1. Introduction
The properties of metal oxides and their behavior in various environments are the subject of consideration of both large review articles [
1,
2] and numerous publications in scientific periodicals. The research of the behavior of aluminum and its anodic oxide (AOA) in various environments is of great practical importance, since aluminum is used as a structural material and its corrosion resistance is substantially important [
3,
4]; aluminum has long been used for the manufacture of electrolytic (oxide) capacitors [
5] and the production of microelectronic products [
6,
7]. The processes are studied, and methods of the electrochemical dissolution of barrier anodic oxide on aluminum (BAOA) in the composition of por-AOA (porous anodic oxide on aluminum) [
8,
9] are developed. Information on AOA chemical dissolution serves as the basis for carrying out assistant operations with its participation [
10,
11,
12]. Por-AOA, the formation of which occurs due to the course of dissolution processes, is a matrix or template for the formation of various objects of nanotechnology [
13,
14,
15]. In addition, investigations of the aluminum dissolution during anodization in combination with other results, supplementing the data on the processes of AOA nucleation and development [
16,
17,
18], are of fundamental importance and have not yet been fully resolved.
Traditional models of AOA formation assumed the almost complete absence of the dissolution of the formed oxide in the case of the BAOA formation and its dissolution in the case of the por-AOA formation [
19,
20]. In the second case (the formation of por-AOA), at the beginning of the porous anodization process, at the stage of the linear growth of the anodic potential, dissolution does not occur at all, the BAOA is formed, and dissolution begins simultaneously with a slowdown in the growth of the anodic voltage and continues further throughout the anodization time. It was also believed that there are so-called non-dissolving electrolytes (solutions of boric acid (BA), borates, or tartrates), where BAOA grows, and dissolving ones, in which por-AOA is formed (e.g., sulfuric acid (SA) solution) [
18]. However, systematic research of the initial anodization stages, undertaken by Surganov and his co-workers, showed that during porous anodization at the substantially initial moments of the anodic process—literally, in the first seconds after its beginning—the nucleation of pores occurs [
21,
22], accompanied by a substantially intense dissolution of aluminum [
23,
24], which significantly contradicted earlier models [
17] and thus supplemented the understanding of the mechanisms of AOA formation.
To obtain even more convincing results, the decision was made to simulate the stage of the linear growth of the anodic potential during galvanostatic anodization using a potentiodynamic mode, setting the required voltage growth rate—“stretching”, thereby, the initial stage of the anodizing process—to certain time limits to make more detailed consideration possible. As a result, specific values of the rates (current densities) of dissolution were found for 0.1 M solutions of inorganic (BA and phosphoric acid (PA)) [
25,
26] and organic (sulfosalicylic (SSA), oxalic (OA), malonic (MA), tartaric (TA), and citric (CA)) acids [
27,
28]. This concentration was chosen to increase the credibility of the investigations. Por-AOA is not formed in BA under any conditions; in the solutions of most other acids at this concentration, BAOA is formed. Nevertheless, under these conditions, a significantly noticeable dissolution of aluminum occurs. Discussing the phenomena occurring at the AOA–electrolyte interface, the authors in the publications [
23,
24,
25,
26,
27,
28] often used the term “dissolution accelerated by an electric field” or “dissolution accelerated by the flow of an electric current”, without making attempts to consistently find out how much the dissolution rate changed during the process compared to purely chemical dissolution in the absence of anodic polarization. The relevance of this research stems from the relevance of electrochemical dissolution research. The features of electrochemical dissolution in different solutions and modes are important for understanding the mechanisms of nucleation and formation of the por-AOA cellular-porous structure. Meanwhile, the study of chemical dissolution should have shown a significant difference in the rates of these processes and dependence on the nature of the electrolyte. Previously, the authors showed [
23,
24,
25,
26,
27,
28] that the etching of the BAOA formed at the initial stages was due to non-trivial chemical dissolution, namely, the flow of electric current through the formed oxide film. One of the highlights of the work [
23,
24,
25,
26,
27,
28] and the proposed investigation that logically follows is that the aluminum dissolution was determined directly from its accumulation in the electrolyte and not assessed indirectly: gravimetrically [
4], using the technique of reanodization [
29,
30], capacitive and impedance measurements [
5,
8,
31], and other methods [
32], which increases the reliability of the research carried out here. The second significant feature of this investigation is that the object of the research was the “real” BAOA and not the BAOA in the composition of the por-AOA, as, for example, in [
8,
9,
10,
11,
12,
29,
30].
