalysts for neutral and large-current-density water electrolysis. Nano Energy 2021, 80, 105545. [Google Scholar] [CrossRef]
- Shi, W.; Yin, G.; Yu, S.; Hu, T.; Wang, X.; Wang, Z. Atomic precision tailoring of two-dimensional MoSi2N4 as electrocatalyst for hydrogen evolution reaction. J. Mater. Sci. 2022, 57, 18535–18548. [Google Scholar] [CrossRef]
- Liu, S.; Ban, J.; Shi, H.; Wu, Z.; Shao, G.; Cao, G.; Hu, J. Near solution-level conductivity of polyvinyl alcohol based electrolyte and the application for fully compliant Al-air battery. Chem. Eng. J. 2022, 431, 134283. [Google Scholar] [CrossRef]
- Kataoka, H.; Saito, Y.; Sakai, T.; Quartarone, E.; Mustarelli, P. Conduction Mechanisms of PVDF-Type Gel Polymer Electrolytes of Lithium Prepared by a Phase Inversion Process. J. Phys. Chem. B 2000, 104, 11460–11464. [Google Scholar] [CrossRef]
- Bai, S.; Wang, C.; Deng, M.; Gong, M.; Bai, Y.; Jiang, J.; Xiong, Y. Surface Polarization Matters: Enhancing the Hydrogen-Evolution Reaction by Shrinking Pt Shells in Pt–Pd–Graphene Stack Structures. Angew. Chem. Int. Ed. 2014, 53, 12120–12124. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Chen, P.; Zhou, T. A Bifunctional Hybrid Electrocatalyst for Oxygen Reduction and Evolution: Cobalt Oxide Nanoparticles Strongly Coupled to B, N-Decorated Graphene. Angew. Chem. Int. Ed. 2017, 56, 7121–7125. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, L.; Zhang, W. Stabilizing sulfur vacancy defects by performing “click” chemistry of ultrafine palladium to trigger a high-efficiency hydrogen evolution of MoS2. Nanoscale 2020, 12, 9943–9949. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, H.; Qu, J. Earth-Rich Transition Metal Phosphide for Energy Conversion and Storage. Adv. Energy Mater. 2016, 6, 1600087. [Google Scholar] [CrossRef]
- Vrubel, H.; Hu, X. Molybdenum Boride and Carbide Catalyze Hydrogen Evolution in both Acidic and Basic Solutions. Angew. Chem. Int. Ed. 2012, 51, 12703–12706. [Google Scholar] [CrossRef]
- Abdelgawad, A.; Salah, B.; Lu, Q.; Abdullah, A.M.; Chitt, M.; Ghanem, A.; Al-Hajri, R.S.; Eid, K. Template-free synthesis of M/g-C3N4 (M = Cu, Mn, and Fe) porous one-dimensional nanostructures for green hydrogen production. J. Electroanal. Chem. 2023, 938, 117426. [Google Scholar] [CrossRef]
- Salah, B.; Abdelgawad, A.; Lu, Q.; Ipadeola, A.K.; Luque, R.; Eid, K. Synergistically interactive MnFeM (M = Cu, Ti, and Co) sites doped porous g-C3N4 fiber-like nanostructures for an enhanced green hydrogen production. Green Chem. 2023, 25, 6032–6040. [Google Scholar] [CrossRef]
- Song, B.; Jin, S. Two Are Better than One: Heterostructures Improve Hydrogen Evolution Catalysis. Joule 2017, 1, 220–221. [Google Scholar] [CrossRef]
- Su, J.; Zhuang, L.; Zhang, S.; Liu, Q.; Zhang, L.; Hu, G. Single atom catalyst for electrocatalysis. Chin. Chem. Lett. 2021, 32, 2947–2962. [Google Scholar] [CrossRef]
- Liu, L.; Wang, C.; Zhang, L.; Liu, C.; Niu, C.; Zeng, Z.; Ma, D.; Jia, Y. Surface Van Hove Singularity Enabled Efficient Catalysis in Low-Dimensional Systems: CO Oxidation and Hydrogen Evolution Reactions. J. Phys. Chem. Lett. 2022, 13, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, G. Recent Advances and Applications of Inorganic Electrides. J. Phys. Chem. Lett. 2020, 11, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Hosono, H.; Kitano, M. Advances in Materials and Applications of Inorganic Electrides. Chem. Rev. 2021, 121, 3121–3185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bao, A.; Mo, G. Effect of multi-walled carbon nanotubes on the electrochemical performance of LiVPO4F cathode material for rechargeable lithium-ion batteries. Solid State Ion. 2014, 264, 45–48. [Google Scholar] [CrossRef]
- Khan, K.; Tareen, A.K.; Aslam, M.; Thebo, K.H.; Khan, U.; Wang, R.; Shams, S.S.; Han, Z.; Ouyang, Z. A comprehensive review on synthesis of pristine and doped inorganic room temperature stable mayenite electride, [Ca24Al28O64]4+(e−)4 and its applications as a catalyst. Prog. Solid State Chem. 2019, 54, 1–19. [Google Scholar] [CrossRef]
- Lu, Y.; Li, J.; Tada, T.; Toda, Y.; Ueda, S.; Yokoyama, T.; Kitano, M.; Hosono, H. Water Durable Electride Y5Si3: Electronic Structure and Catalytic Activity for Ammonia Synthesis. J. Am. Chem. Soc. 2016, 138, 3970–3973. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.-N.; Lu, Y.; Li, J.; Nakao, T.; Yang, H.; Tada, T.; Kitano, M.; Hosono, H. Copper-Based Intermetallic Electride Catalyst for Chemoselective Hydrogenation Reactions. J. Am. Chem. Soc. 2017, 139, 17089–17097. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, Z.; Lei, H.; Toda, Y.; Matsuishi, S.; Kamiya, T.; Ueda, S.; Hosono, H. Two-Dimensional Transition-Metal Electride Y2C. Chem. Mater. 2014, 26, 6638–6643. [Google Scholar] [CrossRef]
