
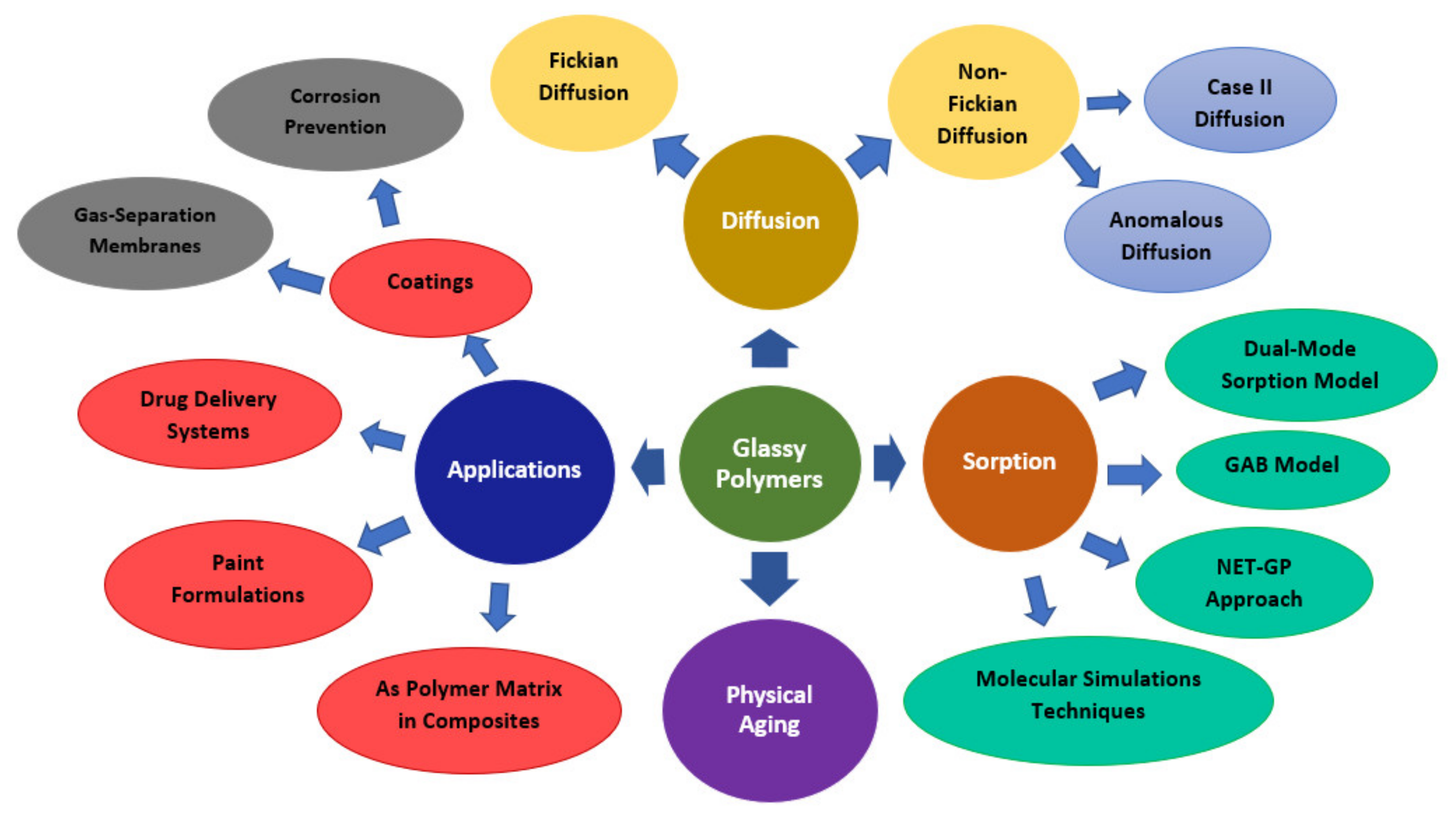
Glassy Polymers—Diffusion, Sorption, Ageing and Applications




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
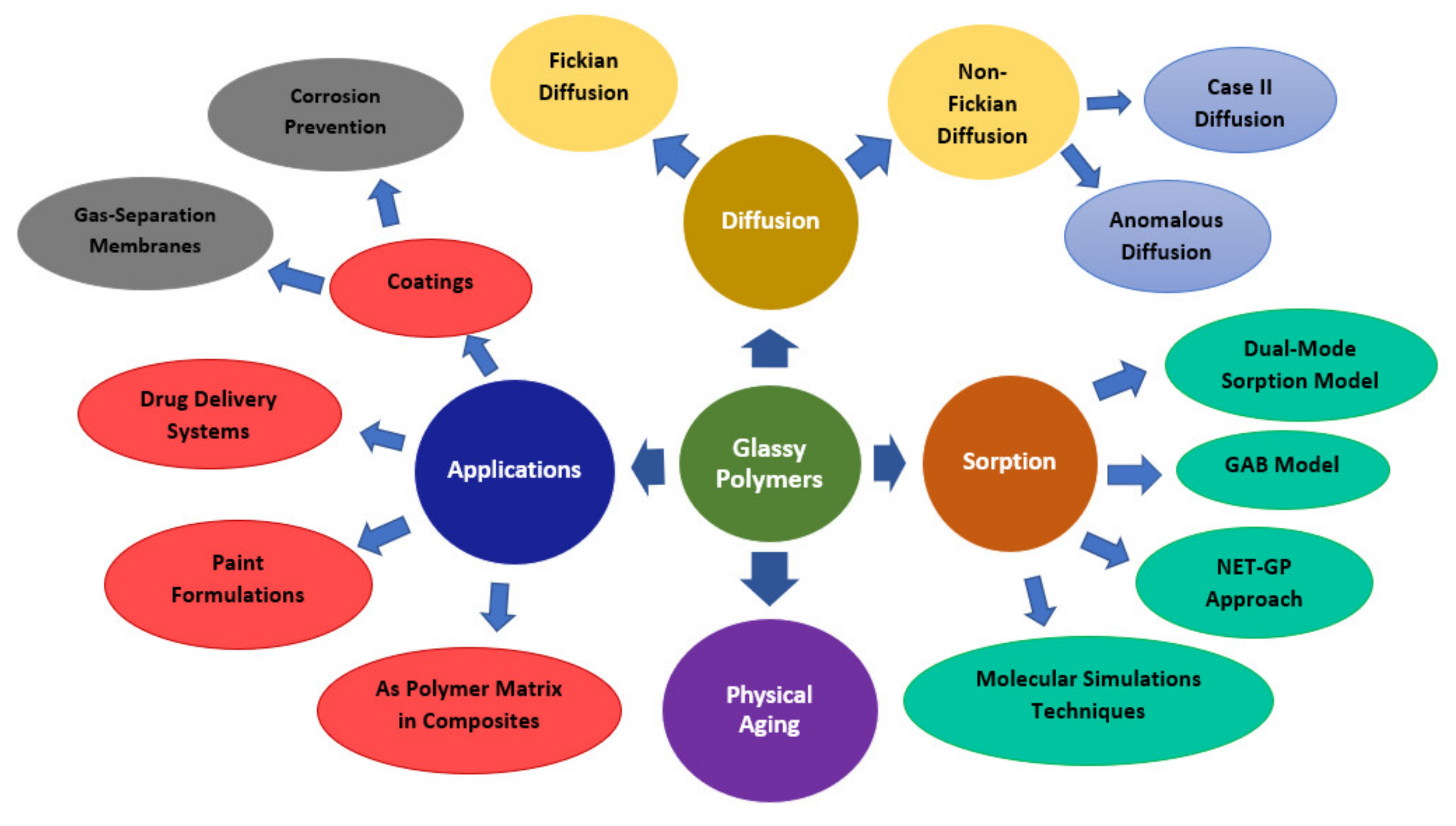
Abstract
:1. Introduction
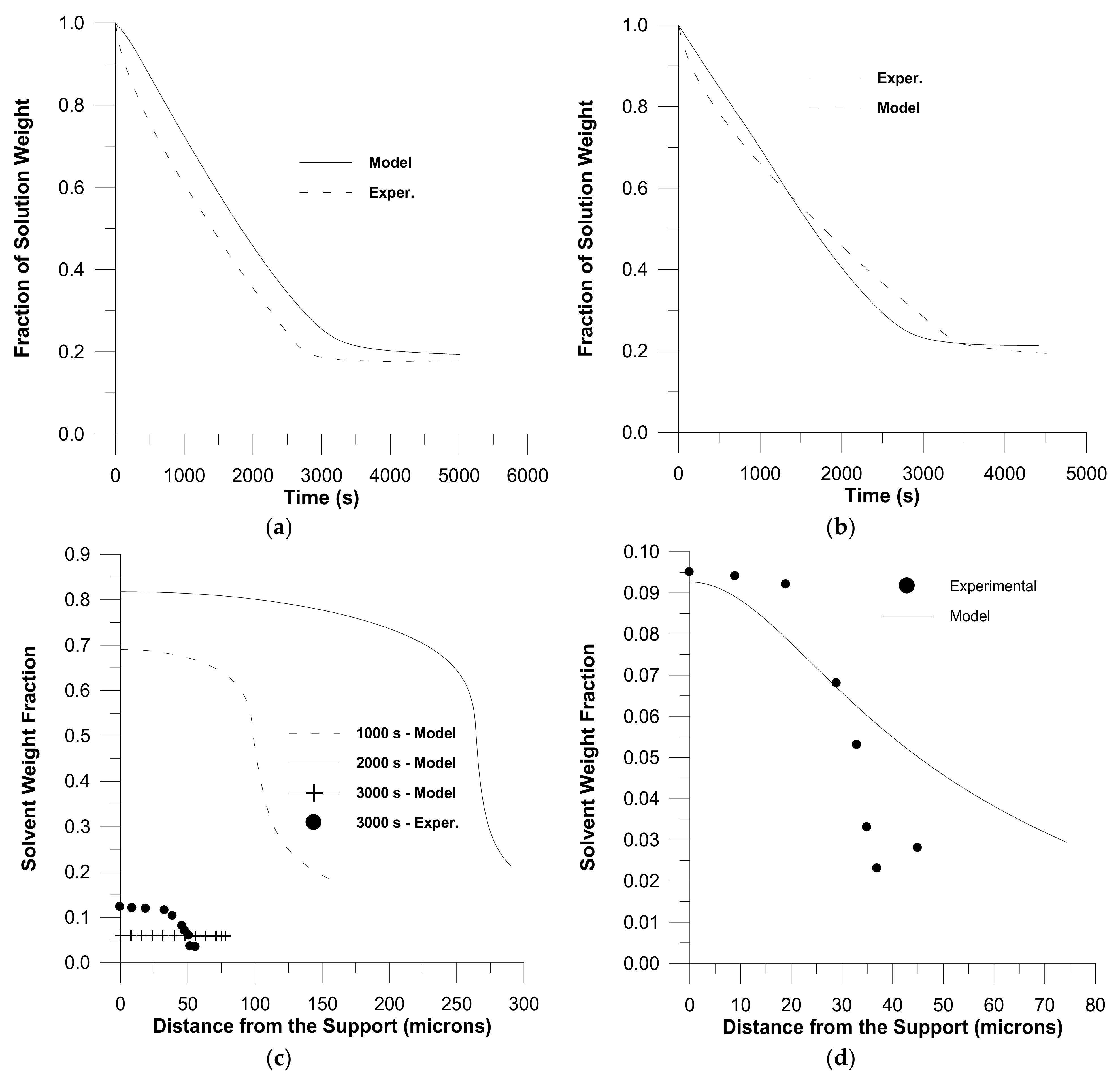
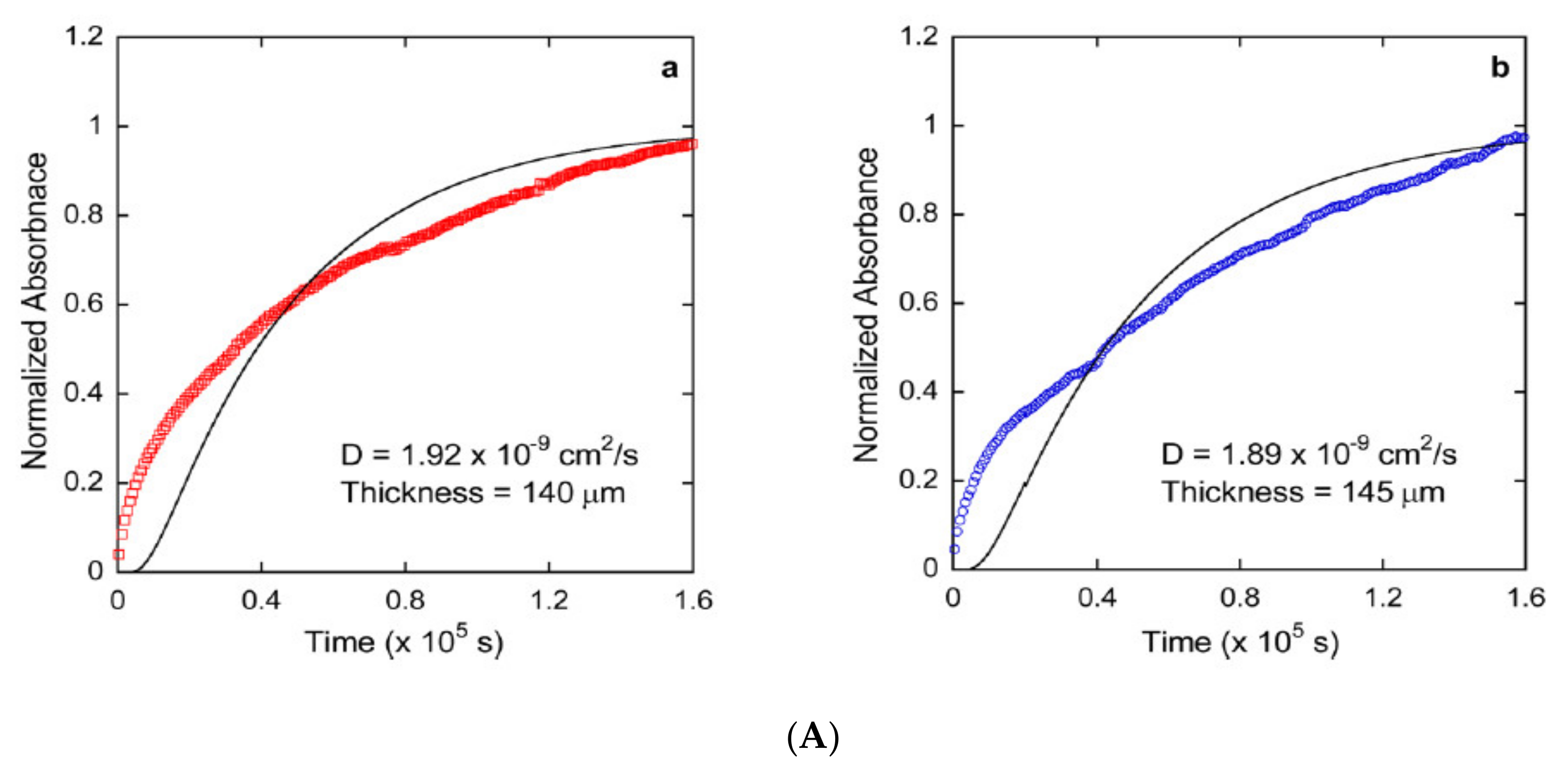
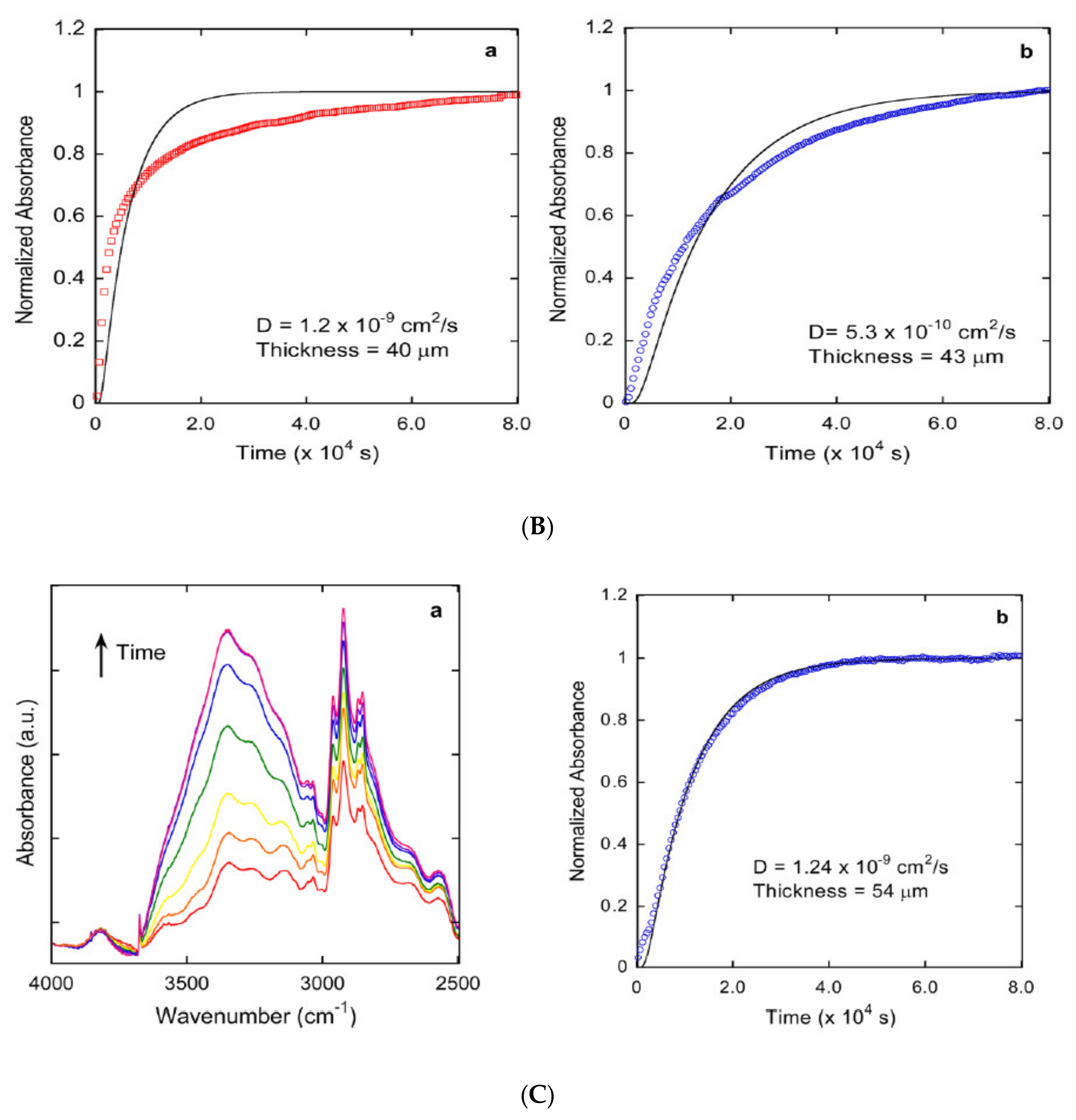
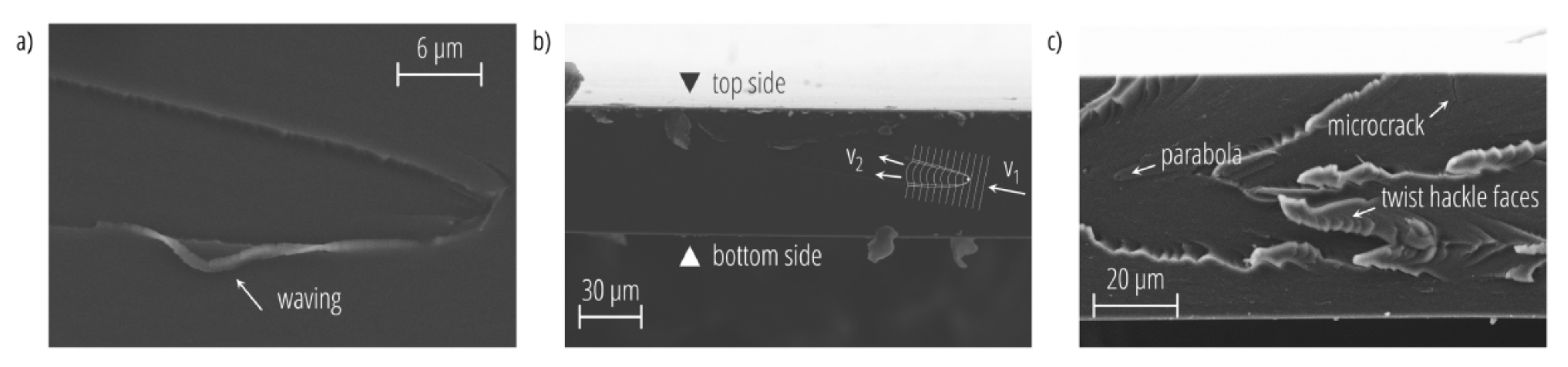
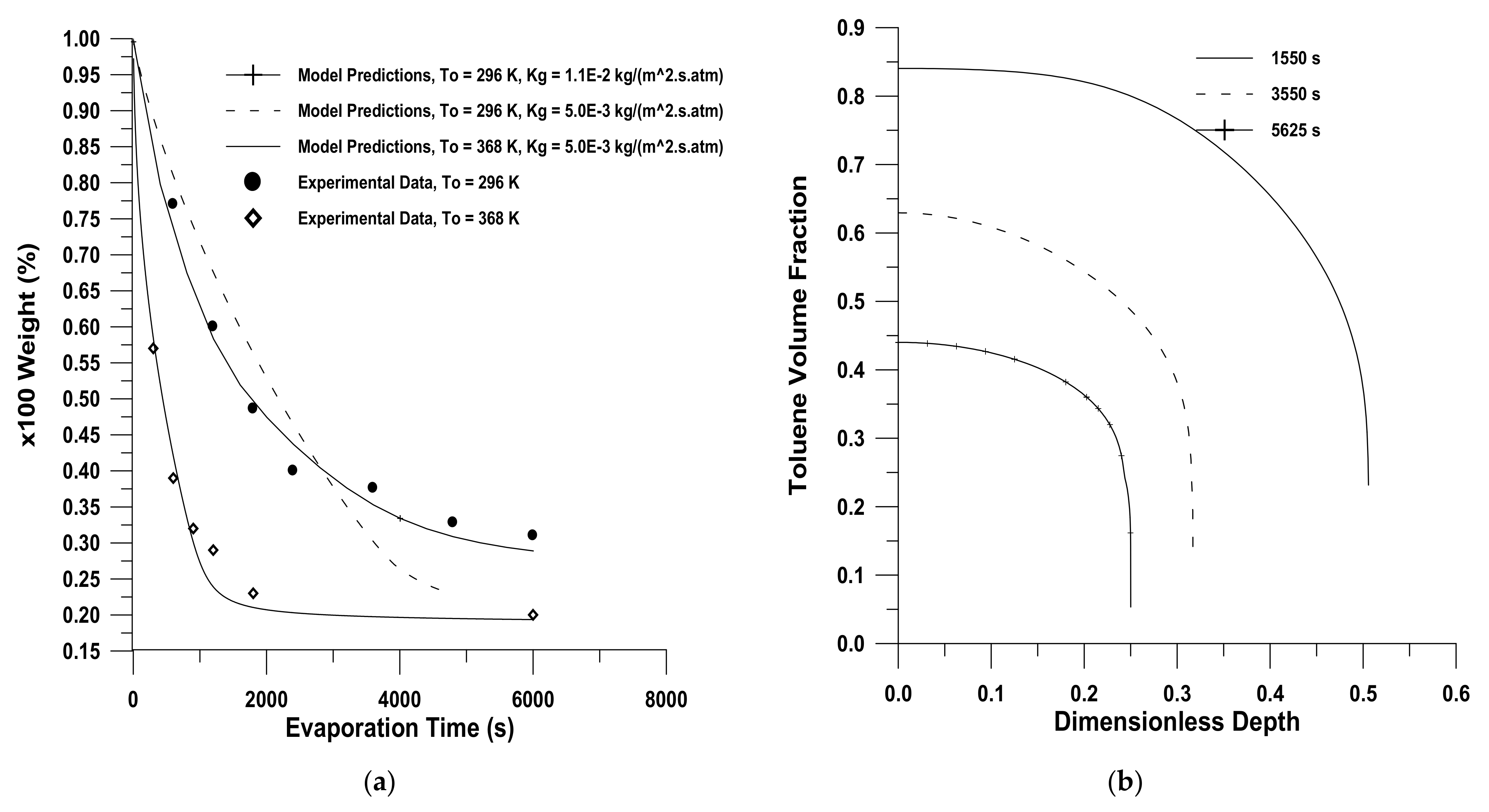
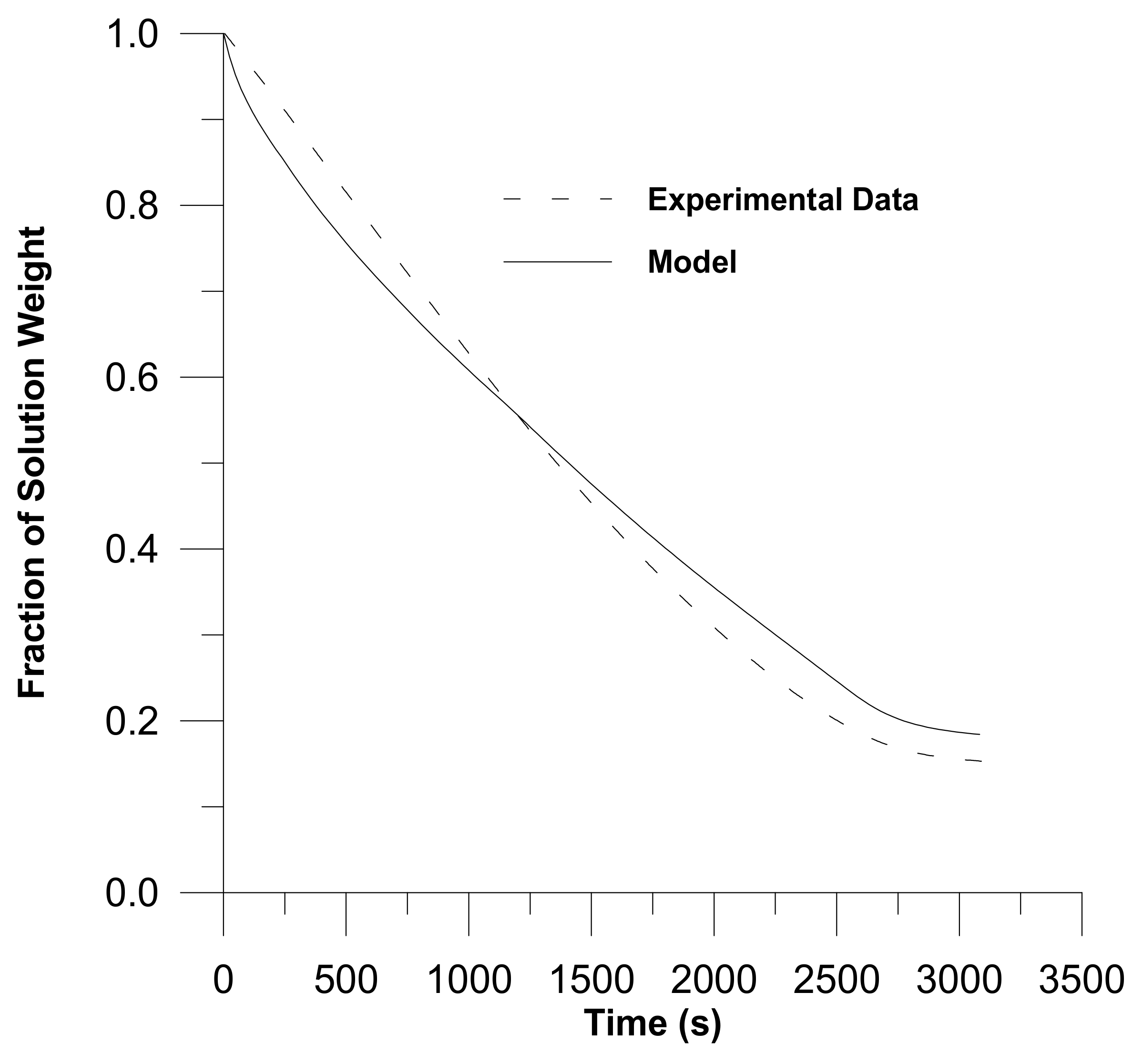
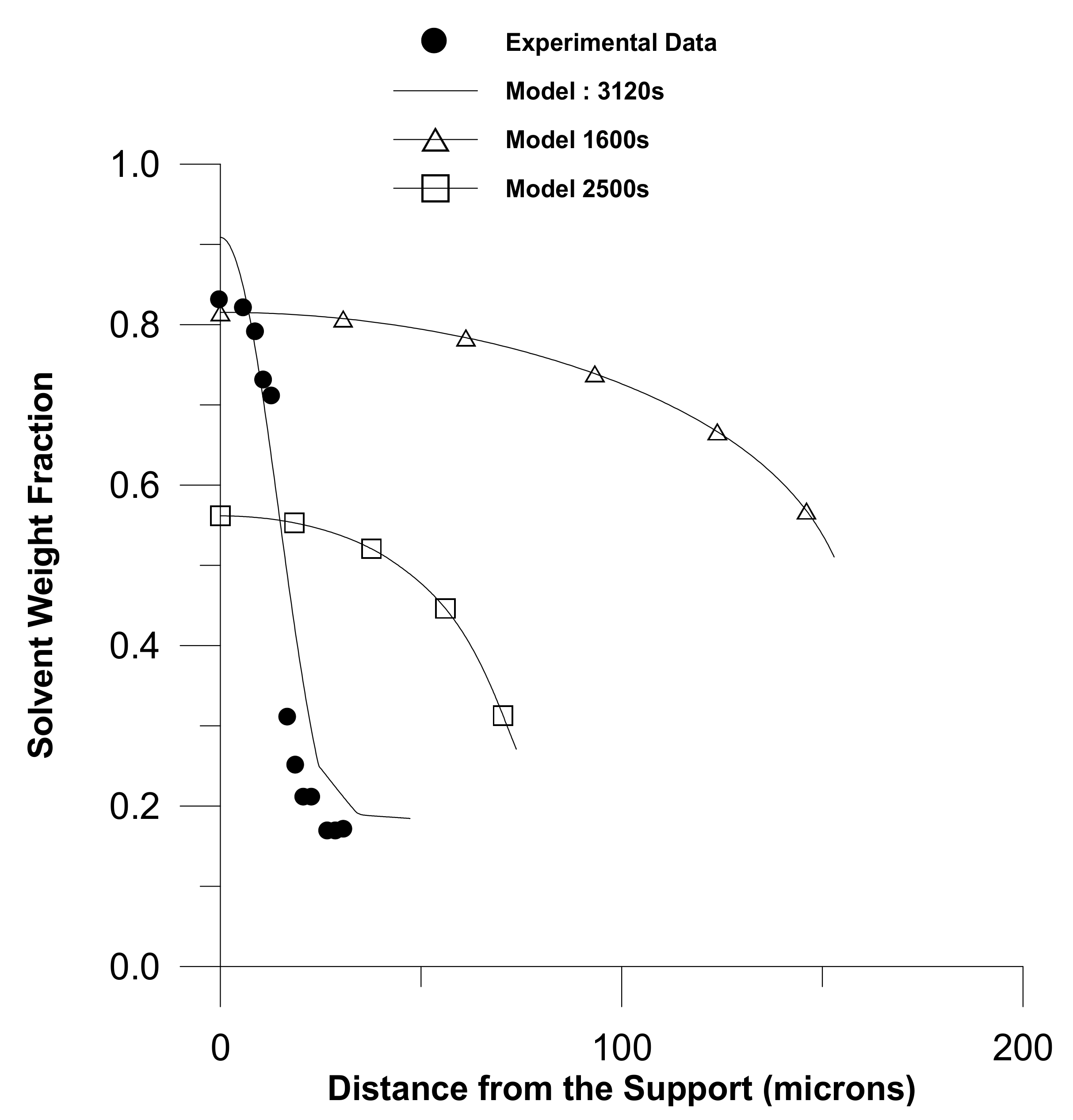
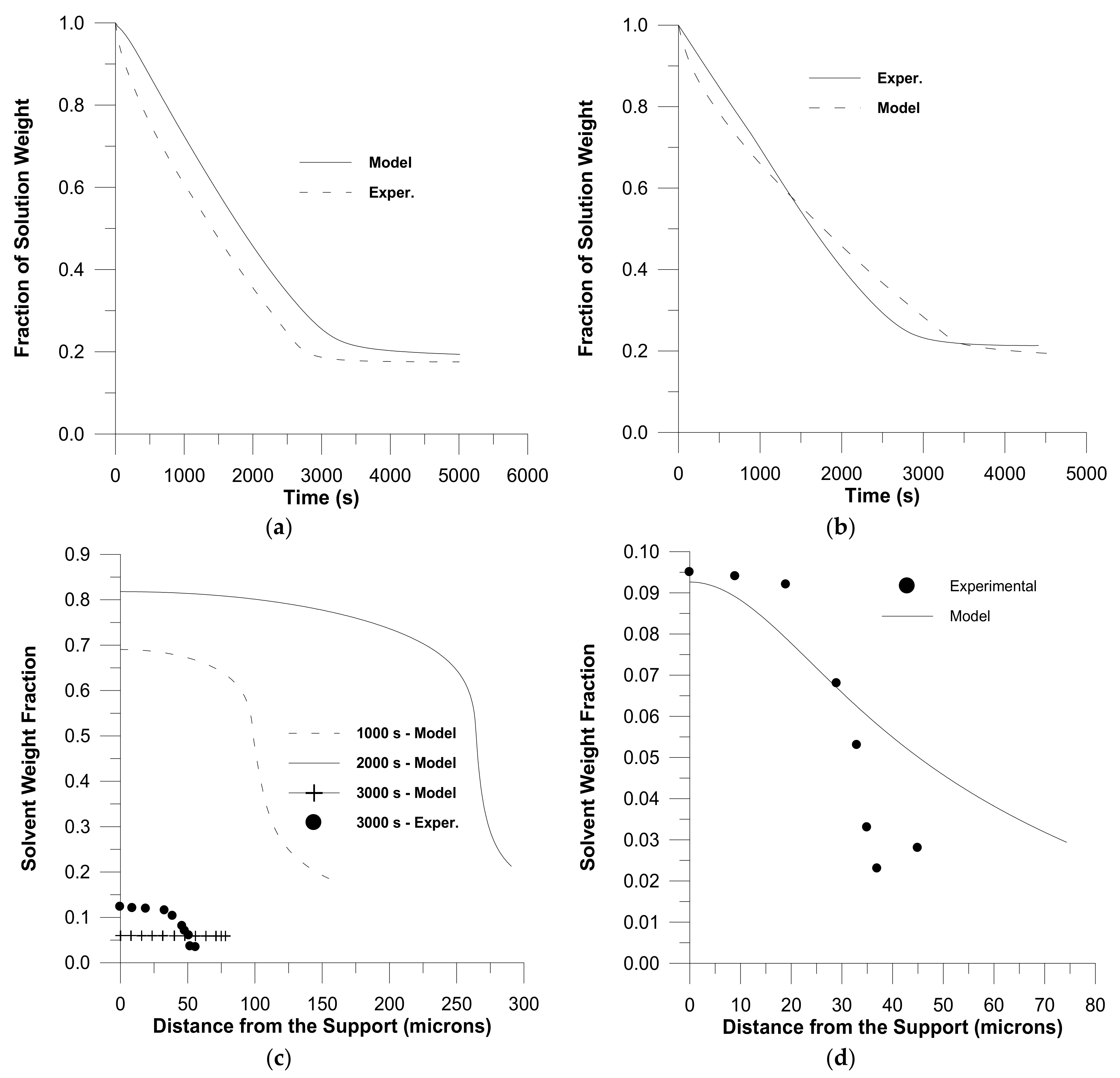
2. Diffusion in Glassy Polymer Systems
3. Sorption in Glassy Polymers
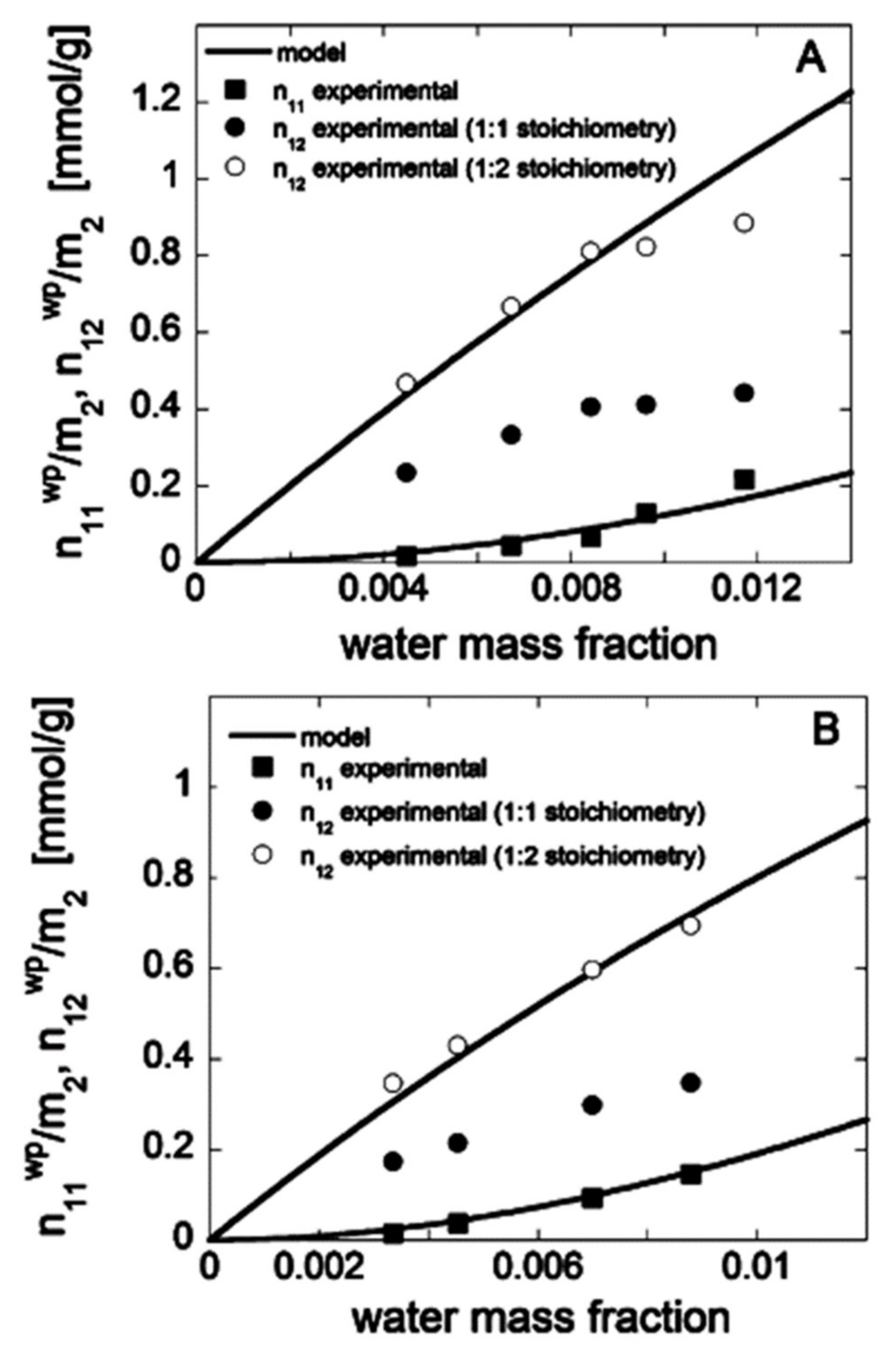
3.1. Different Sorption Models
- In a glassy polymer material, there are two types of sorption sites: one is in the matrix region of the polymer, and the other is in the micro-voids;
- The partition functions of all the molecules in the polymer matrix region are the same;
- GAB sorption occurs in the polymer micro-void site;
- In the micro-void region, the molecules in layers other than the first have the same partition function as those in the matrix region. The new dual-mode sorption equation was created based on these assumptions:
3.2. Hysteresis Effect of Sorption
4. Applications of Glassy Polymers
4.1. Applications in Drug Delivery Systems
4.2. Applications in Coatings
4.2.1. Corrosion Prevention
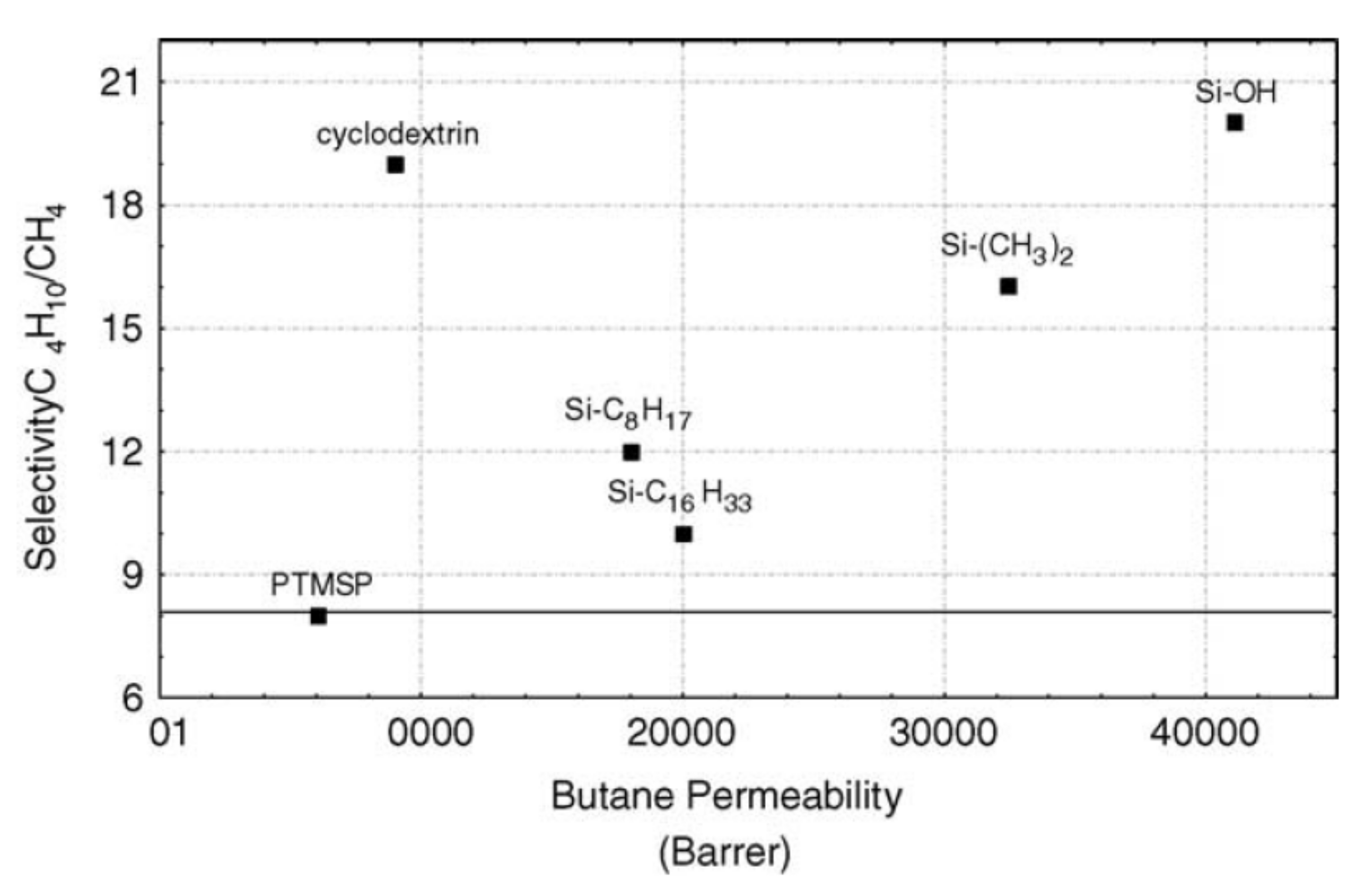
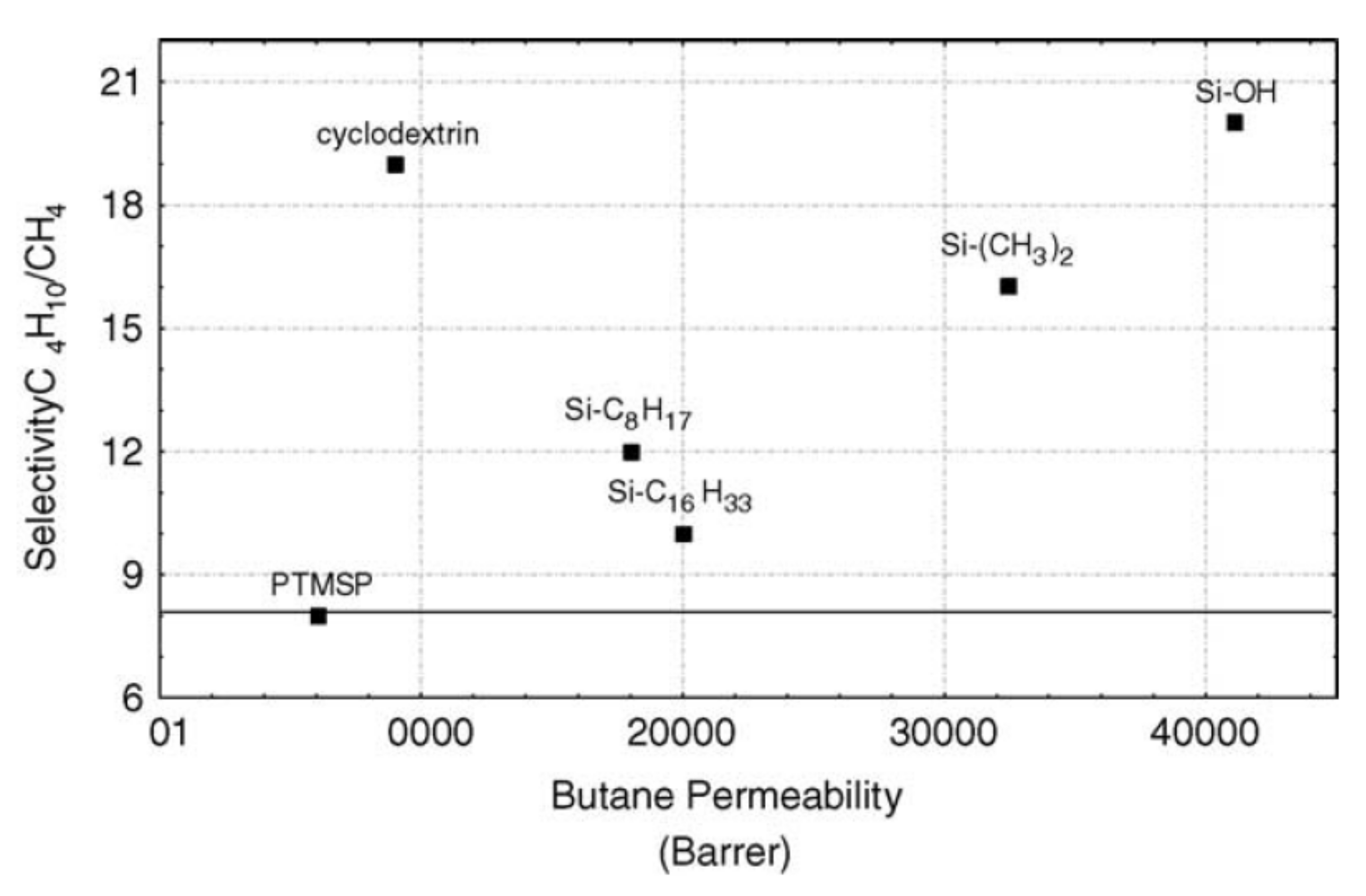
4.2.2. Gas-Separation Membranes
4.3. Applications as Polymer Matrix in Composites
4.4. Miscellaneous Applications of Glassy Polymers
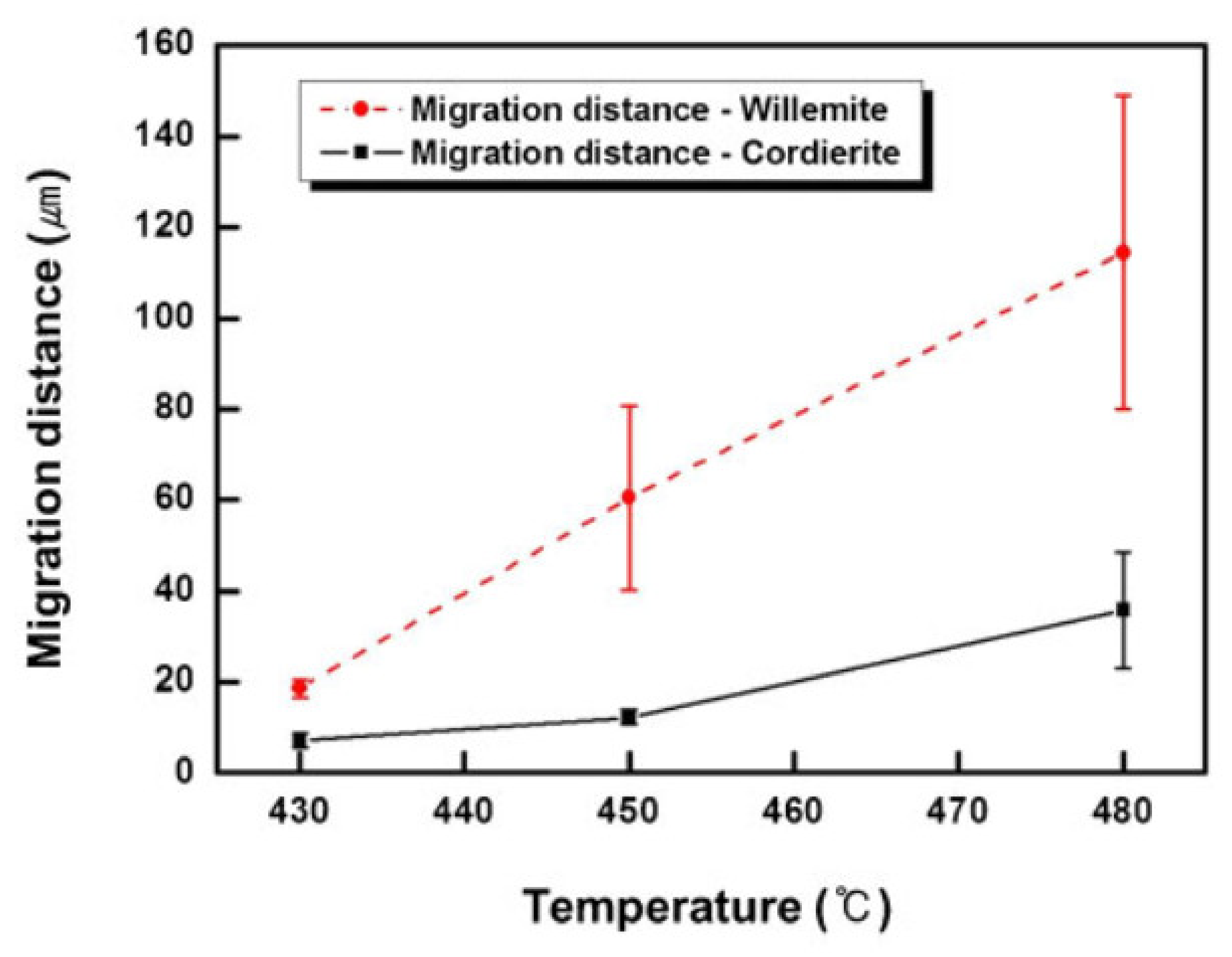
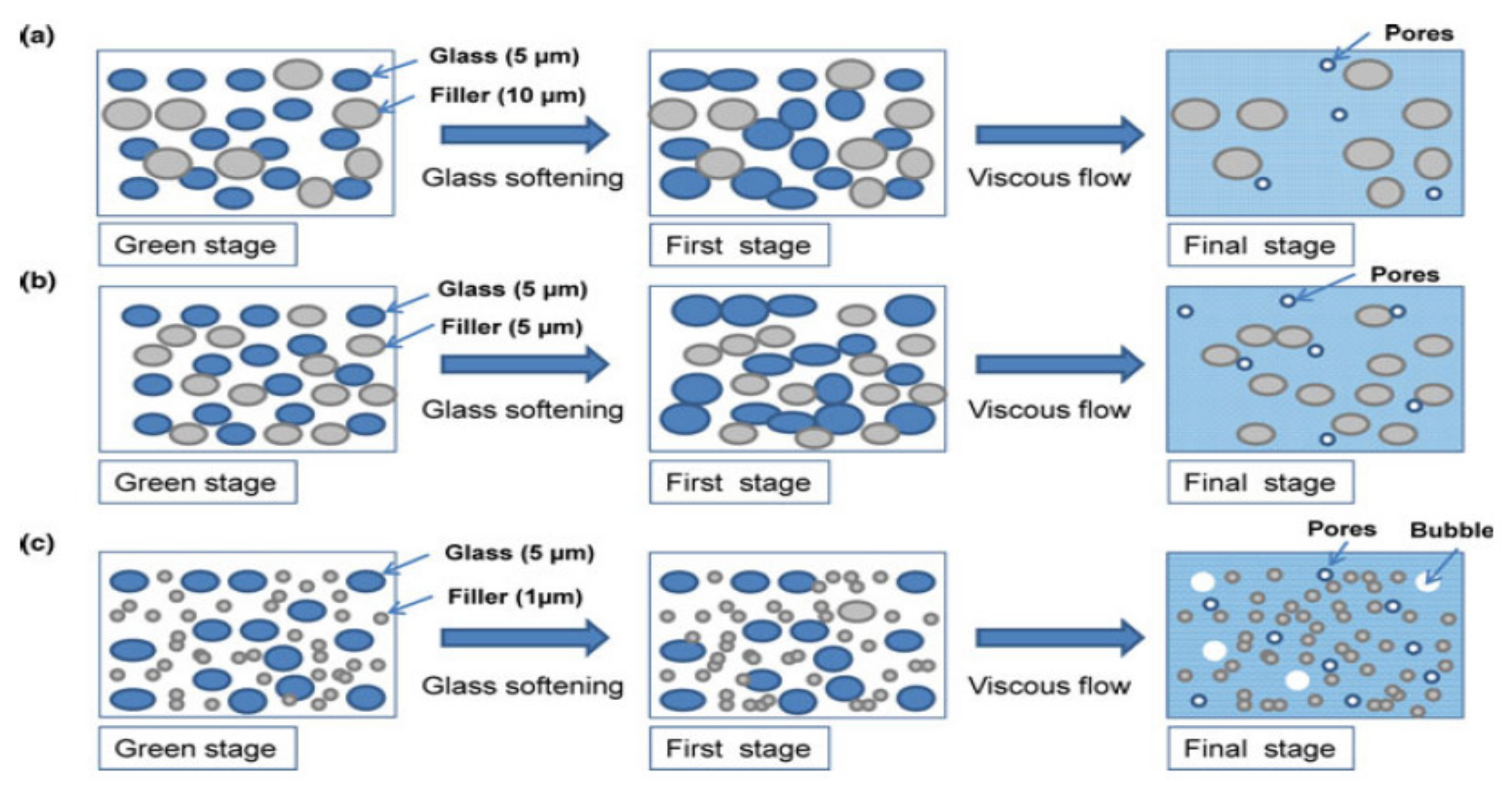
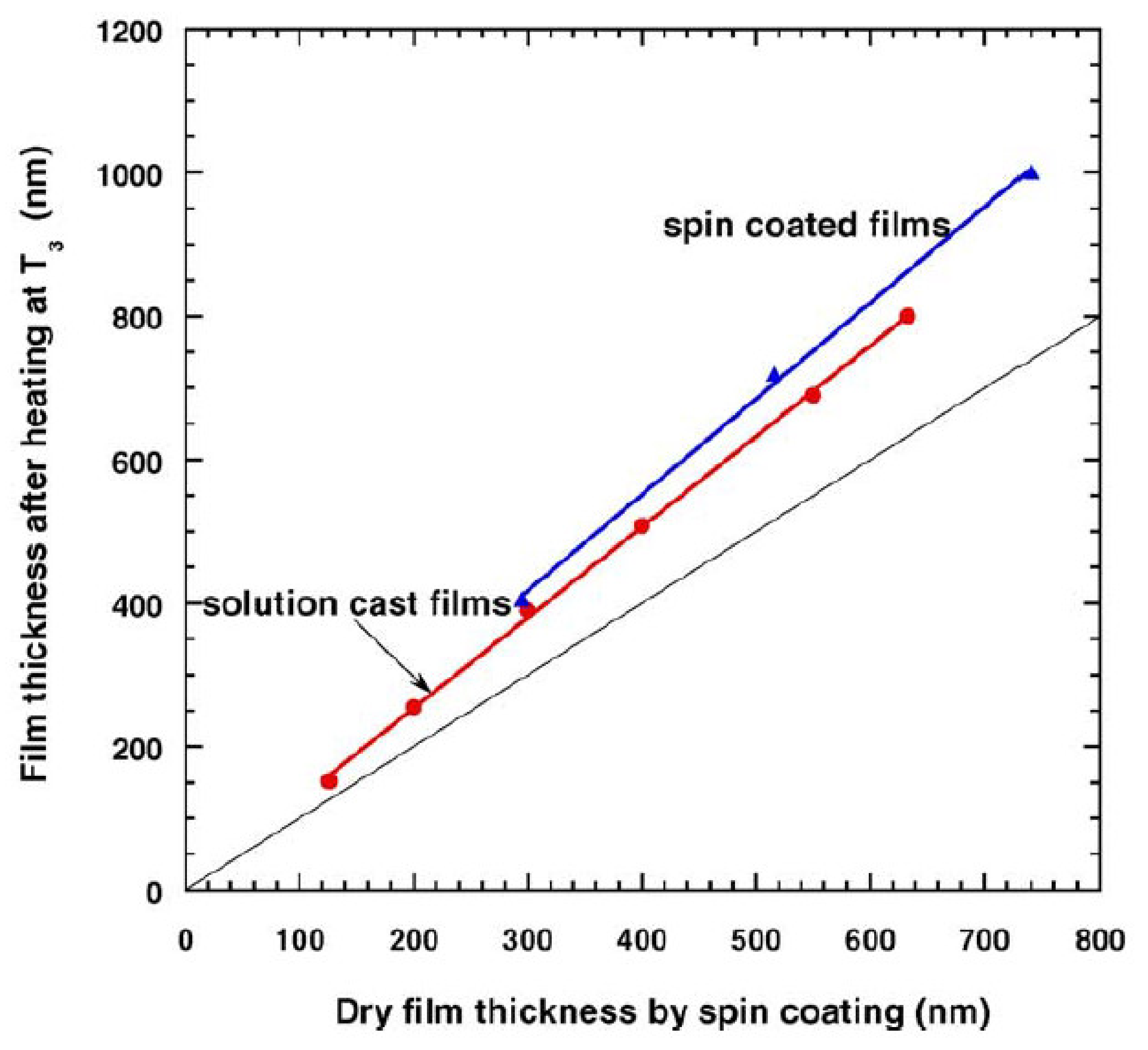
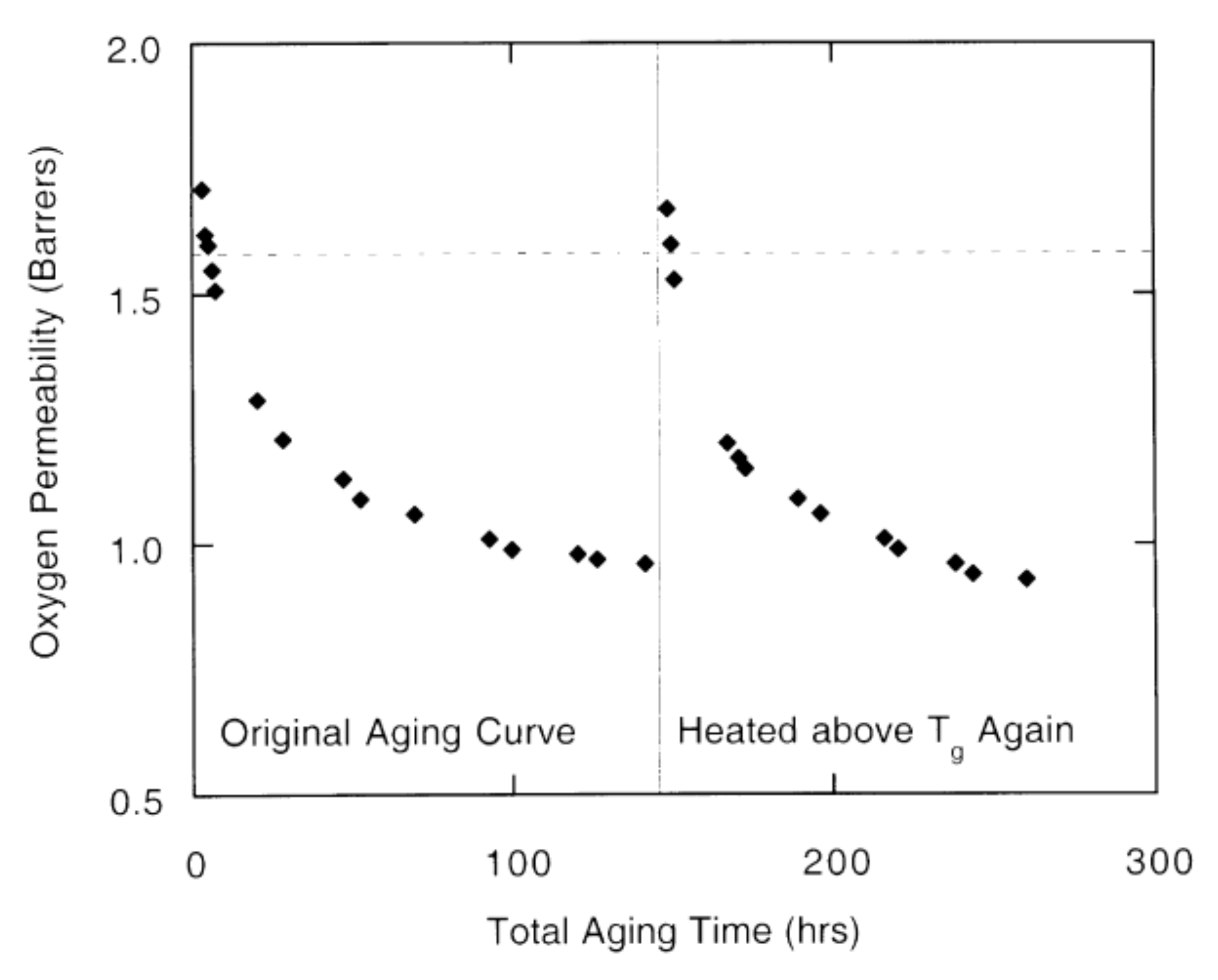
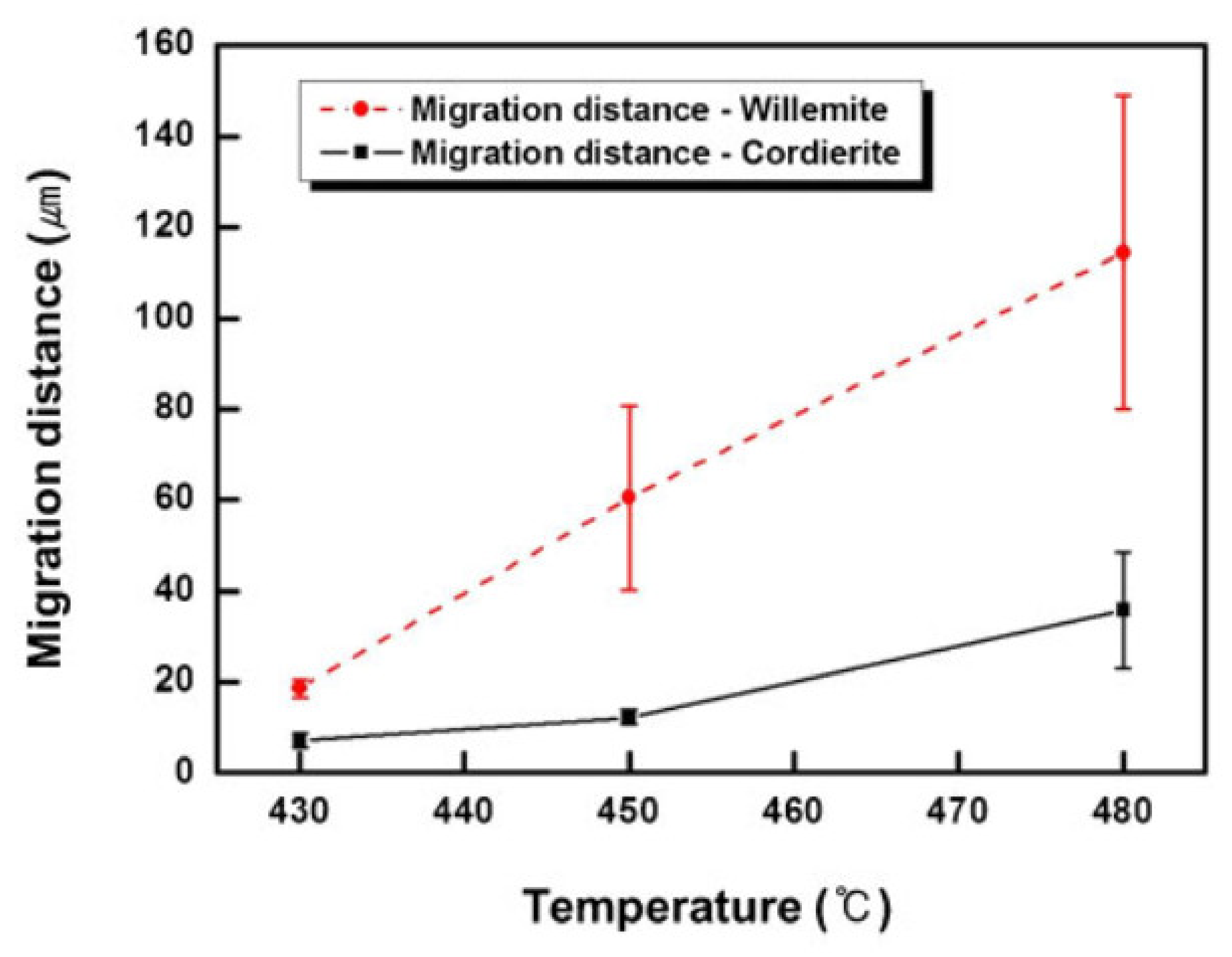
5. Physical Ageing in Glassy Polymers Coatings
6. Conclusions
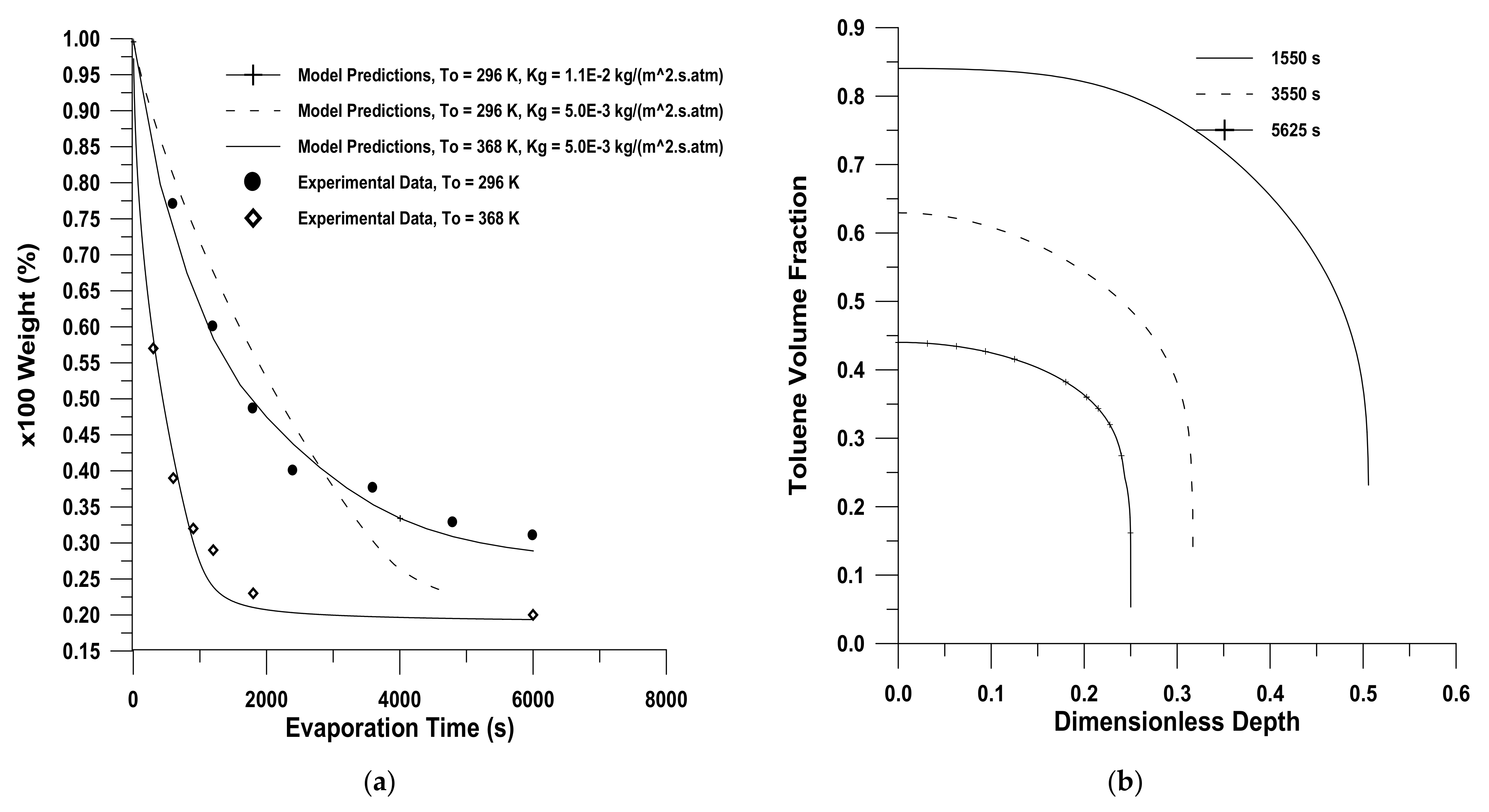
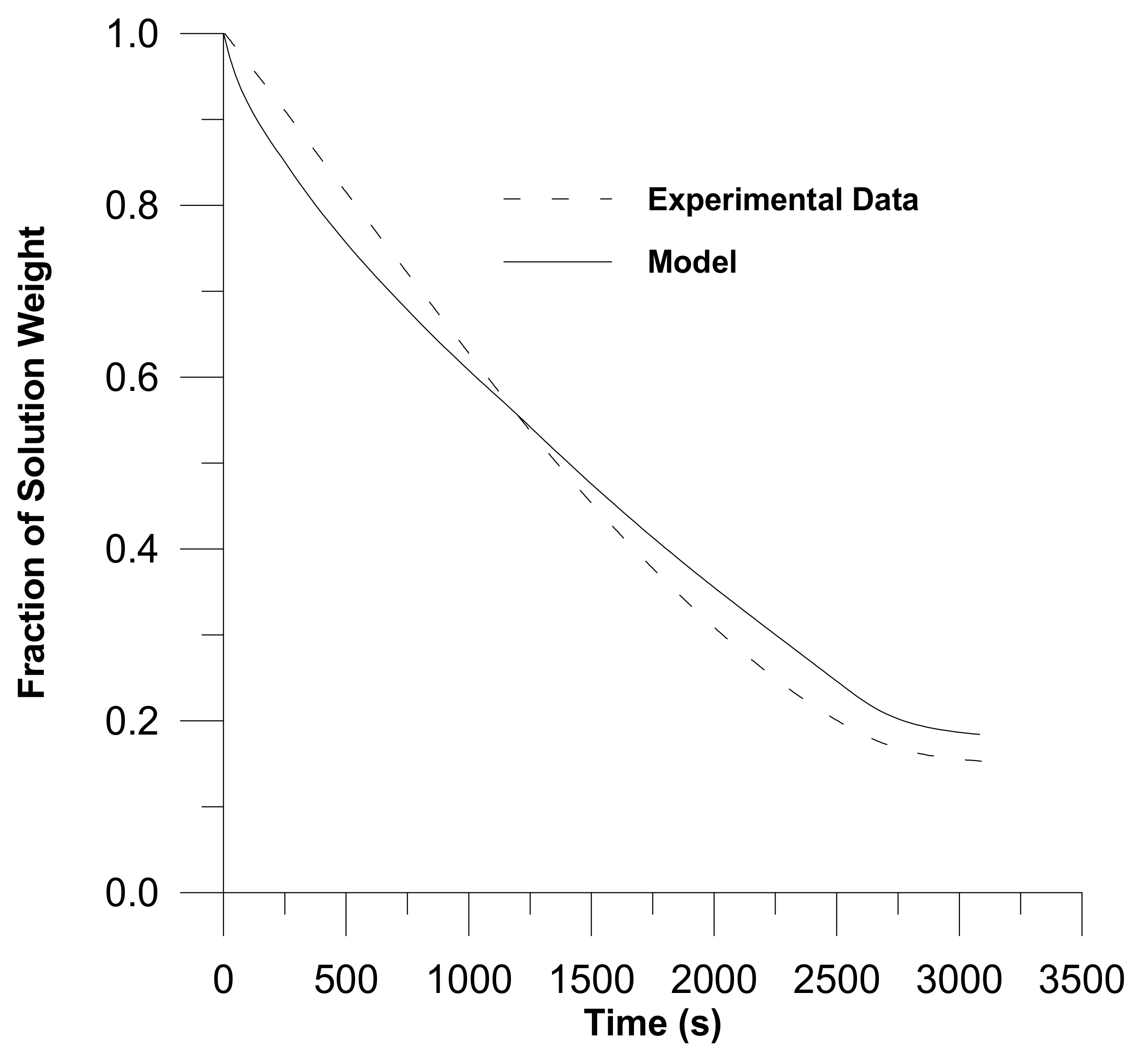
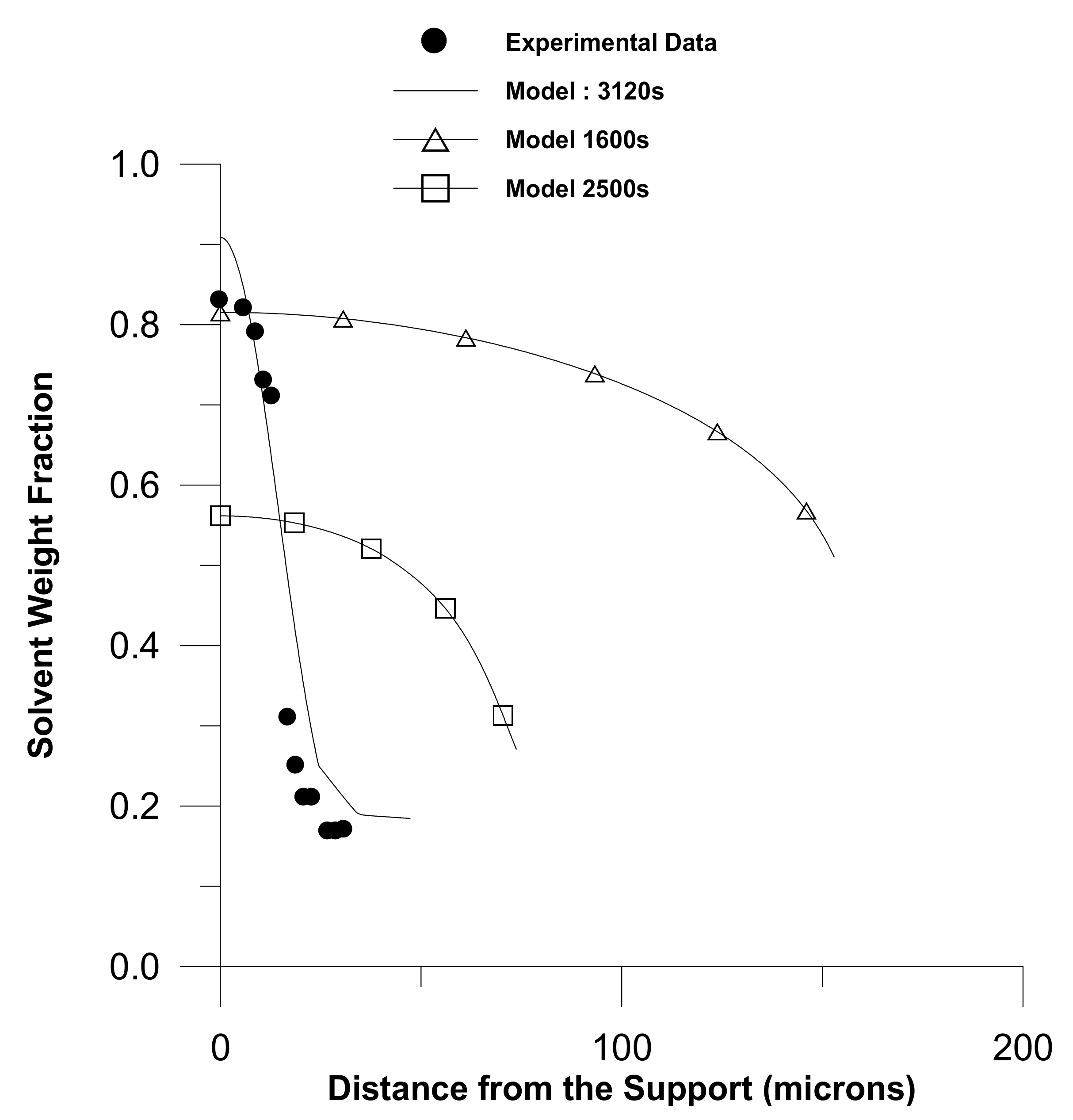
- Sufficient literature is available on diffusion in glassy polymers. Models of diffusion in glassy polymers are governed by Fickian and non-Fickian diffusion, out of which non-Fickian is more common in glassy polymers. The characteristics of the diffusants, the polymer network, and the solvents all have a role in diffusion. When a penetrant is a gas at the required temperature, predicting volumetric behaviour for the glassy–polymer–penetrant system becomes more challenging. However, none of the available models are uniformly applicable in all cases for the entire drying process. Furthermore, comparative studies of various models are missing in the literature;
- The properties of glassy polymers are dependent on sorption isotherms, and to explain the sorption in glassy polymers, there are various models explored in the literature [43,44,45,47,49,50,53,59,63]. The dual-mode sorption model, which is based on sorption sites that obey Henry’s law dissolution and Langmuir-type sorption; the GAB model, which is an extension of the BET method for multilayer adsorption of small molecules in a solid adsorbent; and the NET-GP model, which was developed after investigating non-Fickian diffusion in glassy polymers and molecular simulation techniques, have all been reviewed in this work;
- Characteristics of glassy polymers are dependent on diffusion and sorption; therefore, there is a need for an extension and modification of existing models. There are limitations to the use of physical models, and further research is needed to generalise the models which can be executed efficiently. Machine learning techniques appear to be extremely promising for developing diffusion and sorption models, and they should be further investigated;
- Glassy polymer coatings have wide applications, including membrane separation, drug delivery systems, corrosion prevention, and many more which have been explained in this review. Use of multicomponent coatings, such as binary and tertiary coatings, along with materials such as surfactants, nanoparticles which enhance the properties of glassy polymers need to be explored. The area of green synthesis and biodegradable glassy polymers also needs to be explored to make the applications more environmentally friendly;
