3.1. Single-Stranded DNA Adsorption on GO in the Presence of Mg2+ and Cl− Ions
After the equilibration step, Mg
2+ ions were found adsorbed on the GO surface, near oxidised functional groups or associated with the oligonucleotide negatively charged backbone. Cl
− ions could also interact closely with the DNA molecule and influence its behaviour through electrostatic interactions. Divalent Mg
2+ ions effectively blanketed the negatively charged oxygen atoms present on the GO surface (see
Supplementary Figure S2) and helped stabilize the tertiary structure of the ssDNA molecule. No such specific pattern of distribution was observed in the case of the Cl
− ions. These results were consistent with simulations, indicating that a low-mobility layer of counterions could compensate for the graphenic surface charge and influence oligonucleotide interaction [
28].
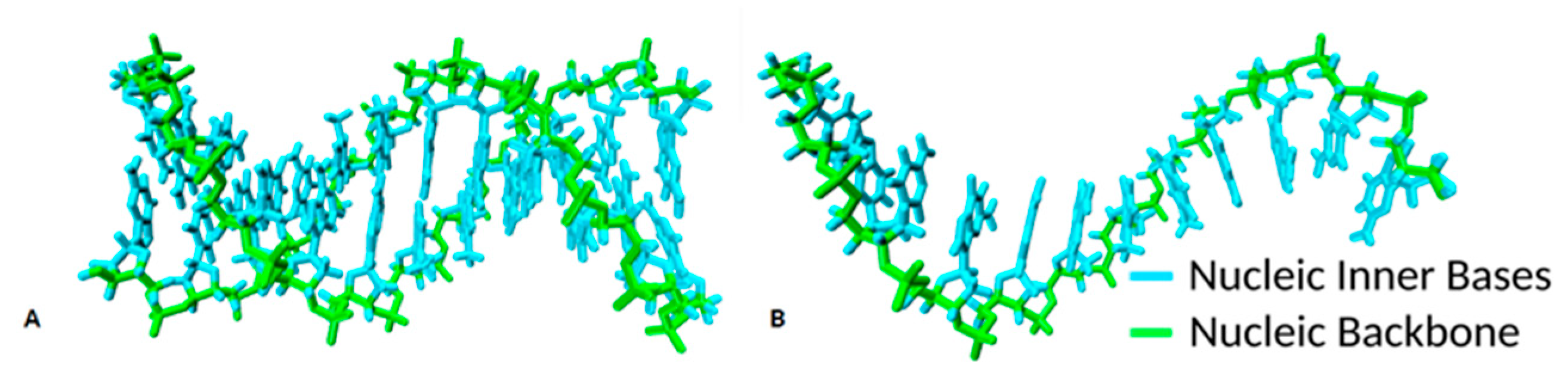
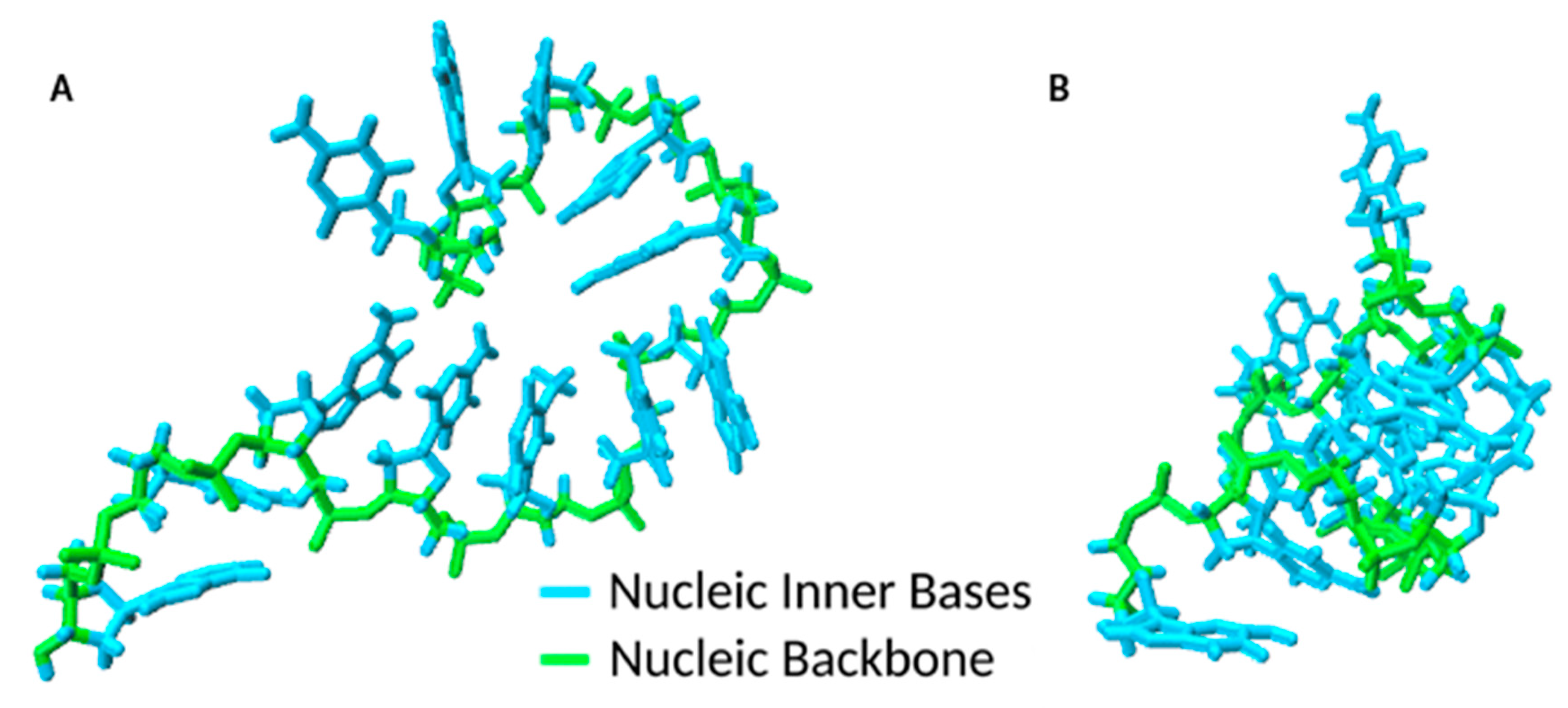
As anticipated, the ssDNA had inherent complexity arising from diverse local conformations such as loops, hairpins and pseudoknots [
29], and different ssDNA molecular conformations (
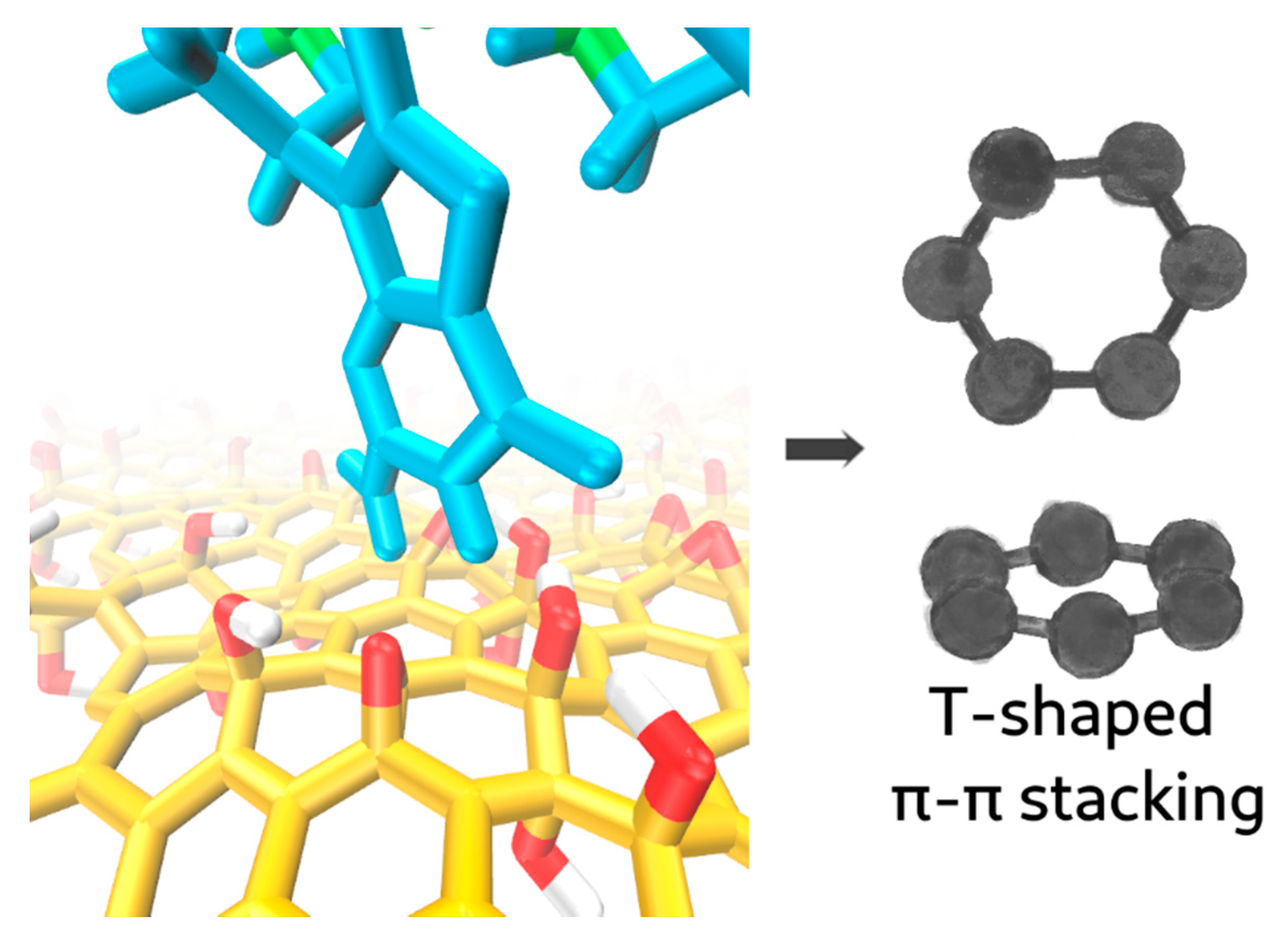
Figure 3) could be adsorbed on GO through a process of dynamic