Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction

Abstract
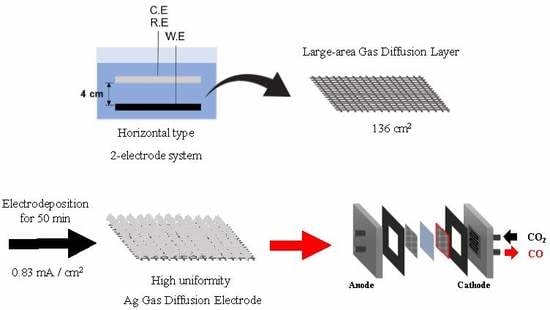
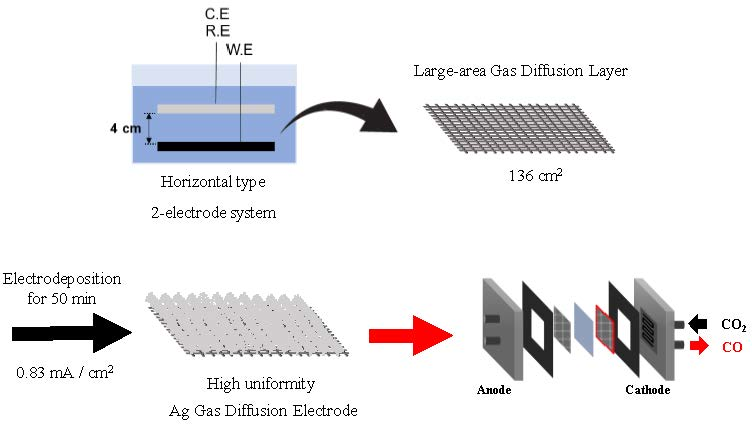{kind=link}
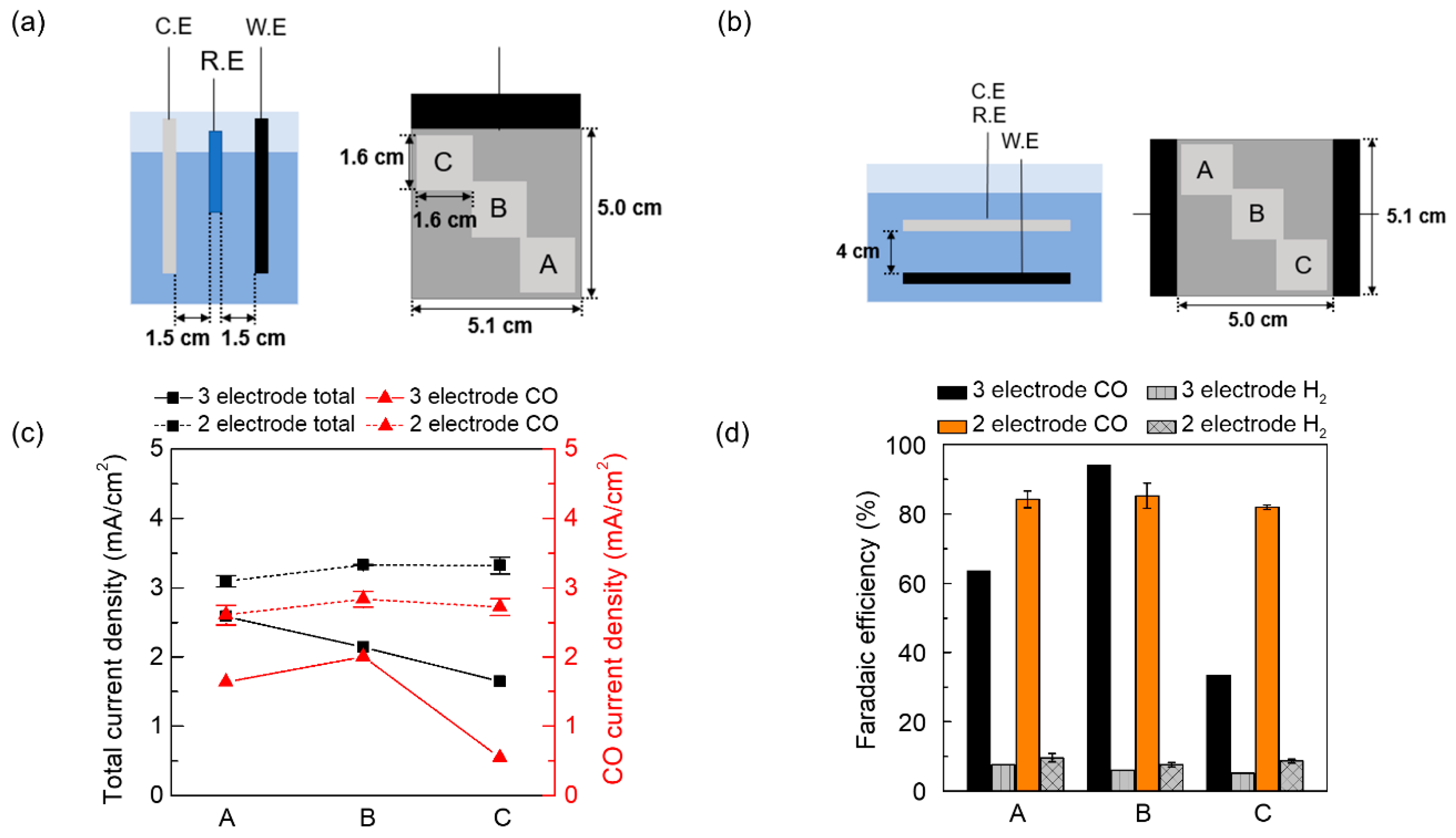{kind=link}
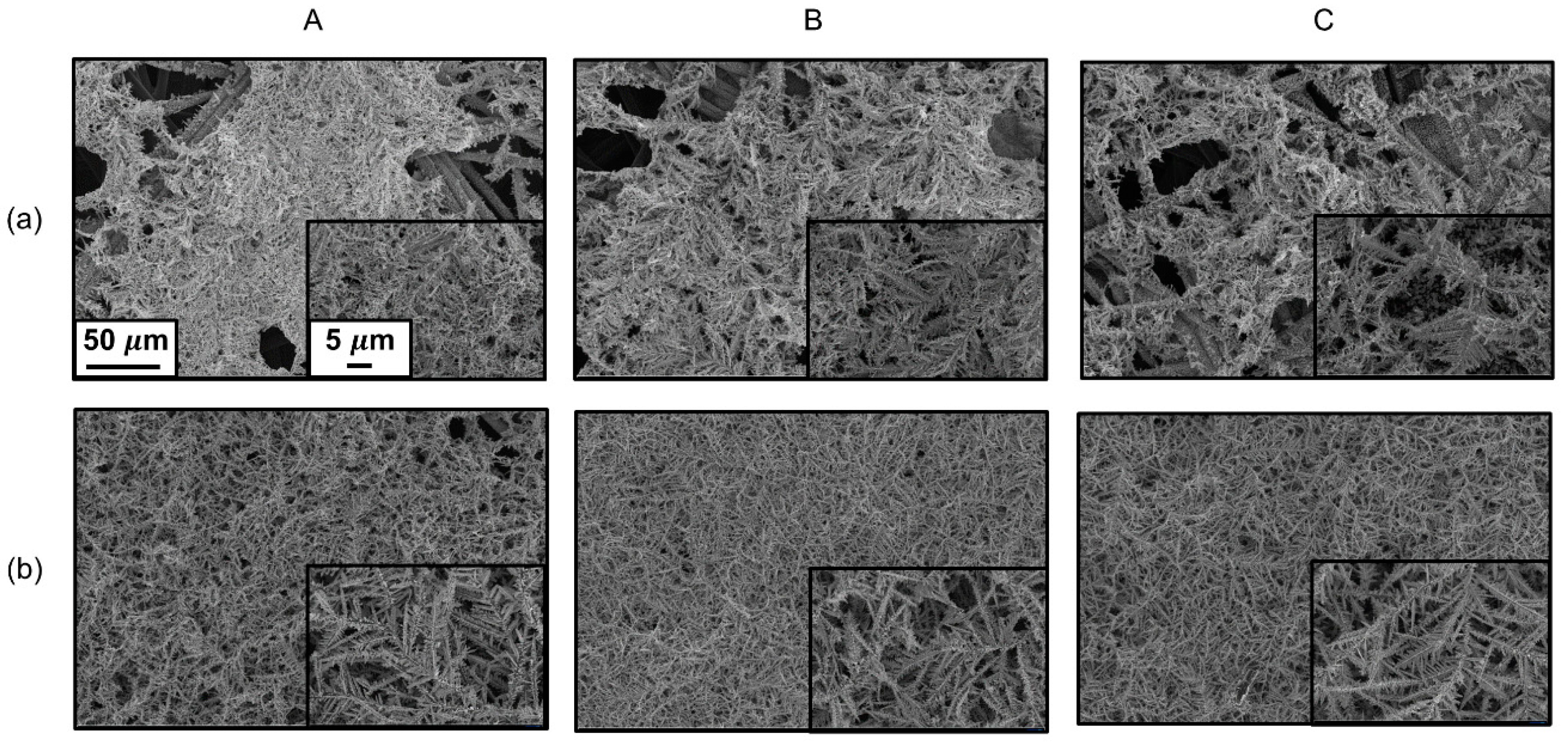{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Equipments
2.2. Fabrication of Ag GDE
2.3. Electrochemical CO2 Reduction to CO in Half-Cell
2.4. Electrochemical CO2 Reduction to CO in MEA-Type Electrolyzer
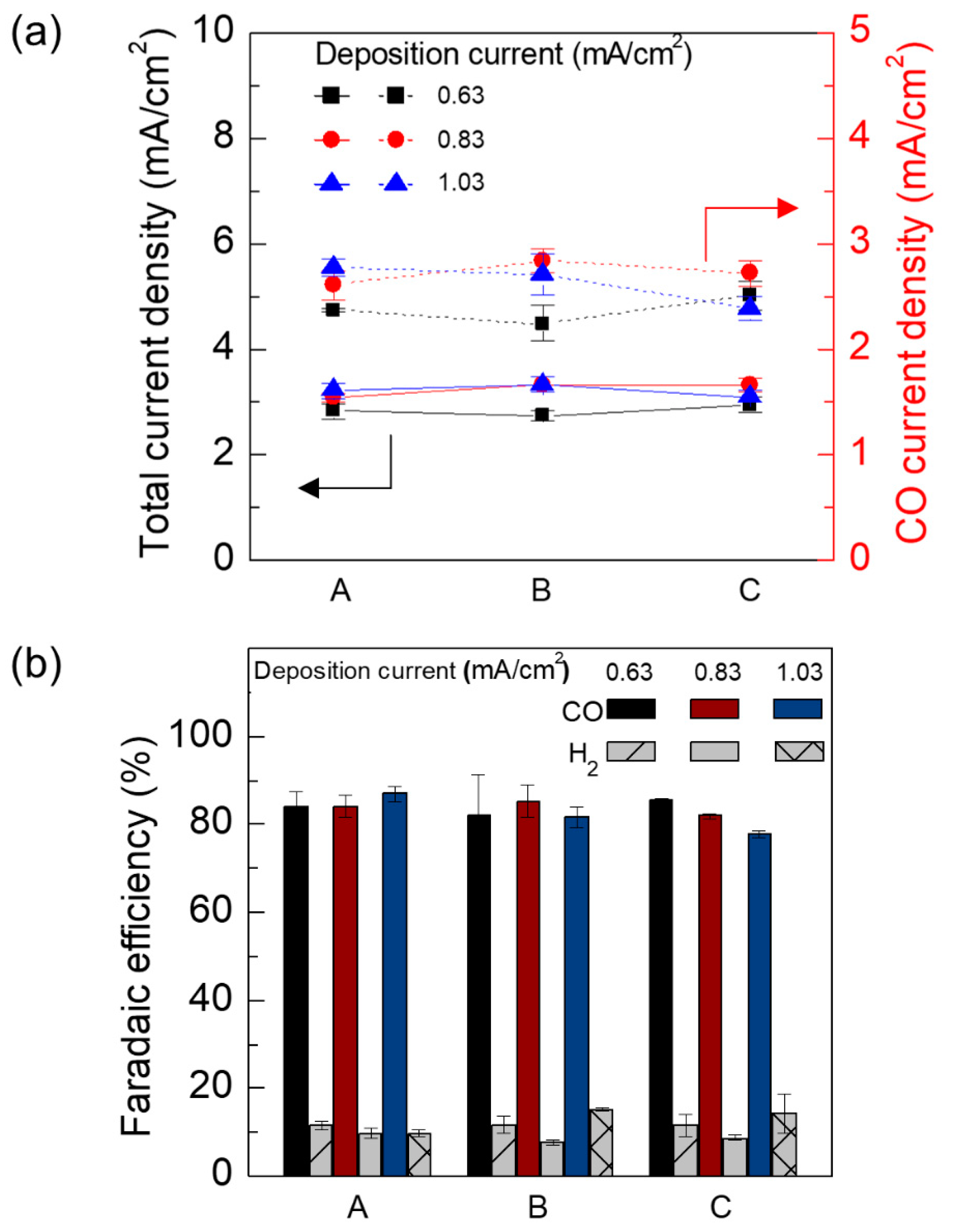
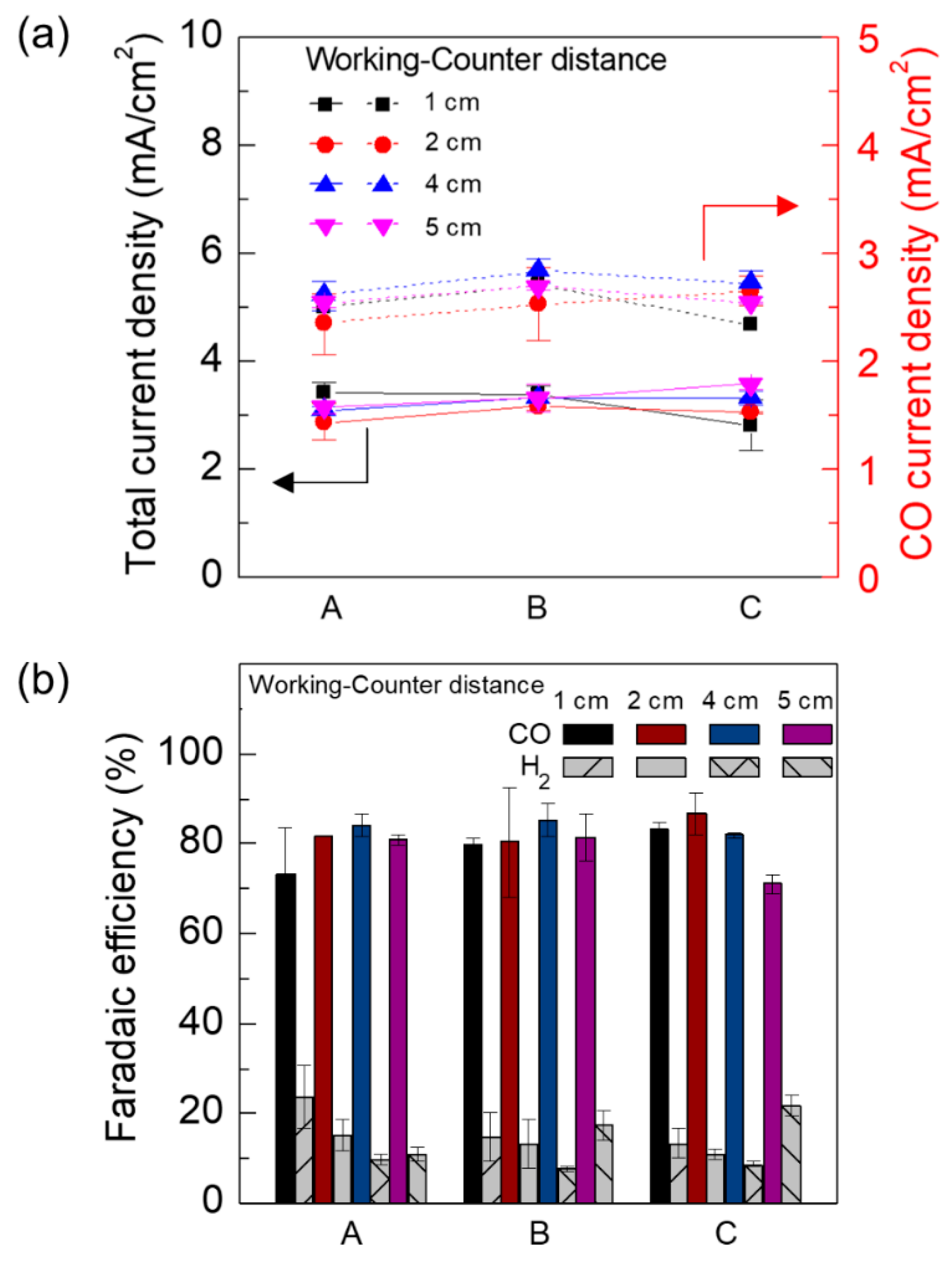
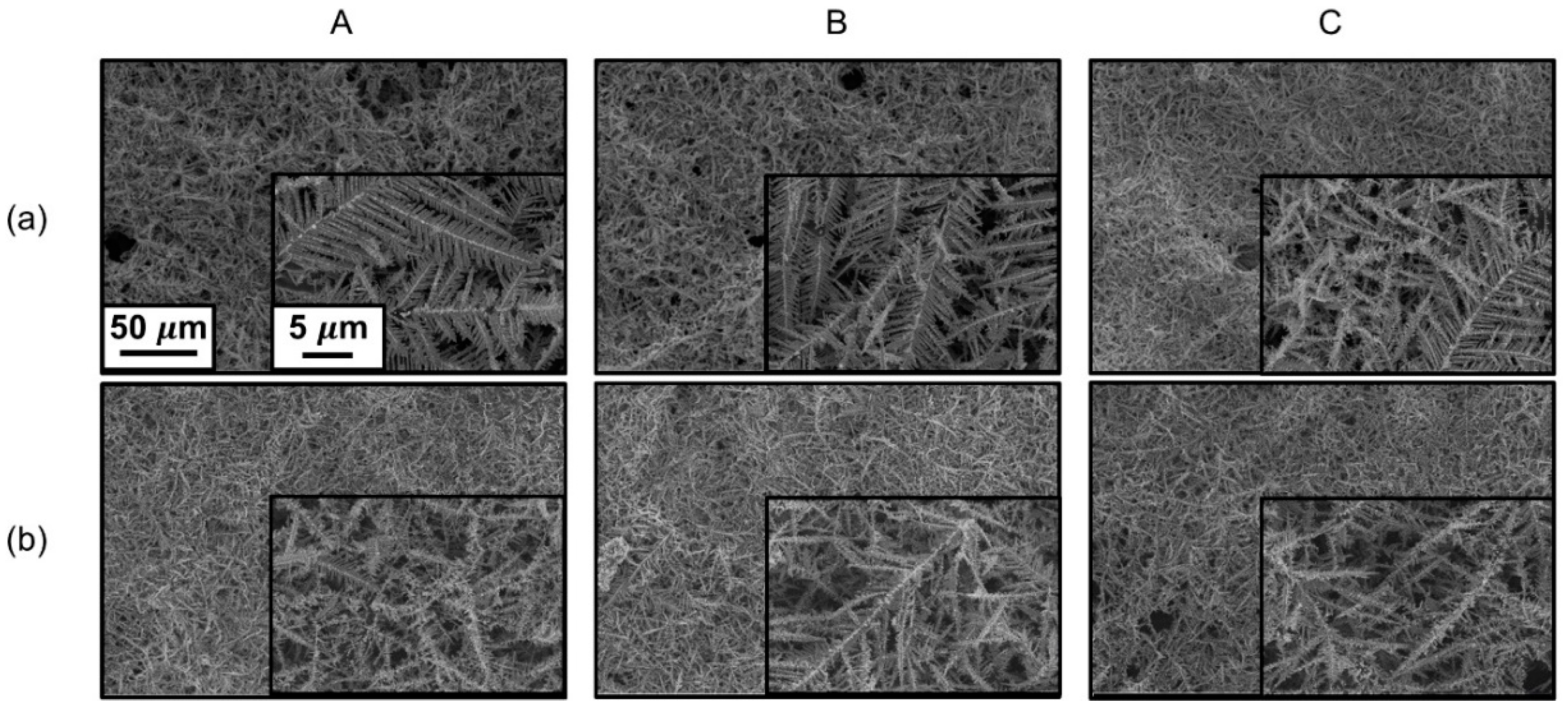
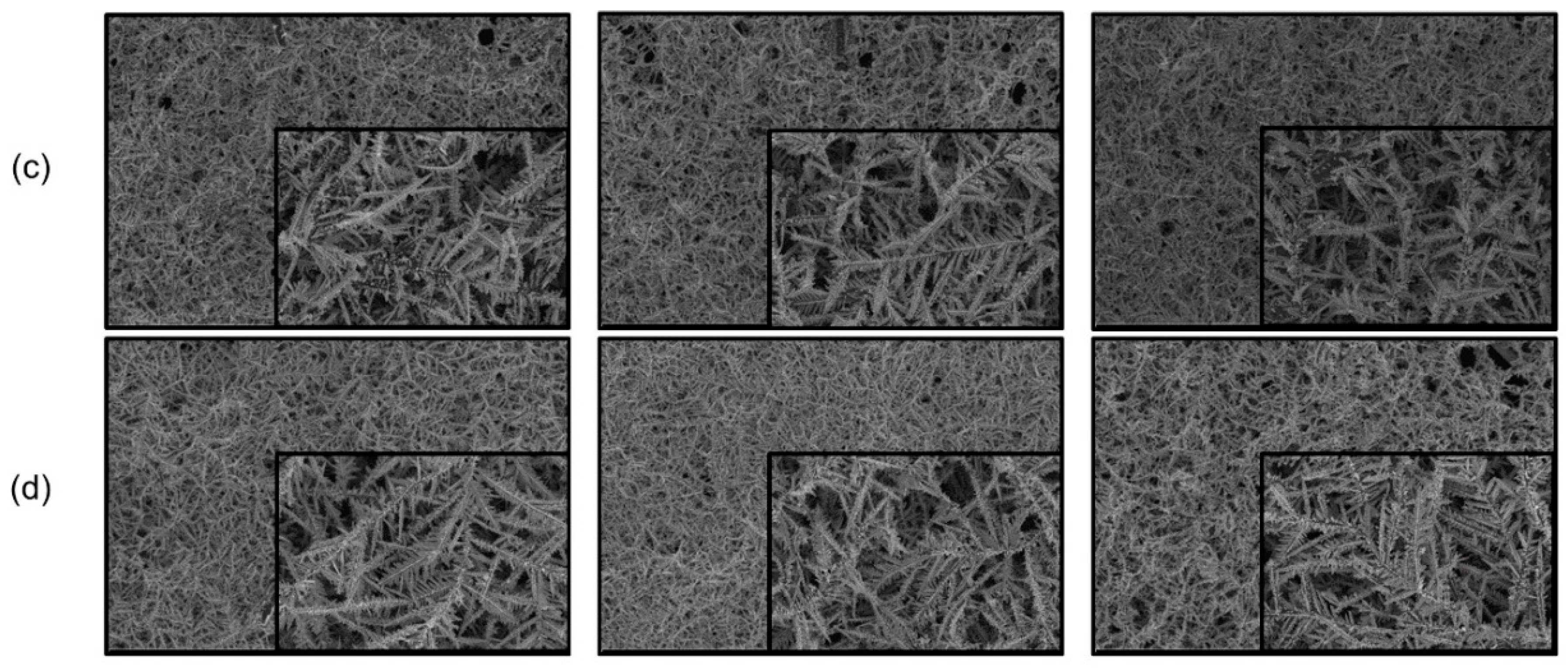
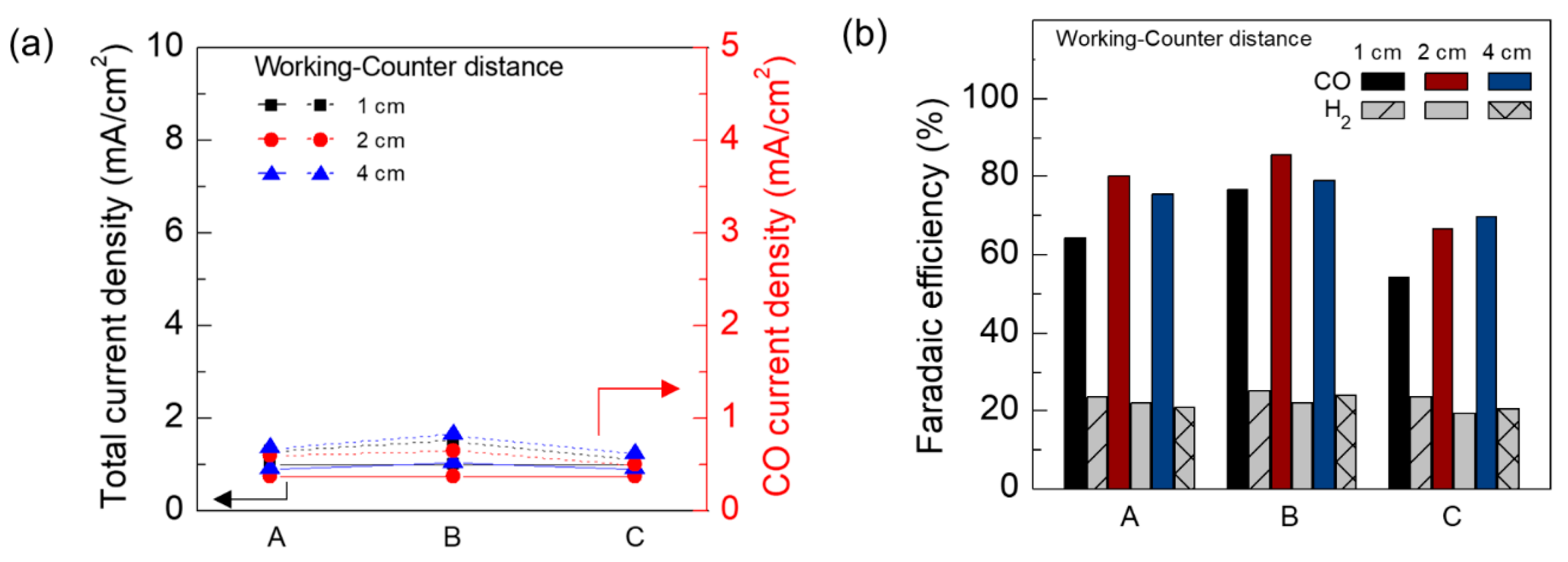
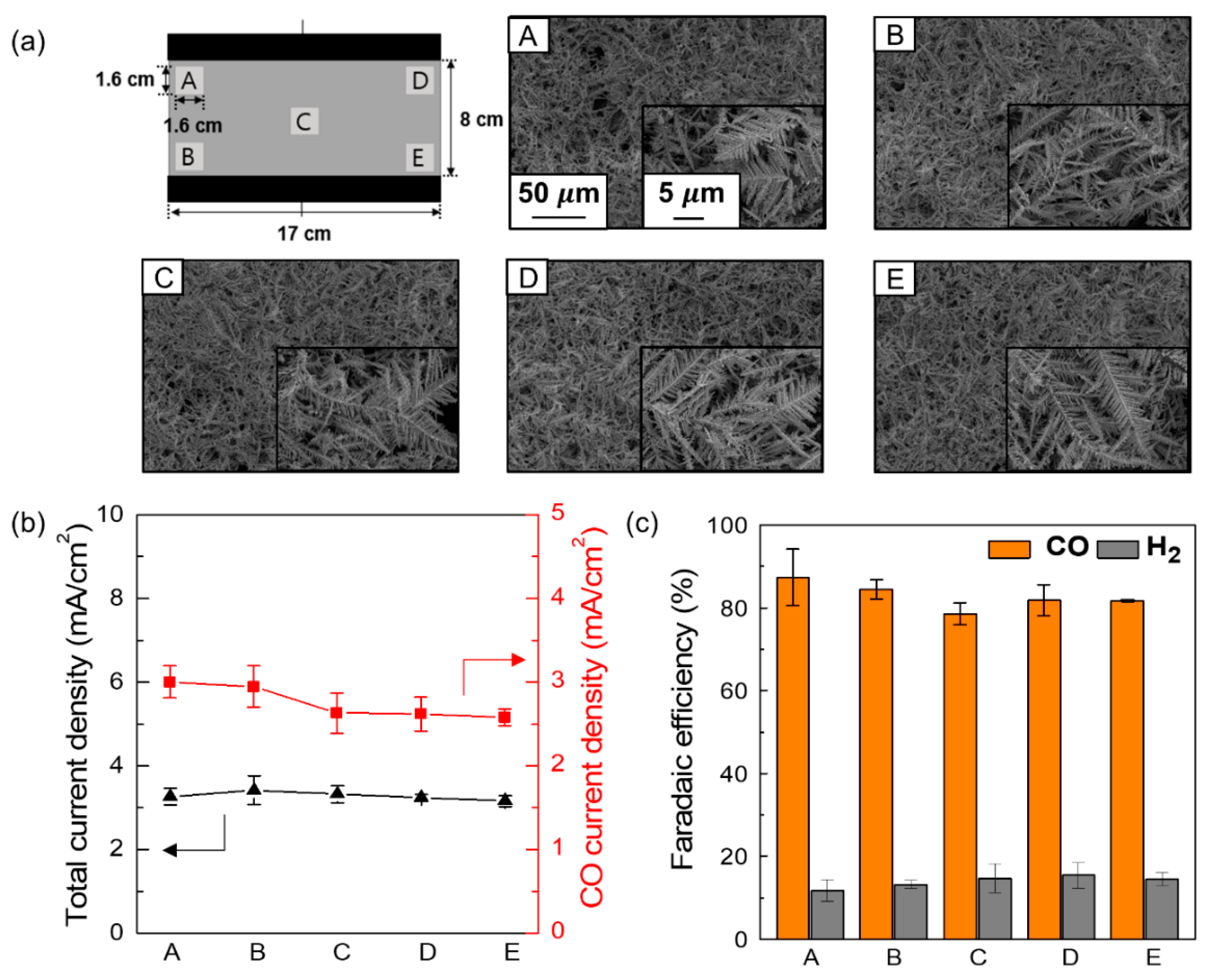
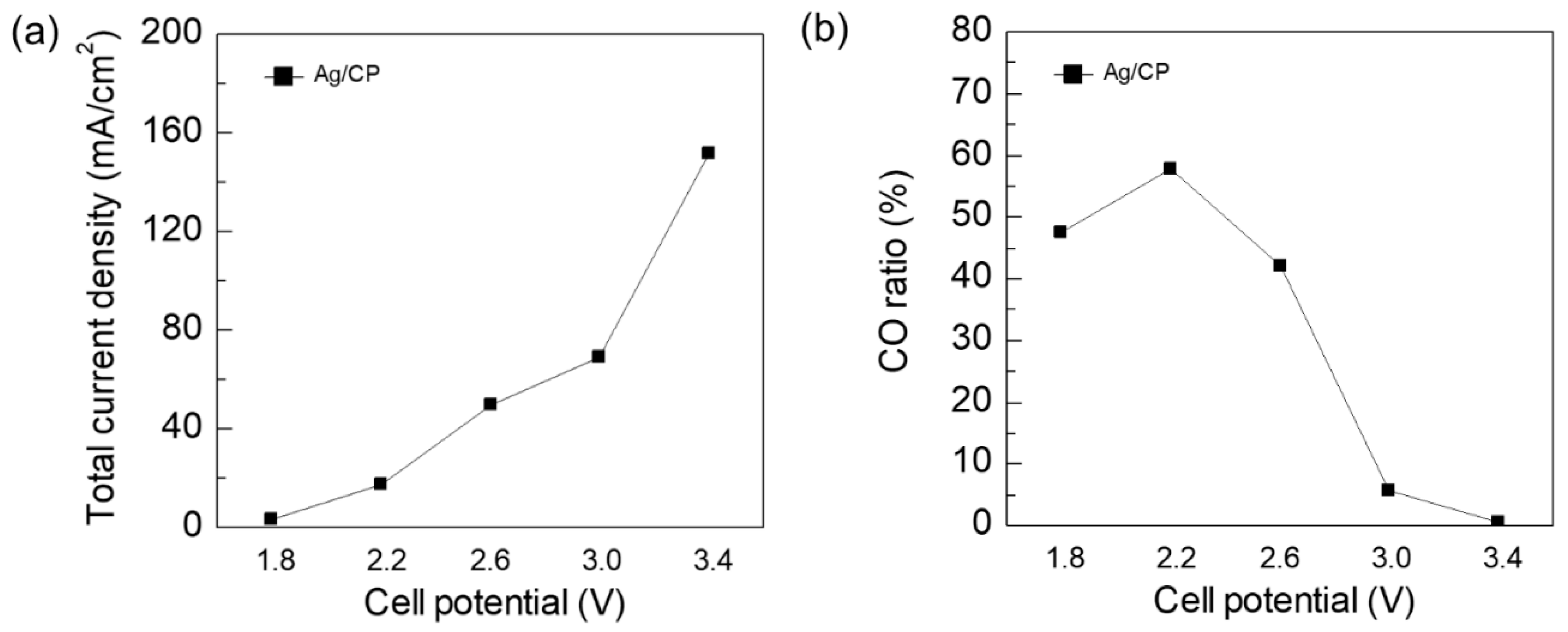
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and Photoelectrochemical Reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jorgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Mendoza, J.A.; Kim, H.K.; Park, H.K.; Park, K.Y. Photocatalytic reduction of carbon dioxide using Co3O4 nanoparticles under visible light irradiation. Korean J. Chem. Eng. 2012, 29, 1483–1486. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.P.; Ma, X.B.; Gong, J.L. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef]