- Despite having so many applications, there is still a challenge for current researchers to simulate real-life coating problems due to issues such as physical ageing which have briefly been described in the review. There is still a scope to explore those glassy polymers which are more durable against physical ageing. Emphasis should be given to the crosslinking of glassy polymers because they are effective against physical ageing.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| a | penetrant activity in a polymer in the GAB model, dimensionless |
| A | temperature-dependent constant in the GAB model, dimensionless |
| A0 | pre-exponential factor of a temperature-dependent constant in the GAB model, dimensionless |
| temperature-dependent constant in the new dual-mode sorption model, dimensionless | |
| b | the Langmuir hole affinity parameter, atm−1 |
| C | the total gas concentration in a glassy polymer, cm3/cm3 polymer |
| CD | the gas concentration based on Henry’s law sorption, cm3/cm3 polymer |
| CH | the gas concentration based on Langmuir sorption, cm3/cm3 polymer |
| the capacity parameter, cm3/cm3 | |
| Cp | monolayer sorption capacity, dimensionless |
| weighted mean value of polymer sorption capacity to sorbate molecules, dimensionless | |
| D01 | pre-exponential factor independent of temperature, m2·s−1 |
| D | binary diffusion coefficient, m2·s−1 |
| D1 | solvent self-diffusion coefficient, m2·s−1 |
| thermodynamic diffusion coefficient for solute mass flux in the solute–polymer mixture, m2·s−1 | |
| HL | heat of condensation of a pure vapor, Kcal/gmol |
| Hm | heat of sorption of monolayer of a vapor, Kcal/gmol |
| Hn | heat of sorption of multi-molecular layers of a vapor, Kcal/gmol |
| diffusive solute flux in the solute-polymer mixture, Kg·m2s−1 | |
| k | temperature-dependent constant in the GAB model, dimensionless |
| Kg | mass transfer coefficient at the coating-air interface (kg·m−2 s−1·atm−1) |
| K0 | pre-exponential factor of a temperature-dependent constant in the GAB model, dimensionless |
| temperature-dependent constant in the new DMS model, dimensionless | |
| kD | the Henry’s law coefficient, cm3/cm3·atm |
| L | Lorentz–Lorenz parameter, dimensionless |
| p | applied gas pressure, atm |
| P | pressure, atm |
| characteristic pressure of pure component 1, atm | |
| characteristic pressure of pure component 2, atm | |
| R | gas constant, Jmol−1·K−1 |
| number of lattice sites occupied by a mole of pure component 1, dimensionless | |
| T | temperature, K |
| Tg | glass transition temperature, K |
| characteristic temperature of pure component 1, K | |
| u1 | solvent volume fraction, dimensionless |
| specific hole-free volume for a diffusional step of component 2, m3/kg | |
| volume occupied by a mole of lattice sites of pure substance, cm3 | |
| the specific hole-free volume, m3/Kg | |
| x∞ | solvent mole fraction at the air phase (bulk) |
Greek Letters
| nonequilibrium solute chemical potential in the solute–polymer mixture, J·kg−1 | |
| μ1 | solvent chemical potential, J·kg−1 |
| solute mass per unit volume in the solute–polymer mixture, kg·m−3 | |
| polymer mass per unit volume in the solute–polymer mixture, kg·m−3 | |
| density of the polymer, kg·m−3 | |
| characteristic density of pure component 2, kg·m−3 | |
| van der Waals volume fractions of the group “i” in the polymer matrix, dimensionless | |
| solute mass fraction in the solute–polymer mixture, dimensionless |
References
- Sharma, J.; Ahuja, S.; Arya, R.K. Effect of molecular weight on morphology and thermal properties of poly (styrene)-poly (methyl methacrylate)-ethylbenzene coatings. Prog. Org. Coat. 2019, 132, 468–474. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An overview of polylactides as packaging materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef] [PubMed]
- Bergström, J.S.; Hayman, D. An overview of mechanical properties and material modeling of polylactide (PLA) for medical. Ann. Biomed. Eng. 2016, 44, 330–340. [Google Scholar] [CrossRef]