Thus, in this work, which has a fundamental novelty, as shown above, the rate of aluminum chemical dissolution and BAOA dissolution in electrolyte solutions often used in anodizing but without the application of the anodic potential was investigated.
Assumptions were made that during the experiment, the following did not occur:
In order to have grounds for such assumptions, when setting up an experiment, the solution volume for the longer dissolution of samples was increased in proportion to the exposure time Τ for the aluminum in solution.
3. Results and Discussion
As a result of investigations, the dissolution of aluminum and its BAOA in appreciable amounts was found to occur in many aggressive environments used in this research. However, it should be noted that a slight dissolution of aluminum and especially BAOA in most 0.1 M acid solutions made the research impossible to continue in dilute solutions for two reasons. Firstly, the concentration of the dissolved aluminum was close to the background concentration for many electrolytes (especially typical for TA and CA, not differing at high purity but slightly dissolving aluminum). Secondly, the concentration of the dissolved aluminum was at the limit of detection.
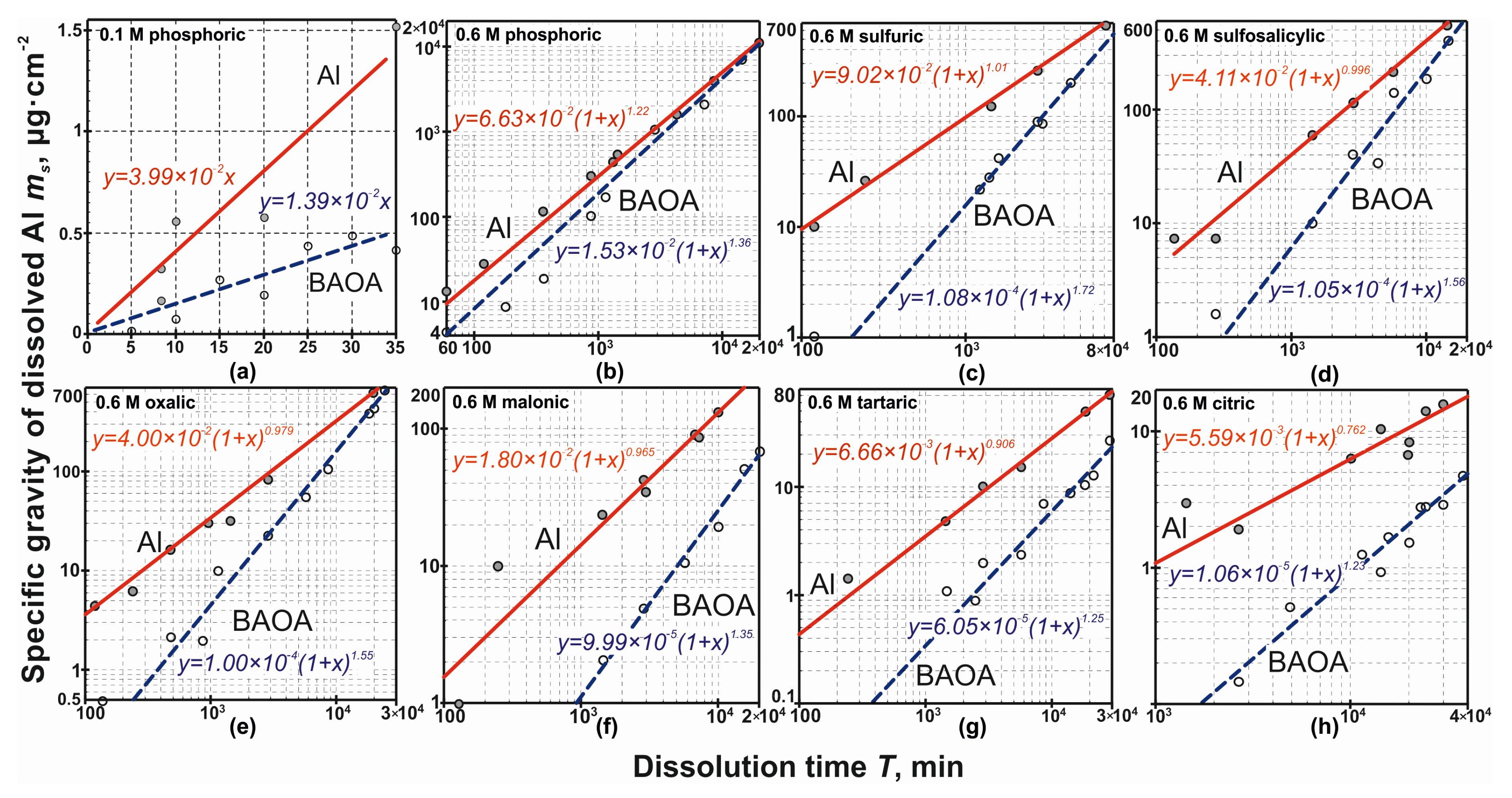
The experimental results for the 0.1 M PA solution and results for a series of experiments conducted in 0.6 M aqueous acid solutions are presented in
Figure 1. The dissolution rate of the unoxidized surface of the aluminum and BAOA in a 0.1 M PA solution was researched, as it turned out, for a too short a period of time Τ (~10 min) in order to detect the possible nonlinearity of the dependence
ms =
f (
Τ). Thus, for this case, the specific gravity of the dissolved aluminum obeys the dependence:
where
A is empirical constant and
Τ is the exposure time of the sample in an acid solution, min.