- Whipple, D.T.; Kenis, P.J.A. Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction. J. Phys. Chem. Lett. 2010, 1, 3451–3458. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes; Springer: New York, NY, USA, 2008; Volume 42, pp. 89–189. [Google Scholar]
- Singh, M.R.; Kwon, Y.; Lum, Y.; Ager, J.W.; Bell, A.T. Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu. ACS Catal. 2016, 138, 13006–13012. [Google Scholar] [CrossRef]
- Sun, K.; Wu, L.; Qin, W.; Zhou, J.; Hu, Y.; Jiang, Z.; Shen, B.; Wang, Z. Enhanced electrochemical reduction of CO2 to CO on Ag electrocatalysts with increased unoccupied density of states. J. Mater. Chem. A 2016, 4, 12616–12623. [Google Scholar] [CrossRef]
- Hirunsit, P.; Soodsawang, W.; Limtrakul, J. CO2 Electrochemical Reduction to Methane and Methanol on Copper-Based Alloys: Theoretical Insight. J. Phys. Chem. C 2015, 119, 8238–8249. [Google Scholar] [CrossRef]
- Peterson, A.A.; Norskov, J.K. Activity Descriptors for CO2 electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Chen, C.S.; Wan, J.H.; Yeo, B.S. Electrochemical Reduction of Carbon Dioxide to Ethane Using Nanostructured Cu2O-Derived Copper Catalyst and Palladium(II) Chloride. J. Phys. Chem. C 2015, 119, 26875–26882. [Google Scholar] [CrossRef]