- Srinivasan, R.; Auvil, S.R.; Burban, P.M. Elucidating the mechanism(s) of gas transport in poly[1-(trimethylsilyl)-1-propyne] (PTMSP) membranes. J. Membr. Sci. 1994, 86, 67–86. [Google Scholar] [CrossRef]
- Ichiraku, Y.; Stern, S.A.; Nakagawa, T. An investigation of the high gas permeability of poly (1-Trimethylsilyl-1-Propyne). J. Membr. Sci. 1987, 34, 5–18. [Google Scholar] [CrossRef]
- Zeng, M.; Feng, Z.; Huang, Y.; Liu, J.; Ren, J.; Xu, Q.; Fan, L. Chemical structure and remarkably enhanced mechanical properties of chitosan-graft-poly(acrylic acid)/polyacrylamide double-network hydrogels. Polym. Bull. 2017, 74, 55–74. [Google Scholar] [CrossRef]
- Morisato, A.; Pinnau, I. Synthesis and gas permeation properties of poly(4-methyl-2-pentyne). J. Membr. Sci. 1996, 121, 243–250. [Google Scholar] [CrossRef]
- Budd, P.M.; Elabas, E.S.; Ghanem, B.S.; Makhseed, S.; McKeown, N.B.; Msayib, K.J.; Tattershall, C.E.; Wang, D. Solution-processed, organophilic membrane derived from a polymer of intrinsic microporosity. Adv. Mater. 2004, 16, 456–459. [Google Scholar] [CrossRef]
- Robeson, L.M.; Liu, Q.; Freeman, B.D.; Paul, D.R. Comparison of transport properties of rubbery and glassy polymers and the relevance to the upper bound relationship. J. Membr. Sci. 2015, 476, 421–431. [Google Scholar] [CrossRef]
- Hadj Romdhane, I.; Price, P.E.; Miller, C.A.; Benson, P.T.; Wang, S. Drying of glassy polymer films. Ind. Eng. Chem. Res. 2001, 40, 3065–3075. [Google Scholar] [CrossRef]
- Vrentas, J.; Duda, J. Diffusion in polymer–solvent systems. I. Reexamination of the free-volume theory. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 403–416. [Google Scholar] [CrossRef]
- Vrentas, J.; Duda, J. Diffusion in polymer–solvent systems. II. A predictive theory for the dependence of diffusion coefficients on temperature, concentration, and molecular weight. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 417–439. [Google Scholar] [CrossRef]
- Yapel, R.A. A Physical Model of the Drying of Coated Films. Master’s Thesis, University of Minnesota, Minneapolis, MN, USA, 1988. [Google Scholar]
- Alsoy, S.; Duda, J. Drying of solvent coated polymer films. Dry. Technol. 1998, 16, 15–44. [Google Scholar] [CrossRef]
- Alsoy, S. Predicting drying in multiple-zone ovens. Ind. Eng. Chem. Res. 2001, 40, 2995–3001. [Google Scholar] [CrossRef] [Green Version]
- Schabel, W.; Scharfer, P.; Kind, M. Measurement and simulation of concentration profiles during drying of thin films with help of confocal-micro-raman spectroscopy. Chem. Ing. Tech. 2003, 75, 1105–1106. [Google Scholar] [CrossRef]
- Schabel, W.; Scharfer, P.; Muller, M.; Ludwig, I.; Kind, M. Measurement and simulation of concentration profiles in the drying of binary polymer solutions. Chem. Ing. Tech. 2003, 75, 1336–1344. [Google Scholar] [CrossRef]
- Scharfer, P.; Schabel, W.; Kind, M. Modelling of alcohol and water diffusion in fuel cell membranes—Experimental validation by means of in situ Raman spectroscopy. Chem. Eng. Sci. 2008, 63, 4676–4684. [Google Scholar] [CrossRef]
- Arya, R.K. Measurement of concentration profiles in thin film binary polymer-solvent coatings using confocal Raman spectroscopy: Free volume model validation. Dry. Technol. 2014, 32, 992–1002. [Google Scholar] [CrossRef]
- Arya, R.K.; Vinjamur, M. Measurement of concentration profiles using confocal Raman spectroscopy in multicomponent polymeric coatings—Model validation. J. Appl. Polym. Sci. 2013, 128, 3906–3918. [Google Scholar] [CrossRef]
- Siebel, D.; Scharfer, P.; Schabel, W. Prediction of diffusion in a ternary solvent–solvent–polymer blend by means of binary diffusion data: Comparison of experimental data and simulative results. J. Appl. Polym. Sci. 2016, 133, 43899. [Google Scholar] [CrossRef]
- Zhang, C.; Cappleman, B.; Defibaugh-Chavez, M.; Weinkauf, D. Glassy polymer-sorption phenomena measured with a quartz crystal microbalance technique. J. Polym. Sci. Part. B Polym. Phys. 2003, 41, 2109–2118. [Google Scholar] [CrossRef]
- Masaro, L.; Zhu, X.X. Physical models of diffusion for polymer solutions, gels and solids. Prog. Polym. Sci. 1999, 24, 731–775. [Google Scholar] [CrossRef]