At the same time, for a series of 0.6 M electrolytes, such nonlinearity is easily detected upon dissolving for a time of 10
2–10
4 min. Further analysis shows that the dependencies from
Figure 1 are best described by the equation:
where
a and
b are empirical values, presented in
Table 1.
The values of the empirical constants A for Equations (3) were obtained as 3.99·10
–2 for aluminum and 1.39·10
−2 for BAOA. Comparing the dependency of the graphs
ms =
f (
Τ) in
Figure 1a,b, one can see that the dissolution rate of the unprotected metal in PA with an increase in the acid concentration by a factor of 6 increases by approximately 1.7 times, and that for BAOA, only 1.1 times. In addition, it was found that the average dissolution rate of BAOA was found to decrease from 2.1×10
−1 to 5.6×10
−5 μg·cm
−2·min
−1 in the following sequence of acid solutions: PA–SA–SSA–OA–MA–TA–CA. The average dissolution rate of the unprotected metal in the same sequence varied from 3.3×10
−1 to 1.0×10
−3 µg·cm
−2·min
−1. At the same time, if the dissolution rate of the metal and oxide in PA differed by approximately 1.6 times, the unprotected aluminum dissolved 180 times faster than the one coated with BAOA. The chemical dissolution rates of the aluminum and its dense alumina oxide increased with an increasing acid dissociation constant. This trend was not a subject to PA; the dissolution rate was higher than would follow from the indicated qualitative rule, which is possibly due to the structure of the acid anion of
PO43−. The reason may also be a possible significant specific sorption of PA and its dissociated anions to varying degrees, leading, firstly, to a local increase in the concentration of PA and its dissociation products in the surface region and, secondly, to an increase in the electric field strength of the double electric layer, which leads to a shift of Equation (10) to the right.