- Aliakbari, A.; Amiri, P.; Salehi, H. First-principles investigation of the structural and dynamical stability, electronic and thermal properties of two-dimensional Yn+1Cn (n = 1, 2, and 3) MXenes. FlatChem 2022, 31, 100328. [Google Scholar] [CrossRef]
- Wang, H.; Choi, J.-H. Hydrogen Adsorption on the Vertical Heterostructures of Graphene and Two-Dimensional Electrides: A First-Principles Study. ACS Omega 2022, 7, 16063–16069. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jin, K.-H.; Zhang, S.; Liu, F. Topological Electride Y2C. Nano Lett. 2018, 18, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Nie, K.; Shao, Z.-J.; Gao, B.; Lin, H.; Zhang, H.; Liu, B.; Wang, Y.; Zhang, Y.; Sun, X.; et al. Phosphorus-Mo2C@carbon nanowires toward efficient electrochemical hydrogen evolution: Composition, structural and electronic regulation. Energy Environ. Sci. 2017, 10, 1262–1271. [Google Scholar] [CrossRef]
- Xu, K.; Ding, H.; Zhang, M.; Chen, M.; Hao, Z.; Zhang, L.; Wu, C.; Xie, Y.J.A.M. Regulating Water-Reduction Kinetics in Cobalt Phosphide for Enhancing HER Catalytic Activity in Alkaline Solution. Adv. Mater. 2017, 29, 1606980. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Yang, K.R.; Lu, Z.; Li, Y.; Xu, W.; Gao, T.; Cai, Z.; Zhang, Y.; Batista, V.S.; Liu, W.; et al. Nitrogen-doped tungsten carbide nanoarray as an efficient bifunctional electrocatalyst for water splitting in acid. Nat. Commun. 2018, 9, 924. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Sa, B.; Li, Y.-L.; Qi, J.; Ahuja, R.; Sun, Z.J.J.O.P.C.C. Strain Engineering for Phosphorene: The Potential Application as a Photocatalyst. J. Phys. Chem. C 2014, 118, 26560–26568. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Mir, S.H.; Chakraborty, S.; Jha, P.C.; Wärnå, J.; Soni, H.; Jha, P.K.; Ahuja, R. Two-dimensional boron: Lightest catalyst for hydrogen and oxygen evolution reaction. Appl. Phys. Lett. 2016, 109, 053903. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Z.; Xiong, R.; Yang, X.; Zhang, Y.; Xie, T.; Wen, C.; Sa, B. Enhancing hydrogen evolution reaction performance of transition metal doped two-dimensional electride Ca2N. Chin. Chem. Lett. 2023, 34, 107643. [Google Scholar] [CrossRef]
- Cao, X.; Bai, H.; Wu, W.; Bao, H.; Li, Y. Improving the catalytic activity of two-dimensional Mo2C for hydrogen evolution reaction by doping and vacancy defects. Int. J. Hydrogen Energy 2022, 47, 38517–38523. [Google Scholar] [CrossRef]
- Gong, Q.; Wang, Y.; Hu, Q.; Zhou, J.; Feng, R.; Duchesne, P.N.; Zhang, P.; Chen, F.; Han, N.; Li, Y.; et al. Ultrasmall and phase-pure W2C nanoparticles for efficient electrocatalytic and photoelectrochemical hydrogen evolution. Nat. Commun. 2016, 7, 13216. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dopant | XC | XT |
|---|---|---|
| N | −0.417 | −1.874 |
| P | 1.215 | −0.835 |
| O | −2.646 | −5.677 |
| S | −0.797 | −3.848 |
| F | −1.444 | −6.028 |
| Dopant | XC | XT |
|---|---|---|
| N | −0.910 | 0.101 |
| P | −0.737 | −0.438 |
| O | −0.876 | −0.492 |
| S | −0.714 | −0.719 |
| F | −0.914 | −0.907 |
| Dopant | XC | XT |
|---|---|---|
| N | −0.670 | 0.341 |
| P | −0.50 | −0.192 |
| O | −0.636 | −0.252 |
| S | −0.474 | −0.478 |
| F | −0.680 | −0.652 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Su, N.; Li, Y. Non-Metal Doping as a First-Principles Study for Promoting the Hydrogen Evolution of Two-Dimensional Electride Y2C Electrocatalysts. Coatings 2024, 14, 657. https://doi.org/10.3390/coatings14060657
Li C, Su N, Li Y. Non-Metal Doping as a First-Principles Study for Promoting the Hydrogen Evolution of Two-Dimensional Electride Y2C Electrocatalysts. Coatings. 2024; 14(6):657. https://doi.org/10.3390/coatings14060657
Chicago/Turabian StyleLi, Chaoqun, Ningning Su, and Yuqiang Li. 2024. "Non-Metal Doping as a First-Principles Study for Promoting the Hydrogen Evolution of Two-Dimensional Electride Y2C Electrocatalysts" Coatings 14, no. 6: 657. https://doi.org/10.3390/coatings14060657
APA StyleLi, C., Su, N., & Li, Y. (2024). Non-Metal Doping as a First-Principles Study for Promoting the Hydrogen Evolution of Two-Dimensional Electride Y2C Electrocatalysts. Coatings, 14(6), 657. https://doi.org/10.3390/coatings14060657