- Yang, K.D.; Ko, W.R.; Lee, J.H.; Kim, S.J.; Lee, H.; Lee, M.H. Morphology-Directed Selective Production of Ethylene of Ethane from CO2 on a Cu Mesopore Electrode. Angew. Chem. Int. Ed. 2016, 56, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Peters, I.; Koper, S.; Kopr, T.M. Electrochemical CO2 reduction to Formic Acid at Low Overpotential and with High Faradaic Efficiency on Carbon-Supported Bimetallic Pd-Pt Nanoparticles. ACS Catal. 2015, 5, 3916–3923. [Google Scholar] [CrossRef]
- Alvarex-Guerra, M.; Quintanilla, S.; Irabien, A. Conversion of carbon dioxide into formate using a continuous electrochemical reduction process in a lead cathode. Chem. Eng. J. 2012, 207–208, 278–284. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Oh, W.; Rhee, C.K.; Han, J.W.; Shong, B. Atomic and Molecular Adsorption on the Bi(111) Surface: Insights into Catalytic CO2 Reduction. J. Phys. Chem. C 2018, 122, 23084–23090. [Google Scholar] [CrossRef]
- Li, W. Electrocatalytic Reduction of CO2 to Small Organic Molecule Fuels on Metal Catalysts; ACS Publications: Washington, DC, USA, 2010; pp. 55–76. [Google Scholar]