interactions. This process reflected the high flexibility of the ssDNA conformations in solution with exposed nucleobases orientated in all directions as the oligonucleotide chain continuously altered its ionic interactions and conformation. In addition, a more peculiar type of π–π stacking (T-shaped) was observed. The event is shown in
Figure 4 and described later in the manuscript.
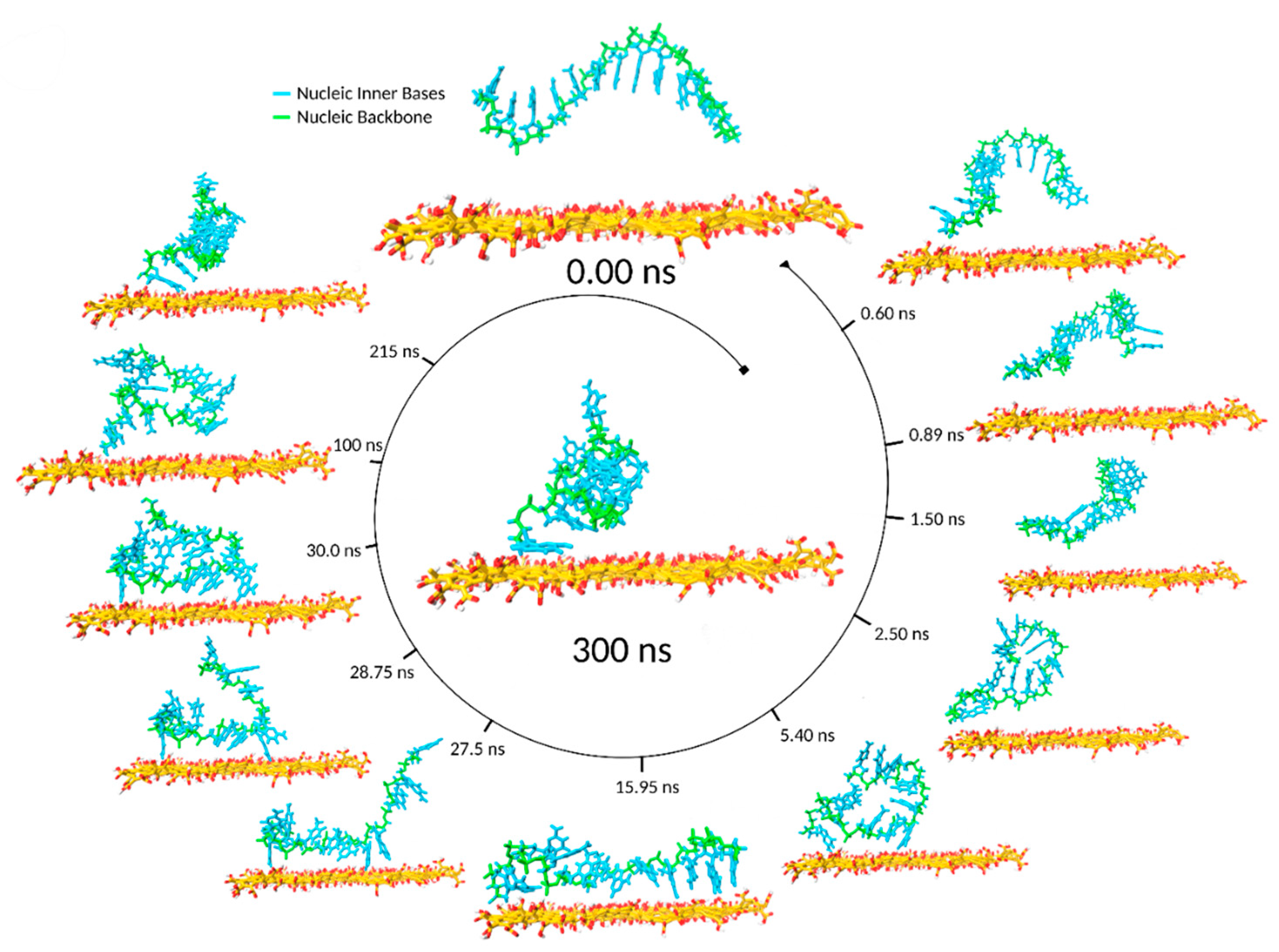