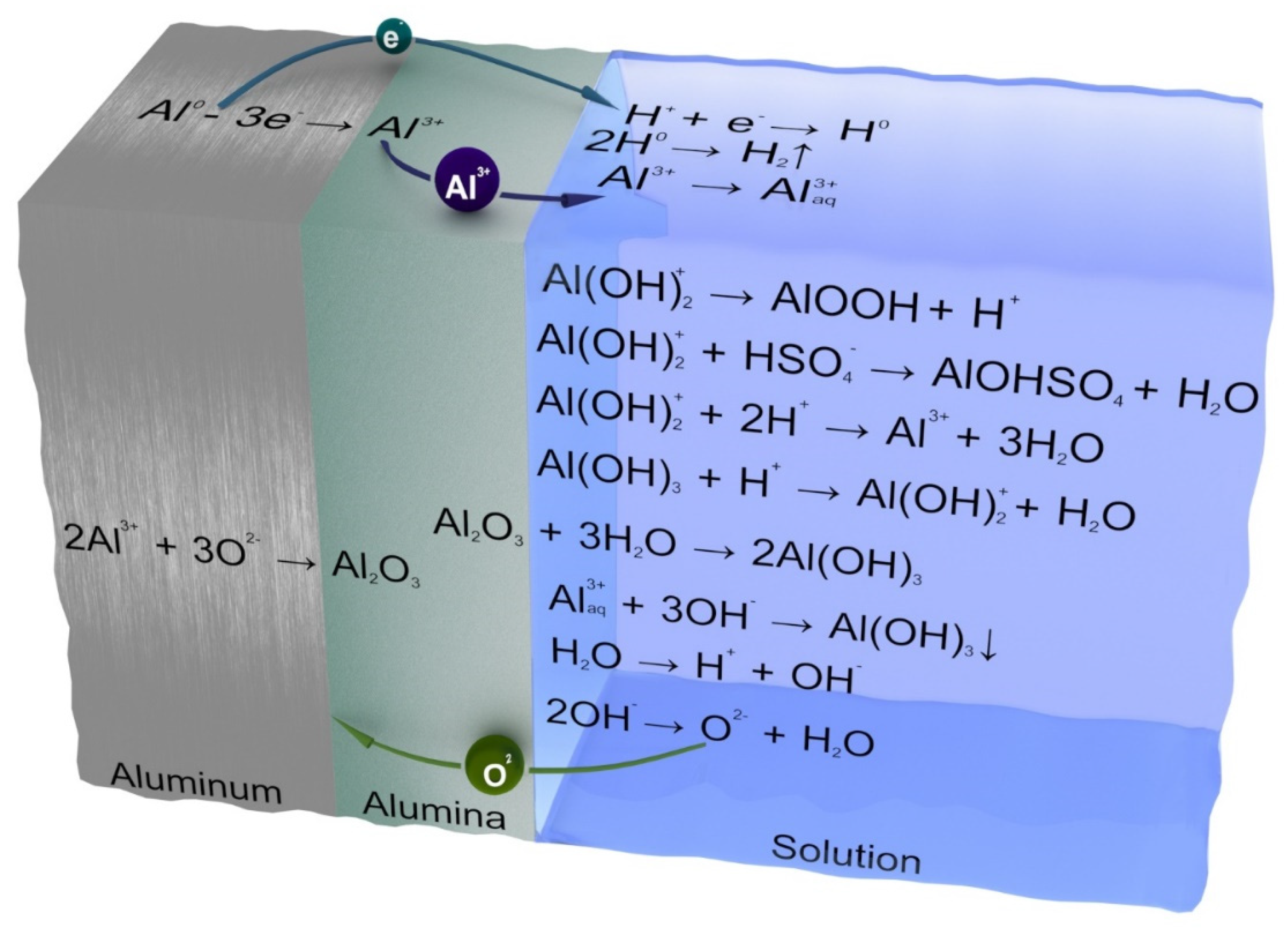
To explain the difference in the dissolution rates of aluminum and its barrier anodic oxide, as well as the nature of the dependence of the dissolution rate on the exposure time, the following model is proposed. It is assumed that in the case of both unprotected aluminum and aluminum coated with a BAOA layer, one has to take into account the oxide film formed on the Al surface (in the case of a non-anodized metal, the thickness of “natural” aluminum oxide is approximately 4.5–10.0 nm [
35,
37], according to the data published in [
38]—20–50 Å—and in the case of a metal coated with alumina oxide, the thickness of the barrier layer was, as mentioned above, 240 and 350 nm for the experiment series in 0.1 and 0.6 M acid solutions, respectively). In this case, the specific gravity of the BAOA depends on the temperature and nature of the electrolyte from 2.69 to 3.25 g·cm
−3 [
34]. A chemical dissolution process for oxide can conditionally be described by the following equations:
The interaction with the anion, for example, Cl
−, can be represented as follows:
However, in an acidic environment, such a continuation is possible:
The complete reaction corresponding to the sum of Equations (5), (6) and (9) is as follows:
In the case when the solvent anion, for example, the oxalate anion, is a good complexing agent with respect to aluminum [
39,
40], the binding of Al
3+ ions should be considered according to the equations
and, in the case of a large excess of oxalic acid,
For the case of a PA electrolyte, the following reaction can also be considered:
This leads, however, to the formation of a complex compound somewhat less stable than that formed in the case of OA [
39]. Reactions (5)–(13), actually removing aluminum ions from the reaction sphere, shift the equilibrium of reaction (10) to the right. On the other hand, reactions (5)–(13) lead to a decrease in the concentration of OA or PA anions, which should be especially pronounced at low acid concentrations and a small solution volume compared to the amount of metal presented.
In the interaction of Al, as well as other metals with