- Keim, W. Carbon monoxide: Feedstock for chemicals, present and future. J. Organomet. Chem. 1989, 372, 15–23. [Google Scholar] [CrossRef]
- Kuhl, K.P.; Hatsukade, T.; Cave, E.R.; Abram, D.N.; Kibsgaard, J.; Jaramillo, T.F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. [Google Scholar] [CrossRef]
- Katayama, Y.; Nattino, F.; Giordano, L.; Hwang, J.; Rao, R.R.; Andreussi, O.; Marzari, N.; Shao-Horn, Y. An In Situ Surface-Enhanced Infrared Absorption Spectroscopy Study of Electrochemical CO2 Reduction: Selectivity Dependence on Surface C-Bound and O-Bound Reaction Intermediates. J. Phys. Chem. C 2019, 123, 5951–5963. [Google Scholar] [CrossRef]
- Kim, C.; Jeon, H.S.; Eom, T.; Jee, M.S.; Kim, H.; Friend, C.M.; Min, B.K.; Hwang, Y.J. Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles. J. Am. Chem. Soc. 2015, 137, 13844–13850. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Senanayake, S.D.; Wenqian, Y.Z.; Xu, W.; Polyansky, D.E. Effect of Chloride Anions on the Synthesis and Enhanced Catalytic Activity of Silver Nanocoral Electrodes for CO2 Electroreduction. ACS Catal. 2015, 5, 5349–5356. [Google Scholar] [CrossRef]