- Grinsted, R.A.; Clark, L.; Koenig, J.L. Study of cyclic sorption-desorption into poly(methyl methacrylate) rods using NMR imaging. Macromolecules 1992, 25, 1235–1241. [Google Scholar] [CrossRef]
- Weisenberger, L.A.; Koenig, J.L. NMR imaging of diffusion processes in polymers: Measurement of the spatial dependence of solvent mobility in partially swollen PMMA rods. Macromolecules 1990, 23, 2445–2453. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. Volumetric behavior of glassy polymer-penetrant systems. Macromolecules 1989, 22, 2264–2266. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Duda, J.L.; Ling, H.C. Antiplasticization and volumetric behavior in glassy polymers. Macromolecules 1988, 21, 1470–1475. [Google Scholar] [CrossRef]
- Laksmana, F.L.; Hartman Kok, P.J.A.; Vromans, H.; Van der Voort Maarschalk, K. Predicting the diffusion coefficient of water vapor through glassy HPMC films at different environmental conditions using the free volume additivity approach. Eur. J. Pharm. Sci. 2009, 37, 545–554. [Google Scholar] [CrossRef]
- Duda, J.L.; Hadj Romdhane, I.; Danner, R.P. Diffusion in glassy polymers—Relaxation and antiplasticization. J. Non-Cryst. Solids 1994, 172, 715–720. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Duda, J.L. Molecular diffusion in polymer solutions. AIChE J. 1979, 25, 1–24. [Google Scholar] [CrossRef]
- Wang, B.-G.; Yamaguchi, T.; Nakao, S.-I. Solvent diffusion in amorphous glassy polymers. J. Polym. Sci. Part. B Polym. Phys. 2000, 38, 846–856. [Google Scholar] [CrossRef]
- Verros, G.D. Application of irreversible thermodynamics to the solvent diffusion in an amorphous glassy polymer: A comprehensive model for drying of toluene-poly(methyl methacrylate) coatings. Can. J. Chem. Eng. 2015, 93, 2298–2306. [Google Scholar] [CrossRef]
- Powers, G.W.; Collier, J.R. Experimental modeling of solvent-casting thin polymer films. Polym. Eng. Sci. 1990, 30, 118–123. [Google Scholar] [CrossRef]
- Arya, R.K. Finite element solution of coupled-partial differential and ordinary equations in multicomponent polymeric coatings. Comput. Chem. Eng. 2013, 50, 152–183. [Google Scholar] [CrossRef]
- Sharma, J.; Arya, R.K.; Verros, G.D. A unified model for the drying of glassy polymer coatings. Prog. Org. Coat. 2019, 134, 219–225. [Google Scholar] [CrossRef]
- Alsoy, S.; Duda, J.L. Modeling of multicomponent drying of polymer films. AIChE J. 1999, 45, 896–905. [Google Scholar] [CrossRef]
- Sharma, J.; Kumar Arya, R.; Verros, G.D. A comprehensive model for the drying of glassy polymer coatings: The low solvent concentration area of the system poly(styrene)/P-xylene. Prog. Org. Coat. 2019, 135, 622–628. [Google Scholar] [CrossRef]
- Davis, E.M.; Minelli, M.; Giacinti Baschetti, M.; Elabd, Y.A. Non-fickian diffusion of water in polylactide. Ind. Eng. Chem. Res. 2013, 52, 8664–8673. [Google Scholar] [CrossRef]
- Tomba, J.P.; Arzondo, L.M.; Carella, J.M.; Pastor, J.M. Liquid-glassy polymer diffusion: Effects of liquid molecular weight and temperature. Macromol. Chem. Phys. 2007, 208, 1110–1121. [Google Scholar] [CrossRef]
- Gallyamov, M.O. Sharp diffusion front in diffusion problem with change of state. Eur. Phys. J. E 2013, 36, 92. [Google Scholar] [CrossRef]
- Santos, M.C.; Bendiksen, B.; Elabd, Y.A. Diffusion of liquid water in free-standing polymer films using pressure-contact time-resolved fourier transform infrared attenuated total reflectance spectroscopy. Ind. Eng. Chem. Res. 2017, 56, 3464–3476. [Google Scholar] [CrossRef]
- Chern, R.T.; Koros, W.J.; Sanders, E.S.; Yui, R. “Second component” effects in sorption and permeation of gases in glassy polymers. J. Membr. Sci. 1983, 15, 157–169. [Google Scholar] [CrossRef]
- Tsujita, Y. Gas sorption and permeation of glassy polymers with microvoids. Prog. Polym. Sci. 2003, 28, 1377–1401. [Google Scholar] [CrossRef]
- Vieth, W.R.; Tam, P.M.; Michaels, A.S. Dual sorption mechanisms in glassy polystyrene. J. Colloid Interface Sci. 1966, 22, 360–370. [Google Scholar] [CrossRef]
- Paul, D.R. Gas sorption and transport in glassy polymers. Ber. Bunsenges. Phys. Chem. 1979, 83, 294–302. [Google Scholar] [CrossRef]