Red-Ox potentials below zero, with water and aqueous solutions of acids and bases, hydrogen reduction and metal oxidation should be observed. However, for many metals (Ta, Ti, and Al), this process is kinetically inhibited to a greater or lesser extent. The reason for this potential barrier is a dense and rather inert oxide film. It is obvious that, for however long aluminum is not in the electrolyte, Al constantly remains covered by an oxide film protecting its surface. The thickness of the film, probably, in the case of an equilibrium steady-state dissolution process in each acid, is constant and depends on the nature and concentration of the acid. Thus, in the case of aluminum, both protected by a BAOA layer and coated with “natural” oxide, one should discuss dissolving a more or less thick oxide film covering the metal. Otherwise, the reaction with aqueous solutions would proceed much faster, as occurs in the case of amalgamated aluminum, which, like alkaline and alkaline-earth metals, interacts exceedingly energetically with water according to the following equation:
Thus, the chemical dissolution process for aluminum, not just that stimulated by an electric field [
41], consists of two competing reactions: the dissolution of the outer surface of the oxide film, which always covers the aluminum, and its continuous growth (restoration of thickness) to a value characteristic of each acid by the oxidation of all the new metal layers.
According to [
1], the dissolution phenomena depend on the following characteristics of the oxide:
Covalence degrees of the oxygen–metal bond;
The existence of a defective lattice;
The anisotropy of the properties associated with the crystal orientation;
The presence of a film surface or bulk oxide;
The presence of impurities in the oxide;
The existence of a long-range or only short-range order.
The presence (or absence) of oxide semiconductor properties located on a metal surface also significantly affects the nature of the processes occurring during immersion. Moreover, when considering, for example, Red-Ox reactions occurring in a metal coated with oxide, three different cases can be distinguished, depending on the thickness of the oxide layer:
The thickness of the oxide is less than 30 Å, when the oxide is sufficiently transparent for electrons due to quantum mechanical tunneling.