- Liu, S.; Tao, H.; Zeng, L.; Liu, Q.; Xu, Z.; Liu, Q.; Luo, J.-L. Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates. J. Am. Chem. Soc. 2017, 139, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Yeom, M.S.; Jung, Y. Active Sites of Au and Ag Nanoparticle Catalysts for CO2 Electroreduction to CO. ACS Catal. 2015, 5, 5089–5096. [Google Scholar] [CrossRef]
- Tornow, C.E.; Thorson, M.R.; Ma, S.; Gewirth, A.A.; Kenis, P.J.A. Nitrogen-Based Catalysts for the Electrochemical Reduction of CO2 to CO. J. Am. Chem. Soc. 2012, 134, 19520–19523. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Forest, R.V.; Moore, A.; Jiao, F. Electrodeposited Zn Dendrites with Enhanced CO Selectivity for Electrocatalytic CO2 Reduction. ACS Catal. 2015, 5, 4586–4591. [Google Scholar] [CrossRef]
- Zhu, W.; Michalsky, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef]
- Luc, W.; Collins, C.; Wang, S.; Xin, H.; He, K.; Kang, Y.; Jiao, F. Ag-Sn Bimetallic Catalyst with a Core-Shell Structure for CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 1885–1893. [Google Scholar] [CrossRef]
- Duan, Z.; Sun, R. Am improved model calculating CO2 solubility in pure water and aqueous NaCl solutions from 273 to 533 K and from 0 to 2000 bar. Chem. Geol. 2003, 193, 257–271. [Google Scholar] [CrossRef]