- Ricci, E.; De Angelis, M.G. Modelling mixed-gas sorption in glassy polymers for CO2 removal: A sensitivity analysis of the dual mode sorption model. Membranes 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Saberi, M.; Rouhi, P.; Teimoori, M. Estimation of dual mode sorption parameters for CO2 in the glassy polymers using group contribution approach. J. Membr. Sci. 2020, 595, 117481. [Google Scholar] [CrossRef]
- Velioğlu, S.; Tantekin-Ersolmaz, S.B. Prediction of gas permeability coefficients of copolyimides by group contribution methods. J. Membr. Sci. 2015, 480, 47–63. [Google Scholar] [CrossRef]
- Feng, H. Modeling of vapor sorption in glassy polymers using a new dual mode sorption model based on multilayer sorption theory. Polymer 2007, 48, 2988–3002. [Google Scholar] [CrossRef]
- Timmermann, E.O. A BET-like three sorption stage isotherm. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 1631–1645. [Google Scholar] [CrossRef]
- Vopička, O.; Friess, K. Analysis of gas sorption in glassy polymers with the GAB model: An alternative to the dual mode sorption model. J. Polym. Sci. Part. B Polym. Phys. 2014, 52, 1490–1495. [Google Scholar] [CrossRef]
- Carlà, V.; Hussain, Y.; Grant, C.; Sarti, G.C.; Carbonell, R.G.; Doghieri, F. Modeling sorption kinetics of carbon dioxide in glassy polymeric films using the nonequilibrium thermodynamics approach. Ind. Eng. Chem. Res. 2009, 48, 3844–3854. [Google Scholar] [CrossRef]
- Scherillo, G.; Galizia, M.; Musto, P.; Mensitieri, G. Water sorption thermodynamics in glassy and rubbery polymers: Modeling the interactional issues emerging from FTIR spectroscopy. Ind. Eng. Chem. Res. 2013, 52, 8674–8691. [Google Scholar] [CrossRef] [Green Version]
- Doghieri, F.; Sarti, G.C. Nonequilibrium lattice fluids: A predictive model for the solubility in glassy polymers. Macromolecules 1996, 29, 7885–7896. [Google Scholar] [CrossRef]
- Panayiotou, C.; Pantoula, M.; Stefanis, E.; Tsivintzelis, I.; Economou, I.G. Nonrandom hydrogen-bonding model of fluids and their mixtures. 1. Pure fluids. Ind. Eng. Chem. Res. 2004, 43, 6592–6606. [Google Scholar] [CrossRef]
- Minelli, M.; Campagnoli, S.; De Angelis, M.G.; Doghieri, F.; Sarti, G.C. Predictive model for the solubility of fluid mixtures in glassy polymers. Macromolecules 2011, 44, 4852–4862. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. An elementary molecular theory of classical fluids. Pure fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
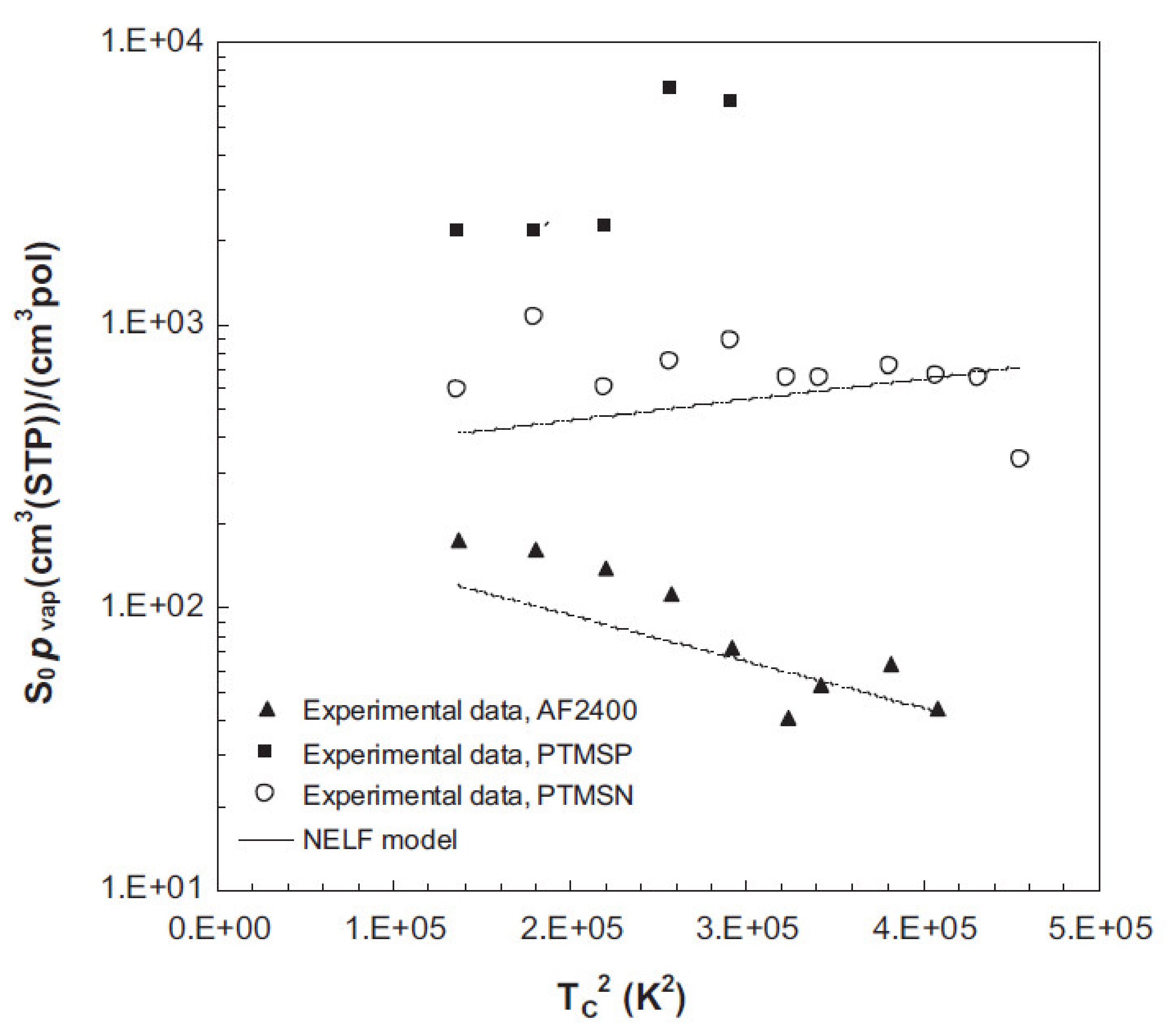
- Galizia, M.; De Angelis, M.G.; Sarti, G.C. Sorption of hydrocarbons and alcohols in addition-type poly(trimethyl silyl norbornene) and other high free volume glassy polymers. II: NELF model predictions. J. Membr. Sci. 2012, 405, 201–211. [Google Scholar] [CrossRef]
- Van der Vegt, N.F.A.; Briels, W.J.; Wessling, M.; Strathmann, H. The sorption induced glass transition in amorphous glassy polymers. J. Chem. Phys. 1999, 110, 11061–11069. [Google Scholar] [CrossRef]
- Kirchheim, R. Sorption and partial molar volume of small molecules in glassy polymers. Macromolecules 1992, 25, 6952–6960. [Google Scholar] [CrossRef]
- Spyriouni, T.; Boulougouris, G.C.; Theodorou, D.N. Prediction of Sorption of CO2 in Glassy Atactic Polystyrene at Elevated Pressures Through a New Computational Scheme. Macromolecules 2009, 42, 1759–1769. [Google Scholar] [CrossRef]
- Boulougouris, G.C.; Economou, I.G.; Theodorou, D.N. On the calculation of the chemical potential using the particle deletion scheme. Mol. Phys. 1999, 96, 905–913. [Google Scholar] [CrossRef]
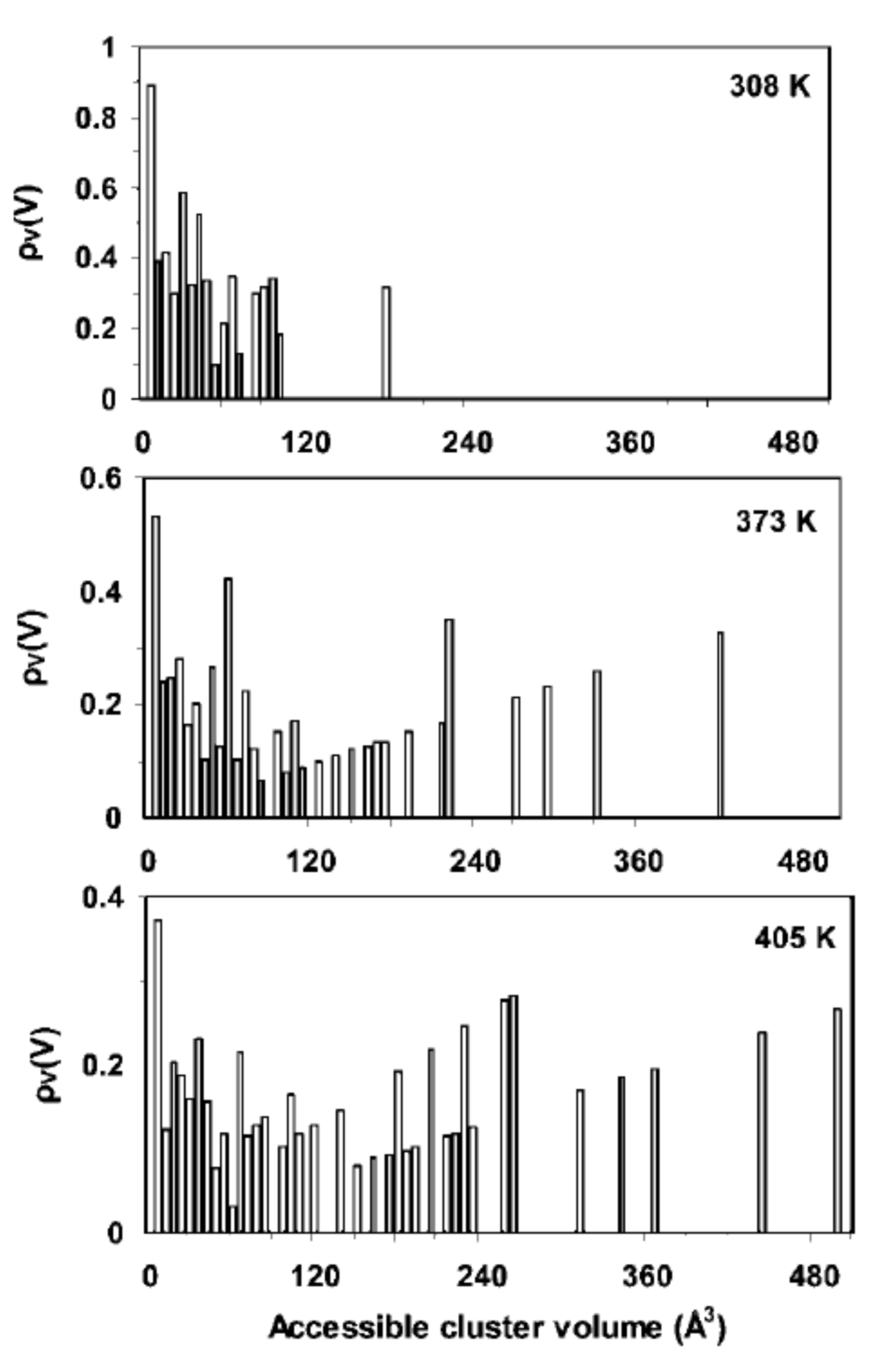
- Neyertz, S.; Brown, D. Single- and mixed-gas sorption in large-scale molecular models of glassy bulk polymers. Competitive sorption of a binary CH4/N2 and a ternary CH4/N2/CO2 mixture in a polyimide membrane. J. Membr. Sci. 2020, 614, 118478. [Google Scholar] [CrossRef]
- Stubbs, J.M.; Chen, B.; Potoff, J.J.; Siepmann, J.I. Monte Carlo calculations for the phase equilibria of alkanes, alcohols, water, and their mixtures. Fluid Phase Equilibria 2001, 183–184, 301–309. [Google Scholar] [CrossRef]