The thickness of the oxide film is more than 100 Å but less than the thickness of the space charge layer. In this case, the rate of Red-Ox reactions will be a function of the properties of the underlying metal, oxide, and two interfaces (metal–oxide and oxide–electrolyte).
The oxide layer on the metal surface has a thickness much greater than the thickness of the space charge layer. The rate of Red-Ox reactions will not depend on the underlying metal but only on the oxide–electrolyte interface and the semiconductive properties of the oxide.
The same trends should be expected for electrochemical dissolution. In practice, this means that, in the general case, the dissolution of a bulk (massive) oxide sample cannot be considered by analogy with the dissolution of the oxide covering the metal surface.
Furthermore, regarding oxide film thickness, its nature also affects the conductivity of the oxide. The electronic conductivity, which is important for electrochemical dissolution, is associated with the structure of the oxide and the nature of the metal–oxygen bond. The more pronounced the ionic nature of an oxide, the greater the band gap and the lower the electron conductivity. The stronger the covalency of an oxide bond, the higher the probability of Red-Ox reaction occurrence, and vice versa—the stronger the ionic nature of the oxide, the greater the likelihood that the dissolution includes the rate-determining ion transport across the interface of the oxide with the electrolyte.
To understand the dissolution processes, the potential distribution at the interface of the oxide with the electrolyte is important to imagine. Such a distribution, in principle, is similar to the potential distribution at the interface of the metal with the electrolyte. The electrolyte interface consists of the inner and outer Helmholtz layers and the diffuse layer.
The potential distribution near the metal–oxide interface of the sample immersed in the electrolyte solution, especially if the oxide is a semiconductor, will be more complicated than the potential distribution at the oxide–electrolyte interface. Such behavior depends on whether the substrate on which the oxide is placed is a metal, a semiconductor, or an insulator—the type and characteristics of the oxide and oxide film thickness. In the presented case, regarding the oxide layer located on the metal surface, the situation with the charge distribution will largely depend on the thickness of the film relative to the Debye length. Moreover, for a thick oxide film, when its thickness is much greater than the Debye length, the potential distribution will be similar to that for a bulk sample. If the oxide thickness is of the Debye length order, then the problem becomes more complicated and the charge distribution near the metal–oxide interface is necessary to take into account. An oxide film on a metal cannot be considered as a simple barrier to charges, both in the case of electrochemical anodic oxidation and its accompanying dissolution, and in the case of dissolution without the application of anode voltage; the properties and nature of the oxide film can be of great importance.
Moreover, one can expect the manifestation of differences in behavior between the samples (Al coated with BAOA and natural oxide layer) immersed in various electrolyte solutions, not only because of the difference in thickness but also because of the different origins of these oxide layers. A different background of “natural” and anodic aluminum oxides implies, at a minimum, the presence of various amounts of various kinds of impurities (for alumina oxide, first of all, significant amounts of electrolyte anions and/or their derivatives built into the crystal structure) and, possibly, differences in the stoichiometry and the crystalline structure of both oxides (the degree of amorphism, crystalline modification, and the presence and nature of defects).
Diggle also pointed out in his review [
1] the influence of pre-treatment modes, which determine the surface under investigation and surface-structure imperfections. For example, dislocations at the crystal surface are preferred points for dissolution, since these are places of facilitated diffusion. Dislocations, due to their increased energy state, are also adsorption centers. Moreover, [
1] shows the contribution of ion adsorption as well as possible complexation reactions for a prepared surface.
There is, in fact, a trivial equation for the interaction of metals with acids:
This consists of several equations describing the various components of the process (which, in turn, also consist of some elementary reactions), namely:
for the oxidation of aluminum,
for the reduction of hydrogen ions present in the solution,
for the formation of an oxide film on a metal, and
for the dissolution of the oxide (according to the mechanism explained by Equations (5)–(9)).
One can determine, for example, the following mechanism of aluminum oxidation, taking into account the abovementioned considerations and implying the restoration of the thickness of a continuously etched oxide film, illustrated in
Figure 2. The above equations and
Figure 2 show that the different stages of the total oxidation process occur near different interfaces.