- Dufek, E.J.; Lister, T.E.; Stone, S.G.; Mcllwain, M.E. Operation of a pressurized system for continuous reduction of CO2. J. Electrochem. Soc. 2012, 159, F514–F517. [Google Scholar] [CrossRef]
- Dufek, E.J.; Lister, T.E.; Mcllwain, M.E. Influence of S Contamination on CO2 Reduction at Ag Electrodes. J. Electrochem. Soc. 2011, 158, B1384–B1390. [Google Scholar] [CrossRef]
- Kim, B.; Ma, S.; Jhong, H.-R.M.; Kenis, J.A. Influence of dilute feed and pH on electrochemical reduction of CO2 to CO on Ag in a continuous flow electrolyzer. Electrochim. Acta 2015, 166, 271–276. [Google Scholar] [CrossRef]
- Delacourt, C.; Ridgway, P.L.; Kerr, J.B.; Newman, J. Design of an Electrochemical Cell Making Syngas (CO + H2) from CO2 and H2O Reduction at Room Temperature. J. Electrochem. Soc. 2008, 155, B42–B49. [Google Scholar] [CrossRef]
- Wu, J.; Risalvato, F.G.; Sharma, P.P.; Pellechia, P.J.; Ke, F.S.; Zhou, X.D. Electrochemical Reduction of Carbon Dioxide II. Design, Assembly, and Performance of Low Temperature Full Electrochemical Cells. J. Electrochem. Soc. 2013, 160, F953–F957. [Google Scholar] [CrossRef]