- Vergadou, N.; Theodorou, D.N. Molecular modeling investigations of sorption and diffusion of small molecules in glassy polymers. Membranes 2019, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Kamiya, Y.; Bourbon, D.; Mizoguchi, K.; Naito, Y. Sorption, dilation, and isothermal glass transition of poly(ethyl methacrylate)-organic gas systems. Polym. J. 1992, 24, 443–449. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Vrentas, C.M. Hysteresis effects for sorption in glassy polymers. Macromolecules 1996, 29, 4391–4396. [Google Scholar] [CrossRef]
- Fleming, G.K.; Koros, W.J. Dilation of polymers by sorption of carbon dioxide at elevated pressures. 1. Silicone rubber and unconditioned polycarbonate. Macromolecules 1986, 19, 2285–2291. [Google Scholar] [CrossRef]
- Hölck, O.; Heuchel, M.; Böhning, M.; Hofmann, D. Simulation of experimentally observed dilation phenomena during integral gas sorption in glassy polymers. J. Polym. Sci. Part. B Polym. Phys. 2008, 46, 59–71. [Google Scholar] [CrossRef]
- Björklund, S.; Kocherbitov, V. Water vapor sorption-desorption hysteresis in glassy surface films of mucins investigated by humidity scanning QCM-D. J. Colloid Interface Sci. 2019, 545, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Paul, D.R. Experimental methods for tracking physical aging of thin glassy polymer films by gas permeation. J. Membr. Sci. 2004, 244, 167–178. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Good, W.R. Diffusion of water soluble drugs from initially dry hydrogels. Polym. Deliv. Syst. 1976, 5, 139–156. [Google Scholar]
- Lee, P.I. Novel approach to zero-order drug delivery via immobilized nonuniform drug distribution in glassy hydrogels. J. Pharm. Sci. 1984, 73, 1344–1347. [Google Scholar] [CrossRef]
- El-Hag Ali Said, A. Radiation synthesis of interpolymer polyelectrolyte complex and its application as a carrier for colon-specific drug delivery system. Biomaterials 2005, 26, 2733–2739. [Google Scholar] [CrossRef] [PubMed]
- Abou Taleb, M.F.; Abdel-Aal, S.E.; El-Kelesh, N.A.; Hegazy, E.-S.A. Adsorption and controlled release of Chlortetracycline HCl by using multifunctional polymeric hydrogels. Eur. Polym. J. 2007, 43, 468–477. [Google Scholar] [CrossRef]
- Mazied, N.A.; Ismail, S.A.; Abou Taleb, M.F. Radiation synthesis of poly[(dimethylaminoethyl methacrylate)-co-(ethyleneglycol dimethacrylate)] hydrogels and its application as a carrier for anticancer delivery. Radiat. Phys. Chem. 2009, 78, 899. [Google Scholar] [CrossRef]
- Mullarney, M.P.; Seery, T.A.P.; Weiss, R.A. Drug diffusion in hydrophobically modified N,N-dimethylacrylamide hydrogels. Polymer 2006, 47, 3845–3855. [Google Scholar] [CrossRef]
- Rudzinski, W.E.; Chipuk, T.; Dave, A.M.; Kumbar, S.G.; Aminabhavi, T.M. PH-sensitive acrylic-based copolymeric hydrogels for the controlled release of a pesticide and a micronutrient. J. Appl. Polym. Sci. 2003, 87, 394–403. [Google Scholar] [CrossRef]
- Brazel, C.S.; Peppas, N.A. Mechanisms of solute and drug transport in relaxing, swellable, hydrophilic glassy polymers. Polymer 1999, 40, 3383–3398. [Google Scholar] [CrossRef]
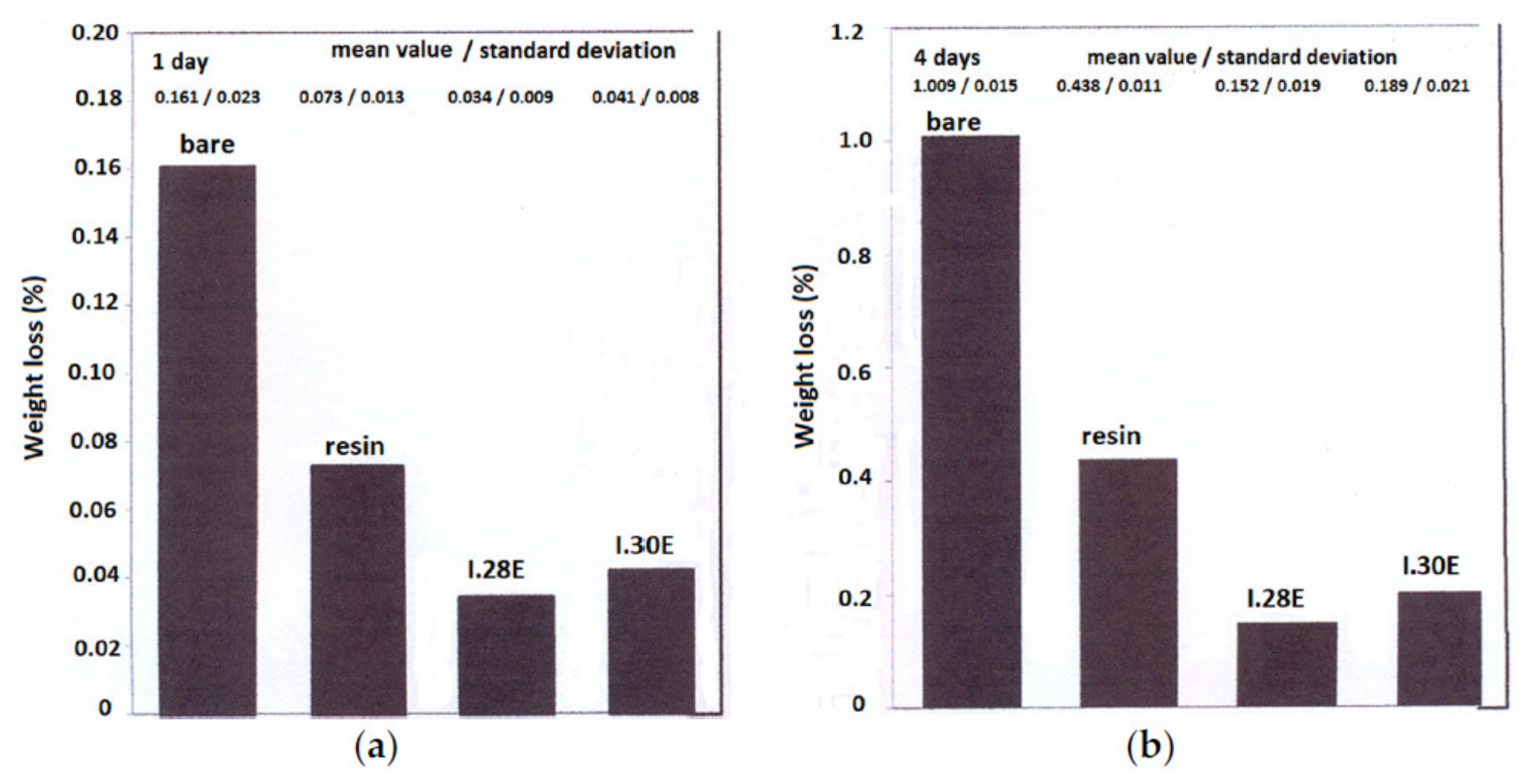
- Hill, A.J.; Thornton, A.W.; Hannink, R.H.J.; Moon, J.D.; Freeman, B.D. Role of free volume in molecular mobility and performance of glassy polymers for corrosion-protective coatings. Corros. Eng. Sci. Technol. 2020, 55, 145–158. [Google Scholar] [CrossRef]
- Vergara, J.H.; La Scala, J.J.; Henry, C.K.; Sadler, J.M.; Yadav, S.K.; Palmese, G.R. The effect of pendant alkyl chain length on the barrier properties of epoxy/amine crosslinked networks. Polymer 2017, 132, 133–142. [Google Scholar] [CrossRef]
- Merachtsaki, D.; Xidas, P.; Giannakoudakis, P.; Triantafyllidis, K.; Spathis, P. Corrosion protection of steel by epoxy-organoclay nanocomposite coatings. Coatings 2017, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Kostina, Z.I.; Krylova, S.A.; Ponurko, I.V. Production and properties of glassy metaphosphate composition for protecting the elements of water-heating systems from corrosion. Glass Ceram. 2016, 73, 71–74. [Google Scholar] [CrossRef]
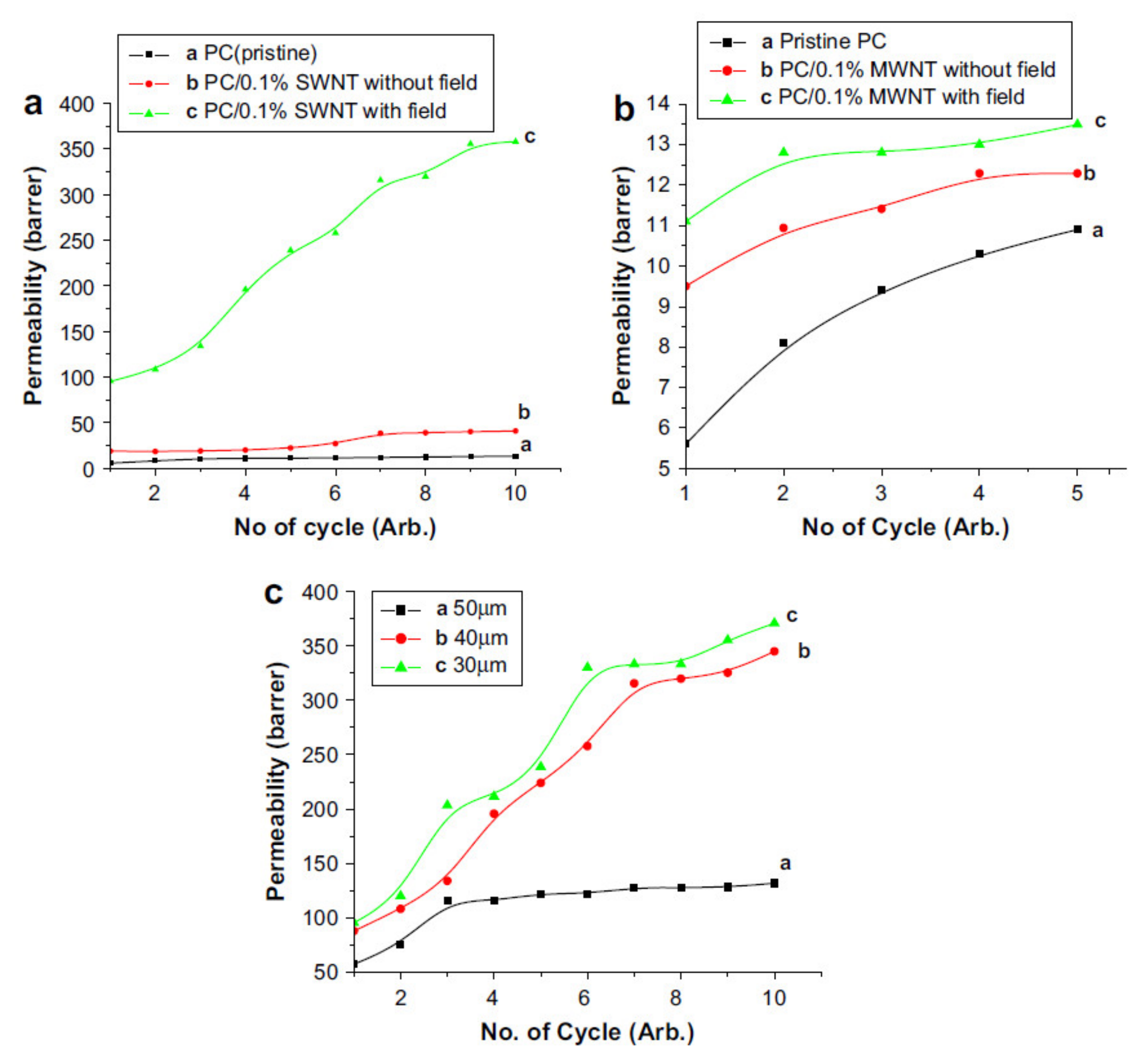