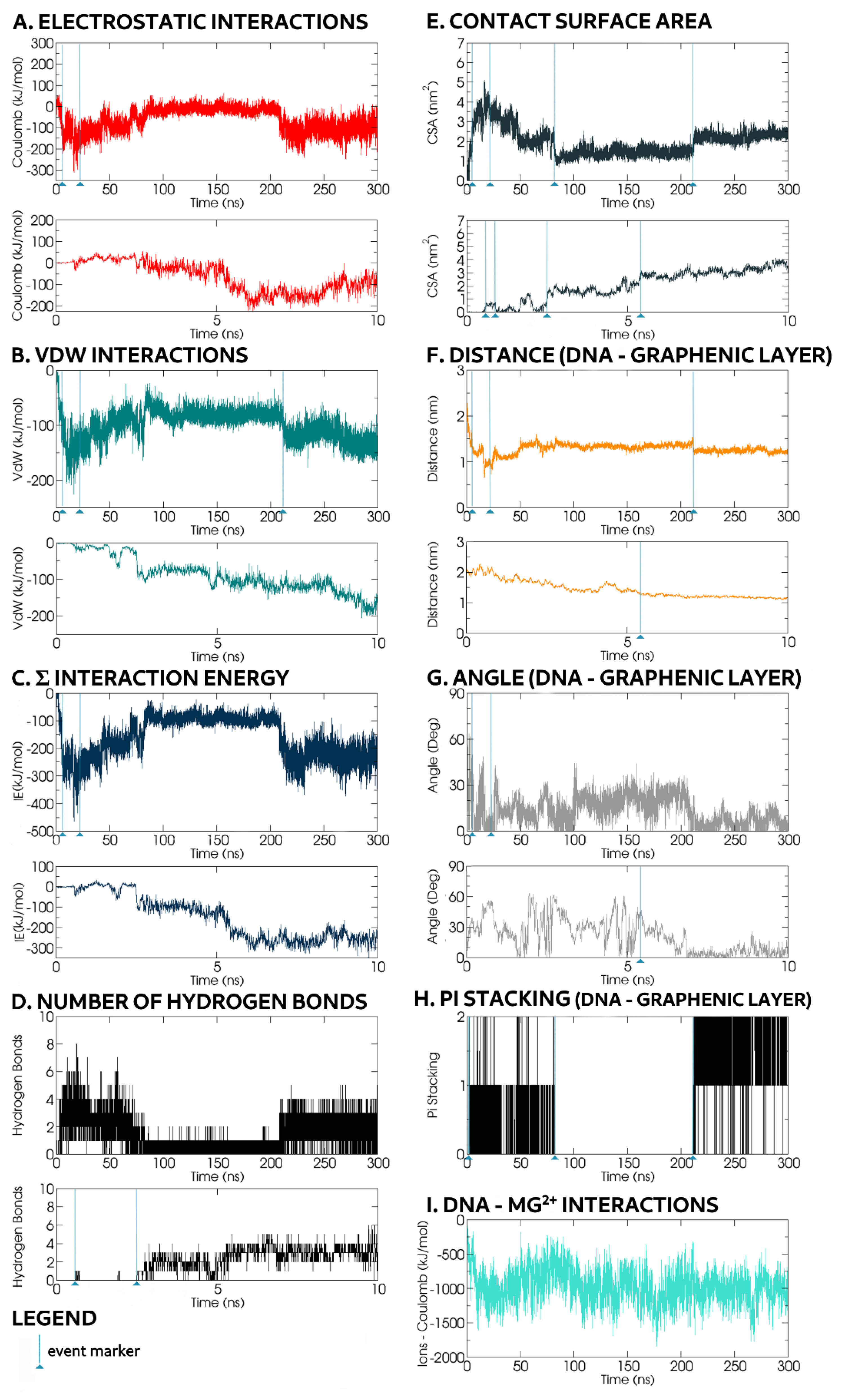
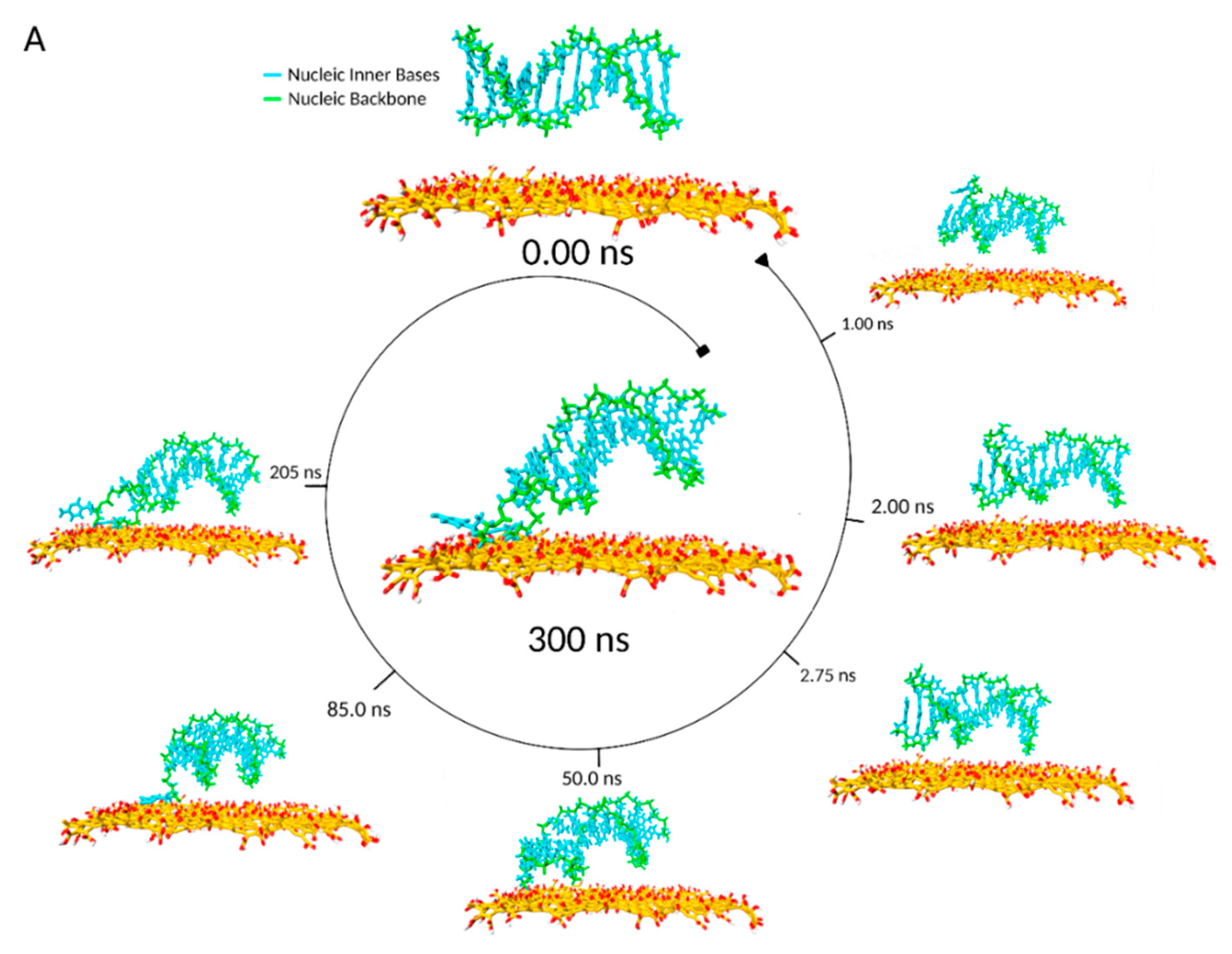
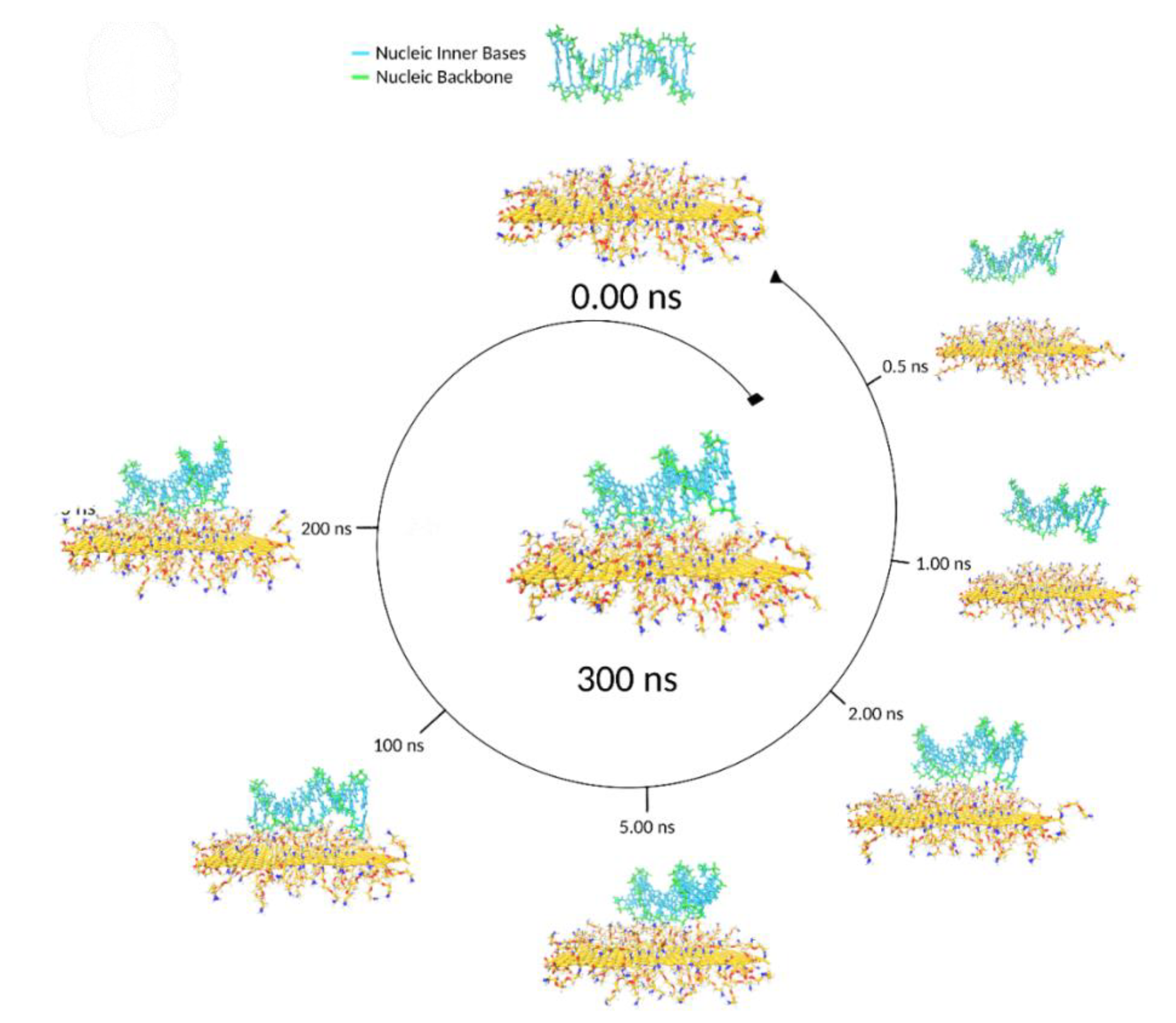
During a typical MD timeline, starting from the ssDNA backbone axis parallel to the GO surface plane (
Figure 5), one or both ssDNA chain ends approached the graphene surface. Given a more energetically favourable location, one end reached the GO surface faster, and at