- Ham, Y.S.; Park, Y.S.; Jo, A.; Jang, J.H.; Kim, S.-K.; Kim, J.J. Proton-exchange membrane CO2 electrolyzer for CO production using Ag catalyst directly electrodeposited onto gas diffusion layer. J. Power Sources 2019, 437, 226898. [Google Scholar] [CrossRef]
- Echendu, O.K.; Okeoma, K.B.; Oriaku, C.I.; Dharmadasa, I.M. Electrochemical Deposition of CdTe Semiconductor Thin Films for Solar Cell Application Using Two-Electrode and Three-Electrode Configurations: A Comparative study. Adv. Mater. Sci. Eng. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Rajkumar, S.R.; Alagar, M.; Kanagasabapathy, M. Current Density Distribution Studies in Manifold Dimensions. J. Chem. Pharm. Res. 2012, 4, 1173–1178. [Google Scholar]
- Park, H.; Choi, J.; Kim, H.; Hwang, E.; Ha, D.-H.; Ahn, S.H.; Kim, S.-K. AgIn dendrite catalysts for electrochemical reduction of CO2 to CO. Appl. Catal. B 2017, 219, 123–131. [Google Scholar] [CrossRef]
- Ham, Y.S.; Choe, S.; Kim, M.J.; Lim, T.; Kim, S.-K.; Kim, J.J. Electrodeposited Ag catalysts for the electrochemical reduction of CO2 to CO. Appl. Catal. B 2017, 208, 35–43. [Google Scholar] [CrossRef]
- Oh, S.; Park, Y.S.; Park, H.; Kim, H.; Jang, J.H.; Choi, I.; Kim, S.-K. Ag-deposited Ti gas diffusion electrode in proton exchange membrane CO2 electrolyzer for CO production. J. Ind. Eng. Chem. 2020, 82, 374–382. [Google Scholar] [CrossRef]