- Sharma, A.; Kumar, S.; Tripathi, B.; Singh, M.; Vijay, Y.K. Aligned CNT/Polymer nanocomposite membranes for hydrogen separation. Int. J. Hydrog. Energy 2009, 34, 3977–3982. [Google Scholar] [CrossRef]
- Park, S.; Bang, J.; Choi, J.; Lee, S.H.; Lee, J.-H.; Lee, J.S. 3-Dimensionally disordered mesoporous silica (DMS)-containing mixed matrix membranes for CO2 and non-CO2 greenhouse gas separations. Sep. Purif. Technol. 2014, 136, 286–295. [Google Scholar] [CrossRef]
- Kramer, P.W.; Murphy, M.K.; Stookey, D.J.; Henis, J.M.; Stedronsky, E.R. Membranes Having Enhanced Selectivity and Method of Producing Such Membranes. U.S. Patent 5,215,554, 1 June 1993. [Google Scholar]
- Liu, C.; Karns, N.K.; Jan, D.Y. High Flux, Cross-Linked, Fumed Silica Reinforced Polyorganosiloxane Membranes for Separations. U.S. Patent 15,685,996, 17 May 2018. [Google Scholar]
- Choi, S.H.; Al-Qahtani, M.S.; Qasem, E.A. Multilayer Aromatic Polyamide Thin-Film Composite Membranes for Separation of Gas Mixtures. U.S. Patent 10,682,606, 16 June 2020. [Google Scholar]
- Salem, A.; Ghoreyshi, A.A. Modelling of water/organic vapor dehydration by glassy polymer membranes. Desalination 2006, 193, 25–34. [Google Scholar] [CrossRef]
- Gomes, D.; Nunes, S.P.; Peinemann, K.-V. Membranes for gas separation based on poly(1-trimethylsilyl-1-propyne)–silica nanocomposites. J. Membr. Sci. 2005, 246, 13–25. [Google Scholar] [CrossRef]
- Martin, M.; Hanagud, S.; Thadhani, N.N. Mechanical behavior of nickel+aluminum powder-reinforced epoxy composites. Mater. Sci. Eng. A 2007, 443, 209–218. [Google Scholar] [CrossRef]
- Jordan, J.L.; Spowart, J.E.; Kendall, M.J.; Woodworth, B.; Siviour, C.R. Mechanics of particulate composites with glassy polymer binders in compression. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2014, 372, 20130215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
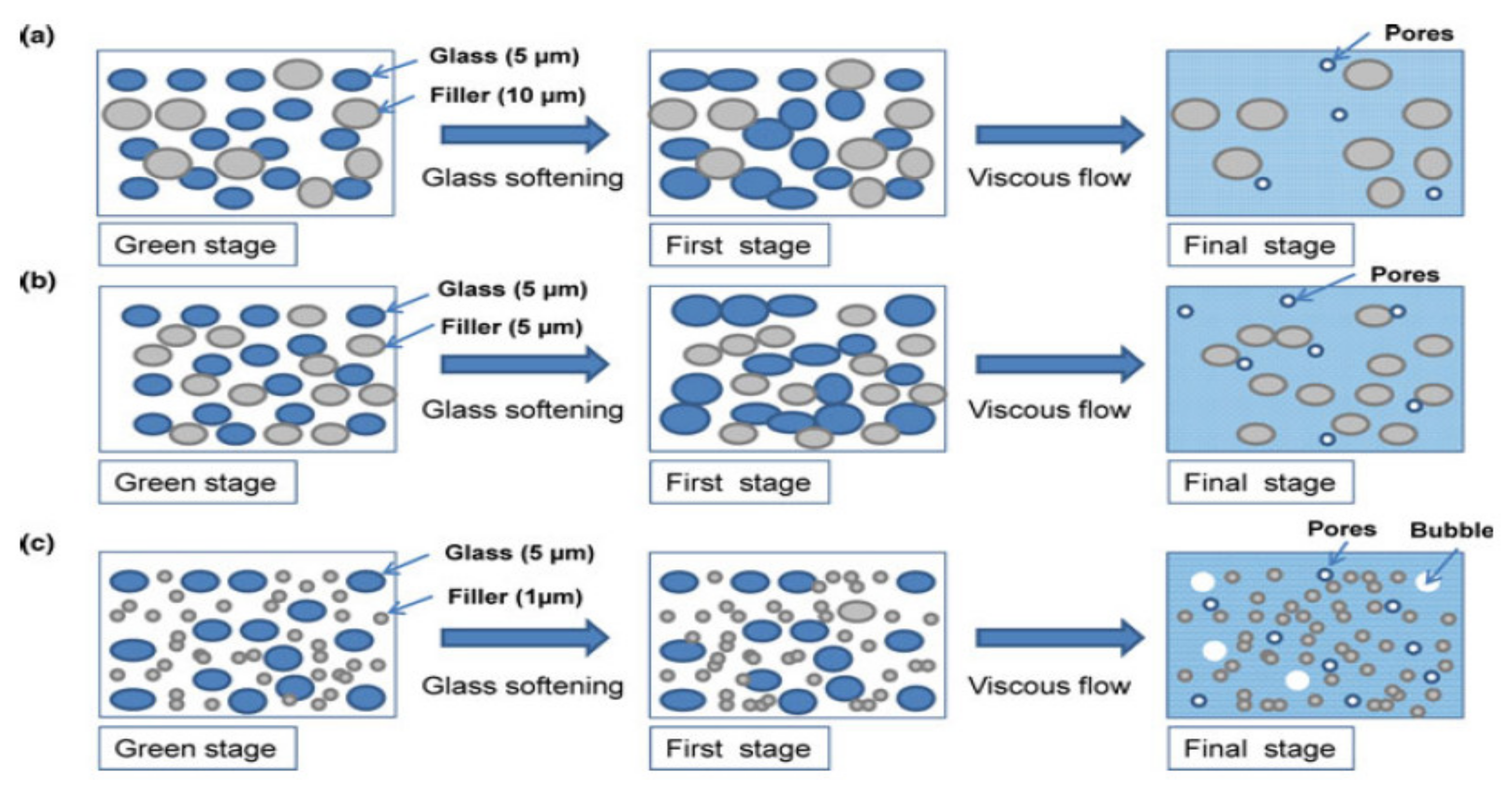
- Seo, J.; Kim, S.; Samal, S.; Kim, H. Viscous behaviour of Bi2O3–B2O3–ZnO glass composites with ceramic fillers. Adv. Appl. Ceram. 2014, 113, 334–340. [Google Scholar] [CrossRef]
- Samal, S.; Kim, S.; Kim, H. Effects of Filler Size and Distribution on Viscous Behavior of Glass Composites. J. Am. Ceram. Soc. 2012, 95, 1595–1603. [Google Scholar] [CrossRef]
- Shin, H.; Kim, S.-G.; Park, J.-S.; An, J.-S.; Hong, K.S.; Kim, H. Co-Additions of TiO2 and SiO2 Crystalline Fillers to Tailor the Properties of BaO–ZnO–B2O3–SiO2 Glass for Application to Barrier Ribs of Plasma Display Panels. J. Am. Ceram. Soc. 2006, 89, 3258–3261. [Google Scholar] [CrossRef]
- Lin, C.W.; Huang, L.C.; Ma, C.C.M.; Yang, A.C.M.; Lin, C.J.; Lin, L.J. Nanoplastic flows of glassy polymer chains interacting with multiwalled carbon nanotubes in nanocomposites. Macromolecules 2008, 41, 4978–4988. [Google Scholar] [CrossRef]
- Merkel, T.C.; Freeman, B.D.; Spontak, R.J.; He, Z.; Pinnau, I.; Meakin, P.; Hill, A.J. Ultrapermeable, reverse-selective nanocomposite membranes. Science 2002, 296, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poslinski, A.J.; Ryan, M.E.; Gupta, R.K.; Seshadri, S.G.; Frechette, F.J. Rheological behavior of filled polymeric systems II. The effect of a bimodal size distribution of particulates. J. Rheol. 1988, 32, 751–771. [Google Scholar] [CrossRef]
- Brouwers, H.J. Particle-size distribution and packing fraction of geometric random packings. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2006, 74, 031309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Yu, S.; Sun, R.; Du, R. Nano- and microsize effect of CCTO fillers on the dielectric behavior of CCTO/PVDF composites. Acta Mater. 2011, 59, 5593–5602. [Google Scholar] [CrossRef]
- Asai, K.; Amao, Y.; Iijima, Y.; Okura, I.; Nishide, H. Novel Pressure-Sensitive Paint for Cryogenic and Unsteady Wind-Tunnel Testing. J. Thermophys. Heat Transf. 2002, 16, 109–115. [Google Scholar] [CrossRef]
- Loui, A.; Ratto, T.V.; Wilson, T.S.; McCall, S.K.; Mukerjee, E.V.; Love, A.H.; Hart, B.R. Chemical vapor discrimination using a compact and low-power array of piezoresistive microcantilevers. Analyst 2008, 133, 608–615. [Google Scholar] [CrossRef]