It can be assumed that in acid solution, provided that its composition is constant, the oxide dissolution rate is constant and does not depend on oxide film thickness. The oxidation rate and, as a consequence, the rate of alumina formation are, ceteris paribus, a function of oxide film thickness, if only because with an increase in the aluminum oxide thickness, its resistance also increases, preventing the flow of both electronic and ion currents necessary for the process of its growth.
All the considerations mentioned above regarding the dissolution rate would be true if the considerations were dealing with an oxide sample under which an unoxidized metal would not be present. Based on the above reasoning, one can make an unambiguous assumption that the dissolution rates for non-oxidized and anodized metals should not differ. Experimental data indicate the exact opposite. For the explanation of the apparent paradox, one more possibility of a triply charged aluminum ion entering the solution should be recalled. The mentioned process is the exit of migrating
Al3+ ions, which did not meet
O2− counter-ions, from the metal–oxide interface to the oxide–electrolyte interface and their subsequent exit directly into the solution, followed by hydration. It is assumed that in the case of the dissolution of aluminum without the application of anode voltage, the phenomenon of ejection is also present, i.e., the direct release of
Al3+ ions into the solution. The assumptions about the permeability of BAOA for aluminum ions were also made in [
1]. However, considering the case when a metal is located under the oxide layer, it can be assumed that the dissolution rate may depend on the thickness of the film covering the oxide precisely due to the presence of a constituent due to the direct migration of aluminum into the solution. This work proves the presence of the impermanence in time of the aluminum dissolution rate, i.e., the dependence of the dissolution rate on oxide thickness. As far as is known, the authors pointed it out in [
5,
42], but, firstly, these studies are of a narrowly applied character. Secondly, as a result, the dissolution of aluminum has been investigated in only one electrolyte. Thirdly, the studies are of a statement (descriptive) character, and the possible dissolution mechanisms have not been considered yet. Fourthly, an explanation of the discovered effect of the dissolution rate versus time has not been given. Since it is not clear either
Al3+ or
O2− ions diffuse predominantly,
Figure 2 shows both processes, and for further discussion, the specific values of the transport numbers for aluminum and oxygen are insignificant.
Then, to Equations (16)–(19), which describe the steps that occur according to the total Equation (15), one more is added that describes the migration of oxidized aluminum from the metal–oxide interface through a thin oxide layer to the oxide–solution interface with the subsequent transition to liquid phase:
Purely theoretically, one can consider the following possibility of the binding of the Al
3+ ion, but the probability of such a process occurring in an acidic environment, and, even more so, in the presence of sufficiently strong complexing agents, is extremely low:
It is much more likely that the cation is bound to an insoluble complex compound with subsequent precipitation at the oxide–solution interface and, as a consequence, a further slowdown of the dissolution process.