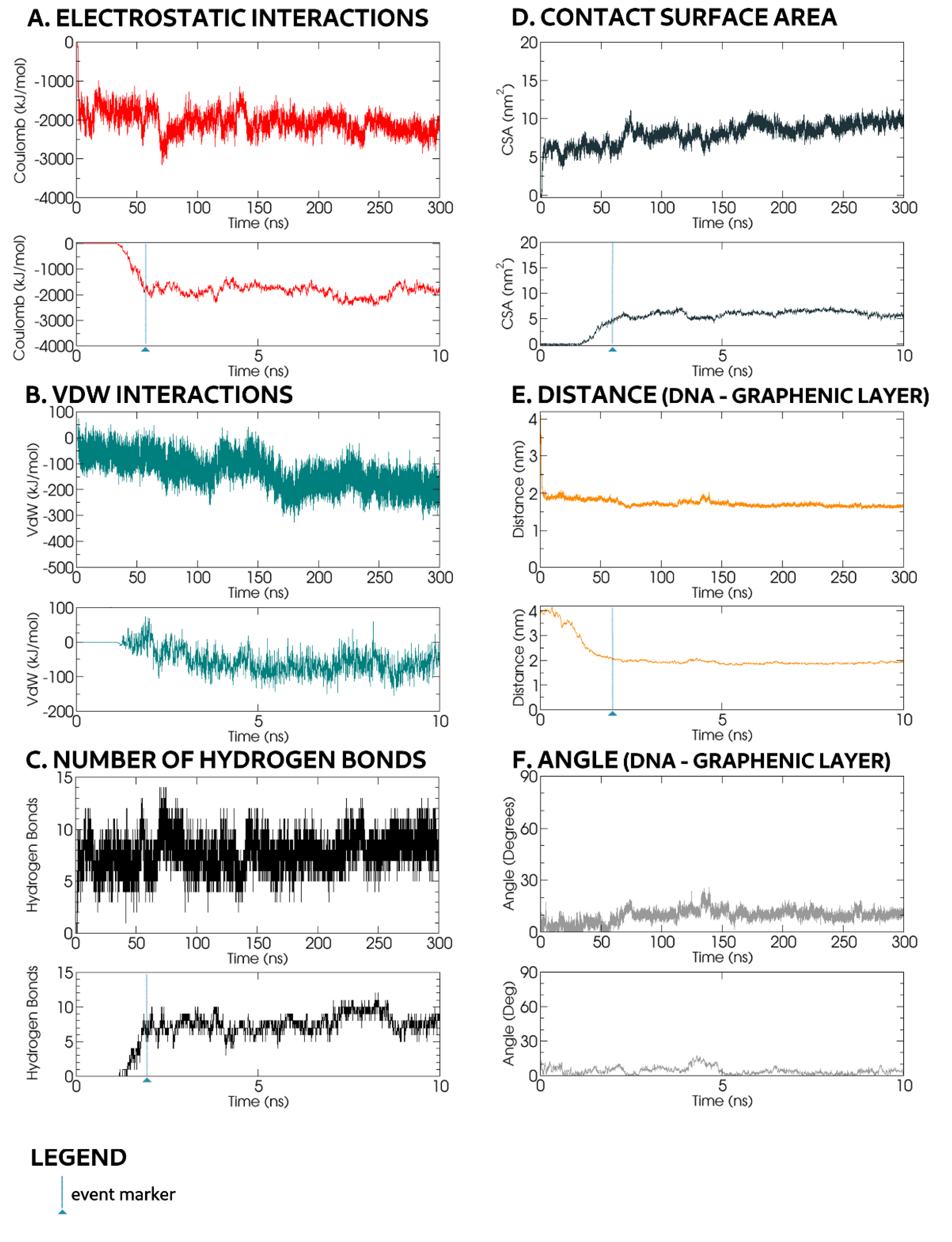
t = 0.6 ns, the first hydrogen bond was formed (
Figure 6D). Concurrently, the contact surface area increased to 0.64 nm
2 (
Figure 6E).
Unless a second hydrogen bond was promptly formed, the existing hydrogen bond broke, and the detached ssDNA returned to a CSA value of 0 nm
2 at
t = 0.89 ns. Temporary hydrogen bond formation occurred whenever a second hydrogen bond was not formed soon enough to support the first. As the Mg
2+ ions competed with each other for the GO oxidised groups, the ssDNA positioned itself preferentially in the vicinity of the unoxidised regions before adsorption. This was in marked contrast to earlier MD studies that simulated GO/ssDNA interactions without Mg
2+ ions, when ssDNA segments preferentially bound to oxidised versus unoxidised regions of a GO surface [
11].