- Hubmann, M.; Kong, X.; Curtis, J.M. Kinetic stabilization of cellulose nanocrystals in a photocurable prepolymer for application as an adhesion promoter in UV-curable coatings. Prog. Org. Coat. 2019, 129, 101–115. [Google Scholar] [CrossRef]
- Viard, A.; Gottardo, L.; Lopez-Ferber, D.; Soleilhavoup, A.; Salameh, C.; Samal, S.; Gueguen, Y.; Rouxel, T.; Motz, G.; Babonneau, F.; et al. Molecular design of melt-spinnable co-polymers as Si–B–C–N fiber precursors. Dalton Trans. 2017, 46, 13510–13523. [Google Scholar] [CrossRef]
- Hutchinson, J.M. Physical aging of polymers. Prog. Polym. Sci. 1995, 20, 703–760. [Google Scholar] [CrossRef]
- Cangialosi, D.; Boucher, V.M.; Alegría, A.; Colmenero, J. Physical aging in polymers and polymer nanocomposites: Recent results and open questions. Soft Matter 2013, 9, 8619–8630. [Google Scholar] [CrossRef]
- Pfromm, P.H.; Koros, W.J. Accelerated physical ageing of thin glassy polymer films: Evidence from gas transport measurements. Polymer 1995, 36, 2379–2387. [Google Scholar] [CrossRef]
- Jérôme, B.; Commandeur, J. Dynamics of glasses below the glass transition. Nature 1997, 386, 589–592. [Google Scholar] [CrossRef]
- Chung, T.-S.; Khean Teoh, S. The ageing phenomenon of polyethersulphone hollow fibre membranes for gas separation and their characteristics. J. Membr. Sci. 1999, 152, 175–188. [Google Scholar] [CrossRef]
- McCaig, M.S.; Paul, D.R. Effect of film thickness on the changes in gas permeability of a glassy polyarylate due to physical aging Part I. Experimental observations. Polymer 2000, 41, 629–637. [Google Scholar] [CrossRef]
- Kim, J.H.; Koros, W.; Paul, D.R. Effects of CO2 exposure and physical aging on the gas permeability of thin 6FDA-based polyimide membranes: Part 2. With crosslinking. J. Membr. Sci. 2006, 282, 32–43. [Google Scholar] [CrossRef]
- Struik, L.C.E. Volume relaxation in polymers. Rheol. Acta 1966, 5, 303–311. [Google Scholar] [CrossRef]
- Xia, J.; Chung, T.-S.; Paul, D.R. Physical aging and carbon dioxide plasticization of thin polyimide films in mixed gas permeation. J. Membr. Sci. 2014, 450, 457–468. [Google Scholar] [CrossRef]
- Xia, J.; Chung, T.-S.; Li, P.; Horn, N.R.; Paul, D.R. Aging and carbon dioxide plasticization of thin polyetherimide films. Polymer 2012, 53, 2099–2108. [Google Scholar] [CrossRef]
- Tiwari, R.R.; Jin, J.; Freeman, B.D.; Paul, D.R. Physical aging, CO2 sorption and plasticization in thin films of polymer with intrinsic microporosity (PIM-1). J. Membr. Sci. 2017, 537, 362–371. [Google Scholar] [CrossRef]
- Huang, Y.; Paul, D.R. Physical Aging of Thin Glassy Polymer Films Monitored by Optical Properties. Macromolecules 2006, 39, 1554–1559. [Google Scholar] [CrossRef]
- Drozdov, A.D. The effect of temperature on physical aging of glassy polymers. J. Appl. Polym. Sci. 2001, 81, 3309–3320. [Google Scholar] [CrossRef]
- Yong, W.F.; Kwek, K.H.A.; Liao, K.-S.; Chung, T.-S. Suppression of aging and plasticization in highly permeable polymers. Polymer 2015, 77, 377–386. [Google Scholar] [CrossRef]

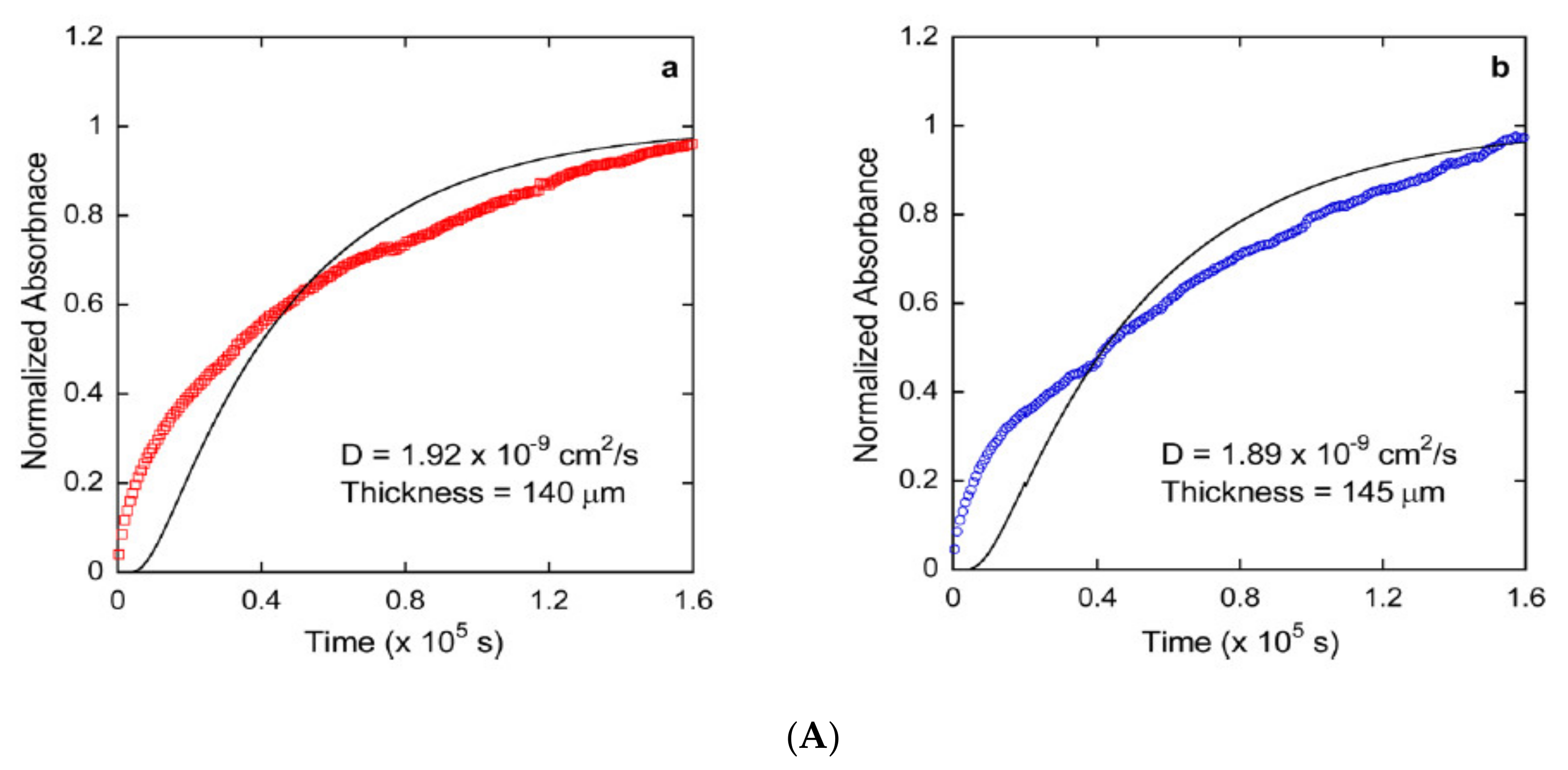
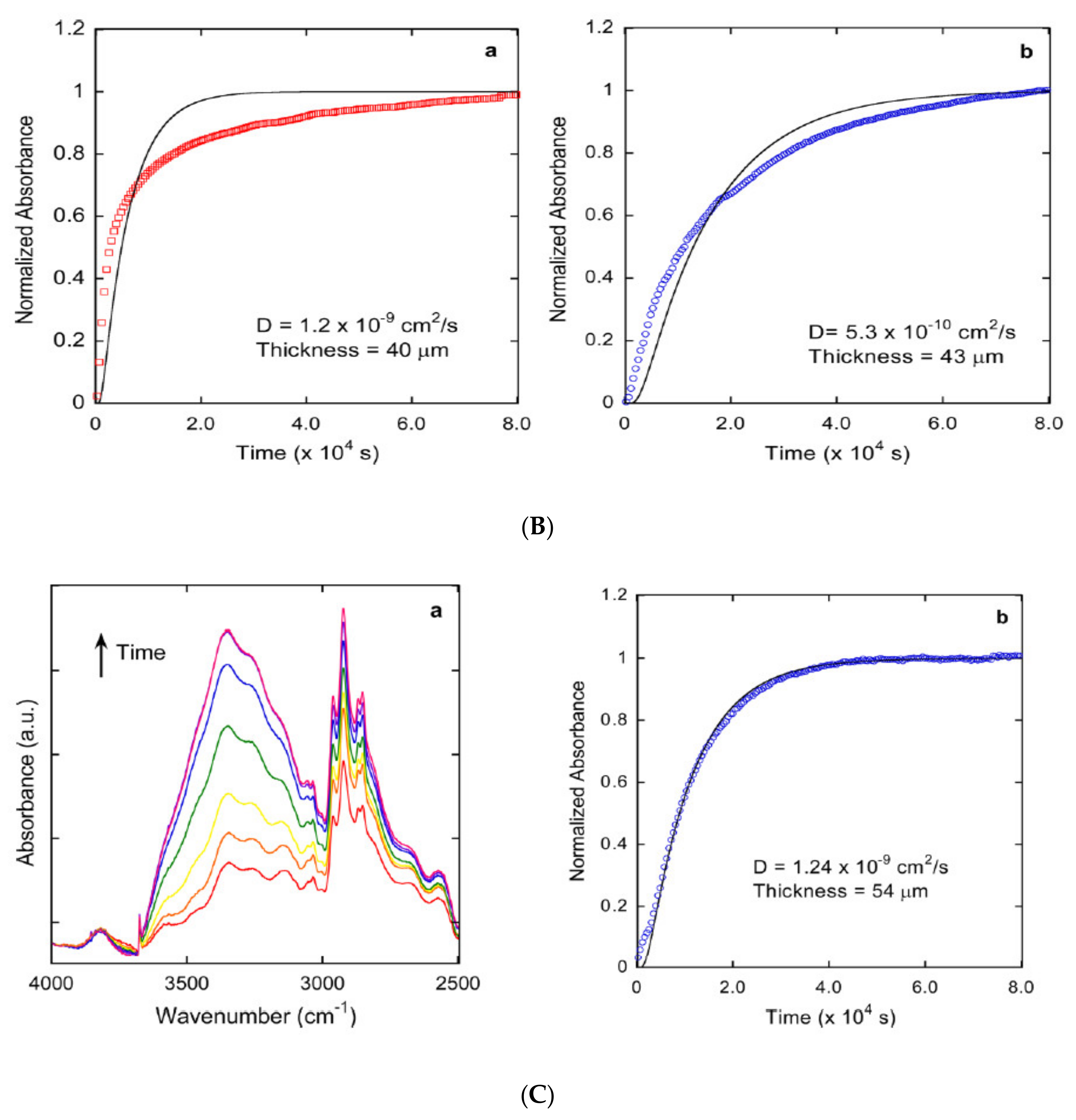
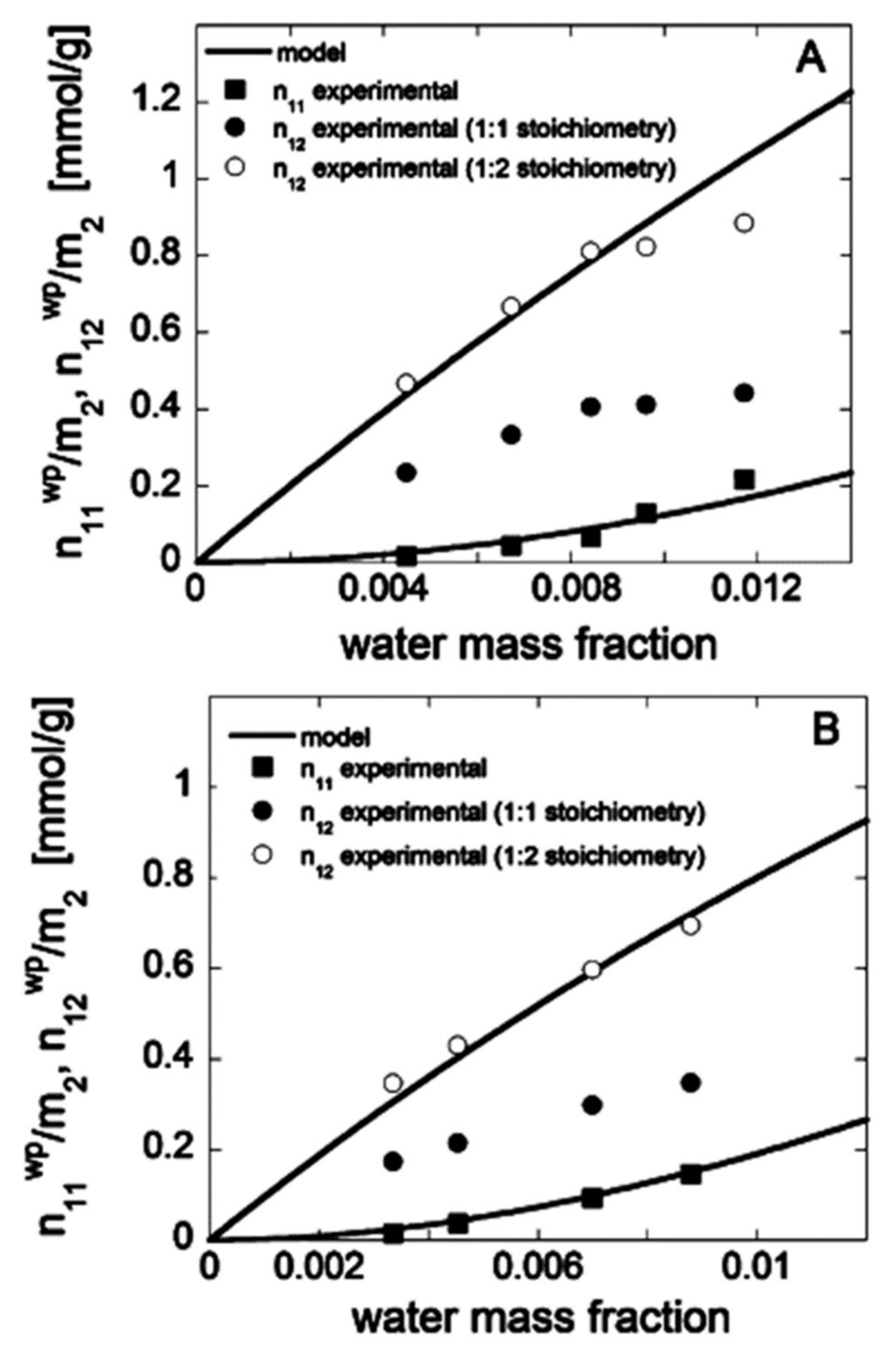
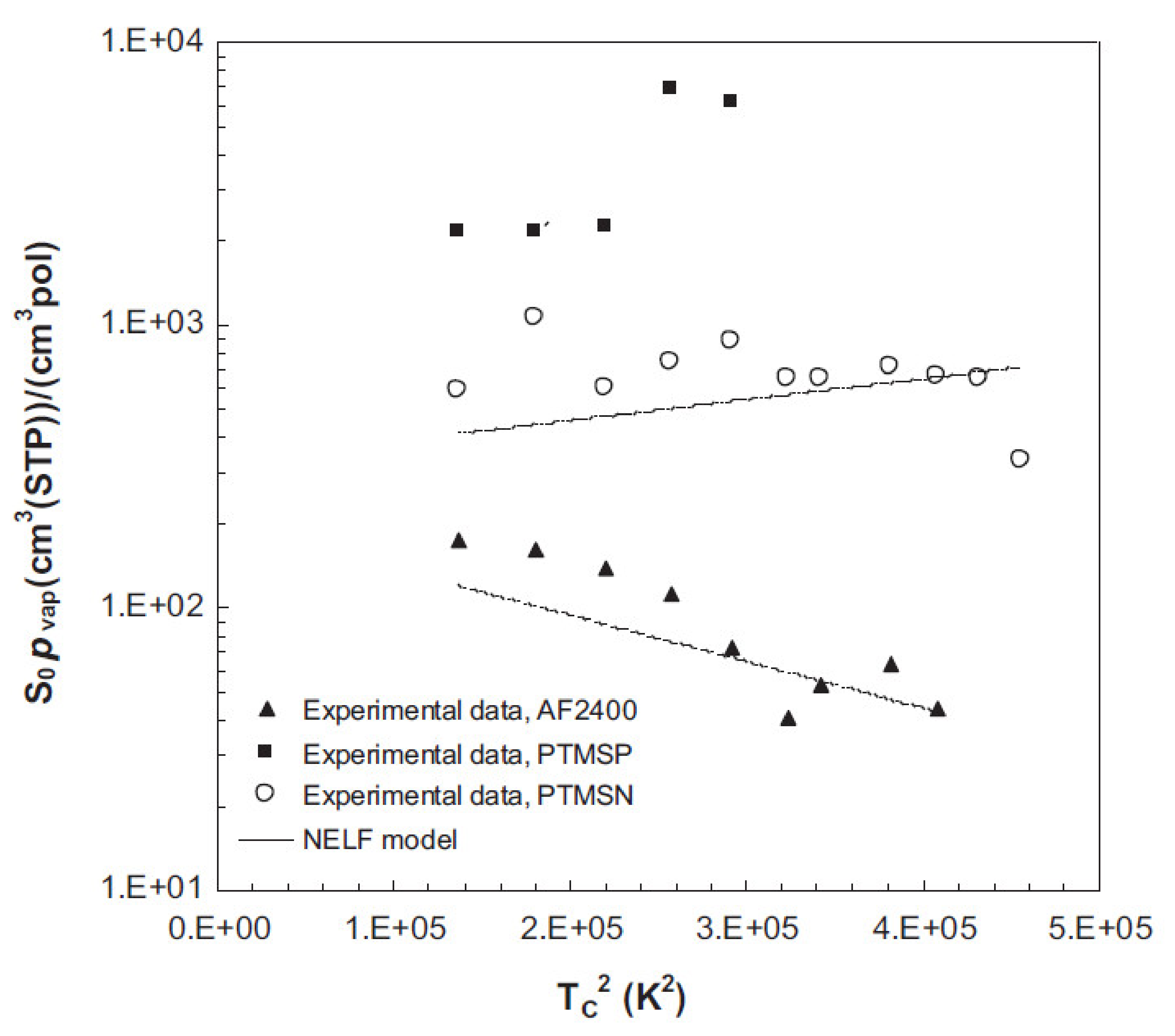
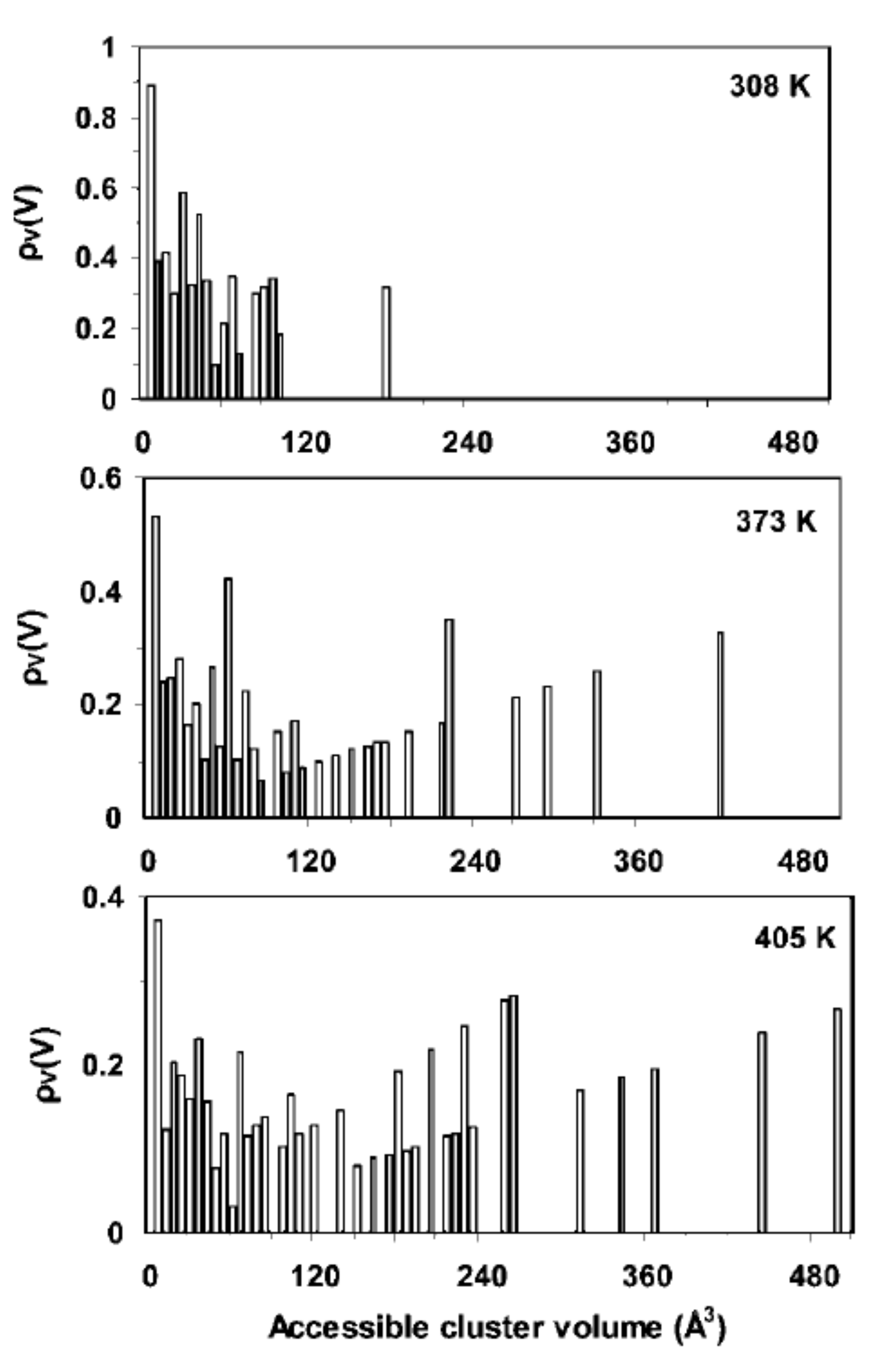
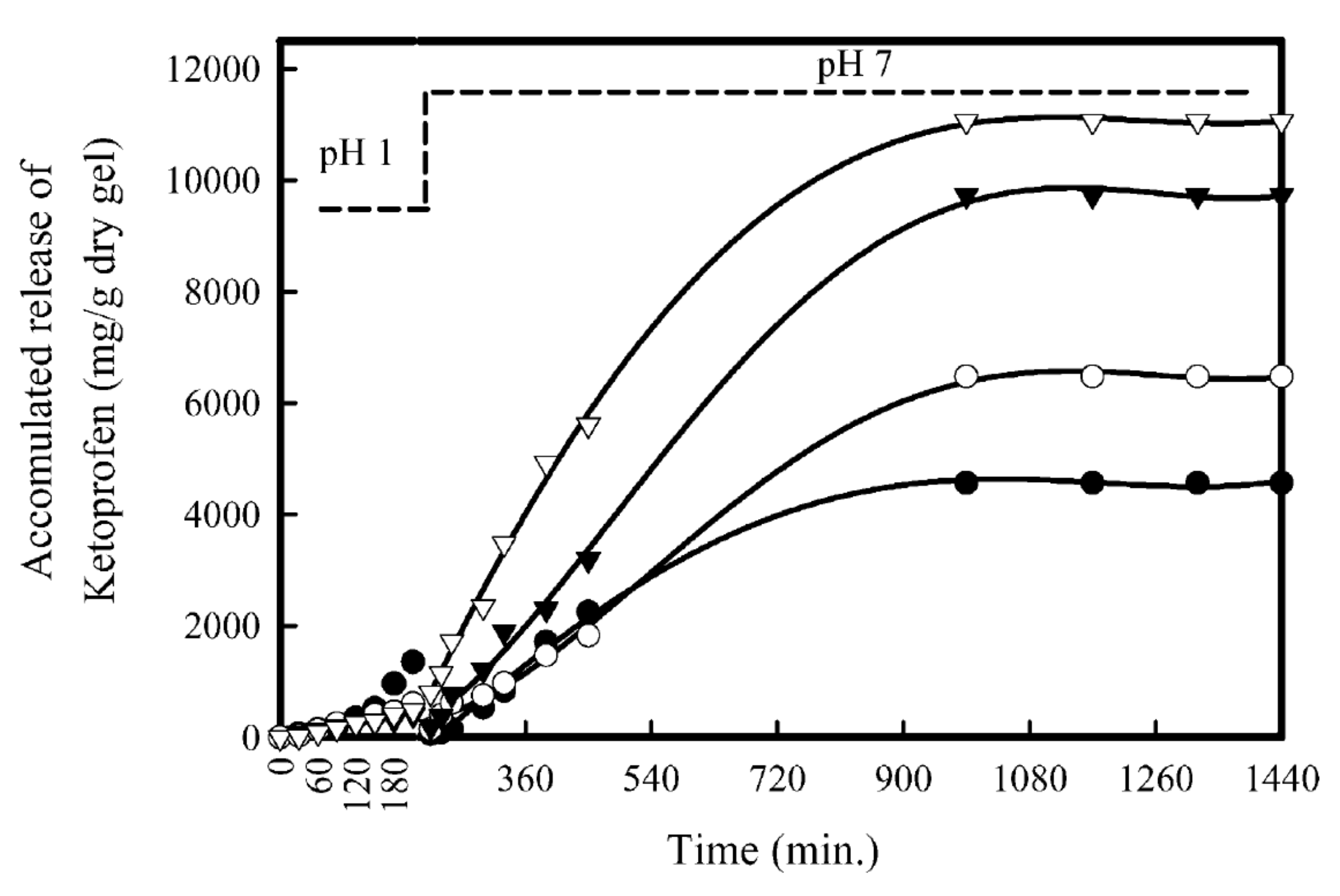
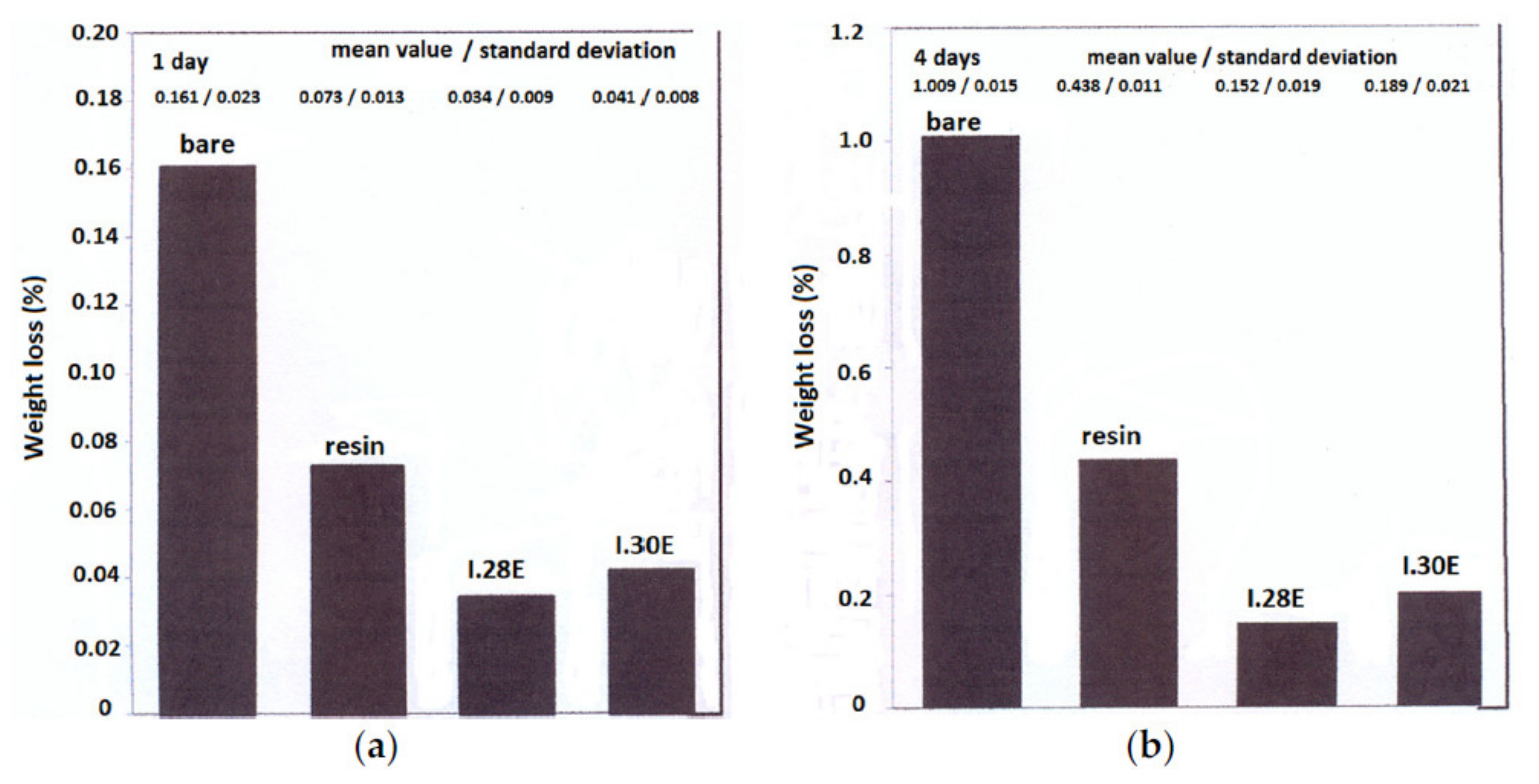
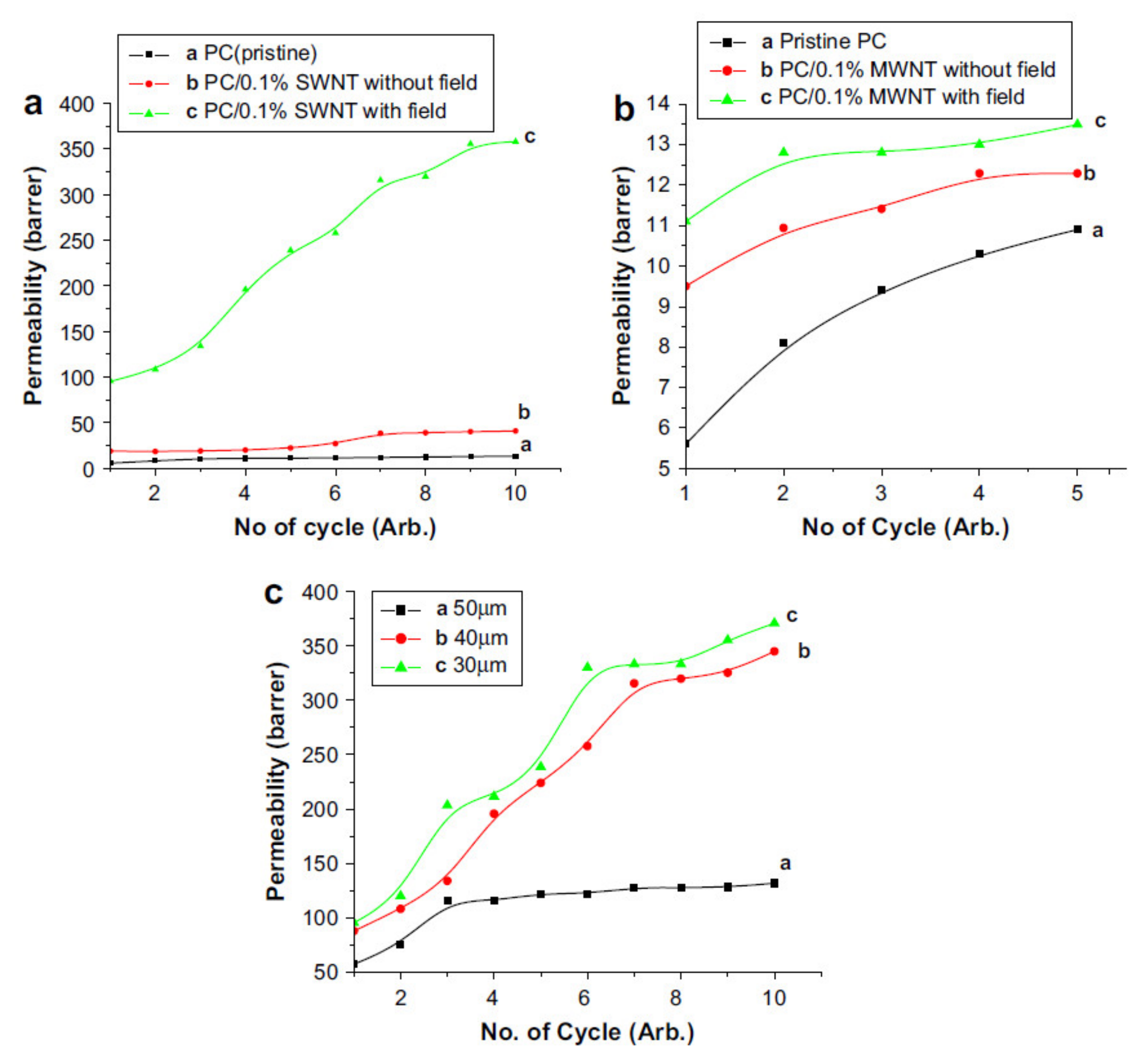
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arya, R.K.; Thapliyal, D.; Sharma, J.; Verros, G.D. Glassy Polymers—Diffusion, Sorption, Ageing and Applications. Coatings 2021, 11, 1049. https://doi.org/10.3390/coatings11091049
Arya RK, Thapliyal D, Sharma J, Verros GD. Glassy Polymers—Diffusion, Sorption, Ageing and Applications. Coatings. 2021; 11(9):1049. https://doi.org/10.3390/coatings11091049
Chicago/Turabian StyleArya, Raj Kumar, Devyani Thapliyal, Jyoti Sharma, and George D. Verros. 2021. "Glassy Polymers—Diffusion, Sorption, Ageing and Applications" Coatings 11, no. 9: 1049. https://doi.org/10.3390/coatings11091049
APA StyleArya, R. K., Thapliyal, D., Sharma, J., & Verros, G. D. (2021). Glassy Polymers—Diffusion, Sorption, Ageing and Applications. Coatings, 11(9), 1049. https://doi.org/10.3390/coatings11091049