- Choi, J.; Kim, M.J.; Ahn, S.H.; Choi, I.; Jang, J.H.; Ham, Y.S.; Kim, J.J.; Kim, S.-K. Electrochemical CO2 reduction to CO on dendritic Ag-Cu electrocatalysts prepared by electrodeposition. Chem. Eng. J. 2016, 299, 37–44. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.; Park, H.; Kim, H.; Park, Y.S.; Ha, M.G.; Jang, J.H.; Kim, S.-K. Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction. Coatings 2020, 10, 341. https://doi.org/10.3390/coatings10040341
Oh S, Park H, Kim H, Park YS, Ha MG, Jang JH, Kim S-K. Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction. Coatings. 2020; 10(4):341. https://doi.org/10.3390/coatings10040341
Chicago/Turabian StyleOh, Seonhwa, Hyanjoo Park, Hoyoung Kim, Young Sang Park, Min Gwan Ha, Jong Hyun Jang, and Soo-Kil Kim. 2020. "Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction" Coatings 10, no. 4: 341. https://doi.org/10.3390/coatings10040341
APA StyleOh, S., Park, H., Kim, H., Park, Y. S., Ha, M. G., Jang, J. H., & Kim, S.-K. (2020). Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction. Coatings, 10(4), 341. https://doi.org/10.3390/coatings10040341