It is substantially likely that the phenomenon described by Equation (20) provides higher dissolution rates for non-anodized metal in comparison with those for a sufficiently thick BAOA-coated metal. From this, it absolutely unambiguously follows that the counter migration of O2− ions in the direction from the solution–oxide interface through the Al2O3 layer to the oxide–metal interface should be negligible. In the case of BAOA dissolution, the dissolution of oxide most likely occurs, at first, rather slowly but constantly accelerates as the thickness of the oxide film decreases, until the thickness of the remaining alumina oxide begins to approach its characteristic value for these conditions, while the rate of aluminum oxidation and the rate of increase (recovery) of the oxide film—due to its still-significant size and, as a result, high electrical resistance—is negligible. After the dissolution of a significant part of the BAOA thickness and when approaching the characteristic value for this solution, the metal oxidation rate begins to increase and the number of Al3+ ions leaving the oxide film also increases, bypassing the stage of oxide formation. It can be assumed that this characteristic thickness is of the order of the Debye length. In the case of natural oxide, its thickness is much less than the Debye length in all electrolytes, except for PA (since in PA, the voltage drop is mainly due to the double electric layer; almost nothing falls on the oxide; therefore, in PA, the natural oxide also dissolves to a thickness corresponding to the Debye length; possible reasons for the special situation of PA are mentioned above). In the case of natural oxide, the oxide grows to a thickness equal to the Debye length. Electron tunnelling becomes impossible, and the oxidation rate slows down (in PA and at the Debye length, it is possible because the length is substantially small; aluminum rapidly oxidizes and dissolves rapidly). When, finally, alumina oxide is so etched that its thickness becomes equal to the characteristic thickness, then, whereas the dissolution rate for the oxide remains equal to that for the protected metal, the number of Al3+ ions entering the solution per unit time reaches a maximum and far exceeds the rate of true chemical dissolution precisely due to ion “migrants”. The aluminum oxidation rate and the “migration” current, which can be defined as the charge transferred by migrating Al3+ ions per unit time, take on constant values. The rates of both competing processes (bleeding and growth) become maximal and equal; the process takes on a stationary and equilibrated character. In this case, the authors consider a certain idealized case, not taking into account the accumulation of Al3+ ions in the solvent and their possible influence on the rate of the Red-Ox reaction and the dissolution of the oxide and the change in the concentration of protons in the solution. Aluminum ions hydrated or bound into complex compounds with acid anions, by varying the dielectric constant of the solvent in the bulk and especially in the anode region, can significantly change the properties of the aggressive environment itself; the same consequences can change the concentration of H+ ions (increase the pH of the solution).
These considerations are confirmed by the calculated values for the dissolution rate of aluminum and its BAOA obtained by differentiating the empirical expressions for specific dissolution given in
Table 1. A graph illustrating the dependence of the dissolution rate for aluminum and its BAOA is shown in
Figure 3. The short dashed line in the same figure shows a hypothetical one, which follows from previous arguments about the mechanisms of aluminum dissolution in acid solutions, as well as from the dependence logic
, a change in the rate of the appearance of
Al3+ ions in the electrolyte for the unstudied time domain dissolution.
Based on the results of this experiment, a quantitative criterion can be proposed for assessing the solubility of acid solutions: the exponent in the expression ms = f (Τ) (Equation (4)) for a metal coated with atmospheric oxide. Strongly dissolving electrolytes should include a solution of PA and SA (the exponent is greater than unity, which means the characteristic, equilibrium thickness of the oxide coating the metal in the solution is less than that of atmospheric oxide). Electrolytes of medium solubility can be attributed to solutions of SSA, OA, and MA: the exponent is close to unity (0.996, 0.979, and 0.965 for solutions of SSA, OA, and MA, respectively). The thickness of the oxide coating the metal in such solutions with prolonged exposure is slightly higher than that of the “atmospheric” one. Solutions of TA and CA should be classified as weakly soluble aluminum electrolytes. In terms of the dependence of the dissolved metal’s mass on the exposure time of the sample, the exponent is already significantly less than unity; when the sample not protected by BAOA is immersed, a noticeable increase in the thickness of the oxide film occurs.
Thus, in the first approximation, it can be assumed that dissolution consists of two processes: chemical dissolution, similar to the dissolution of a massive oxide sample, the rate of which does not depend on whether the metal is under it or not but depends only on the nature and concentration of the electrolyte and oxide properties; the second process is the oxidative dissolution of aluminum, which is under the oxide layer, due to the course of migration processes and depending, in addition to the above reasons, on the thickness of the oxide. It can be reasonably assumed that the presence of metal under the oxide layer can also indirectly affect the rate of chemical dissolution. The reason for this may be the presence of a positive charge on the surface of the metal electrode, the appearance of a double electric layer, e.g., a local change in the concentration of the electrolyte in the near-electrode region.






{kind=link}
{kind=link}
{kind=link}