As the oligonucleotide chain remained close to the GO, the CSA value increased up to 2 nm
2 at
t ≈ 2.50 ns, corresponding to the formation of three hydrogen bonds and a T-shaped π–π stacking in which the rings adopt a perpendicular geometry [
30], as shown in
Figure 4. This type of π-stacking has been previously observed in DNA–protein interactions [
31]. Despite respecting our π–π stacking criteria, the GO region involved was heavily oxidised, and this oxidation could shield and significantly weaken the π-stacking energy. The T-shaped stacking broke apart at
t = 82ns.
At
t ≈ 5.4 ns, the CSA value increased to 3.15 nm
2, corresponding to the formation of one more hydrogen bonds. For the rest of the simulation, at least one hydrogen bond persisted between the ssDNA and the GO surface. Within this
t = 5.4 ns timeframe, the distance between the backbone of the ssDNA and the graphene surface carbon atoms narrowed from 2.25 to 1.30 nm. The oligonucleotide conformation, resembling
Figure 3A, had a terminal nucleobase forming hydrogen bonds with oxidised groups that anchored the rest of the oligonucleotide chain to the GO surface. The ssDNA backbone region that connected to the GO anchored terminal base was bridged by Mg
2+ ions and lay flat parallel to the plane of the graphene. The rest of the ssDNA was looped over this GO-associated oligonucleotide chain, allowing Mg
2+ ions to reside in between these two oligonucleotide backbone regions. During the next 17 ns, almost all of the ssDNA backbone was bridged by Mg
2+ ions. Towards the end of this 17 ns duration, the backbone was flattened with a small axis angle (0–10°) onto the GO surface plane (
Figure 6G). Throughout this transitional interaction, most of the parameters were at or near maximal or minimal absolute values; VdW energy peaked at −200 kJ mol
−1, Coulomb energy: ≈300 kJ mol
−1, CSA reached ≈5 nm
2 and intermolecular distance ranged between 0.70 and 0.75 nm. Despite Mg
2+ ion-mediated oligonucleotide backbone interactions with the GO surface, some nucleobases formed hydrogen bonds with GO oxidised moieties, and were thus oriented towards the GO surface.
This lying-flat adsorption conformation was not necessarily stable. In the representative simulation, by
t = 27.5 ns, the region of the ssDNA backbone carrying free terminal nucleobases was quickly associated with floating Mg
2+ ions in preference to adsorption onto the GO surface. In the following 7 ns, the molecule returned to a condensed conformation stabilised by intramolecular π–π stacking (
Figure 3B).
During the next 200 ns, the ssDNA migrated from site to site on the GO surface, displaying a CSA value close to 1.5 nm
2 until, at
t = 212 ns, the contacting base bonded to an oxidised group through hydrogen bonds and further relaxed its conformation forming a π–π stacking interaction with an adjacent graphene ring. This correlated well to CSA jumping from 1.5 to 2.5 nm
2 (
Figure 6E) and a decreased VdW interaction energy from −75 kJ mol
−1 to −125 kJ mol
−1 (
Figure 6B). The distance, VdW, Coulomb and CSA values remained constant for the rest of the simulation. The overall molecular alignment of the oligonucleotide chain nucleobases was towards the centre of the backbone loop, away from nearby Mg
2+ ions attracted to the negatively charged phosphate backbone. Small periodic oscillations resulted in slight fluctuations of the angle (0–20°) between the ssDNA phosphate backbone and the GO (
Figure 6G).
In summary, the ssDNA molecule was adsorbed on the surface of the GO with part of its oligonucleotide chain bridged by the Mg
2+ ions to the GO surface, the remainder adopted a folded conformation with its oligonucleotide backbone looped onto itself (
Figure 3A,B). The π–π stacking interactions played a key role in allowing the oligonucleotide to anchor, rather than migrate, on the GO surface with binding energies influenced by the presence of hydrogen bonds and electrostatic interactions between the GO surface adsorbed Mg
2+ ions and the ssDNA. Given the very dynamic conformational changes of ssDNA, we do not exclude the possibility that other adsorption mechanisms may exist. Future exploration of different sets of ion parameters, such as CUFIX [
32], and the use of force fields that allow continuous bond formation and breakage, such as ReaxFF [
33], may improve accuracy for biosensor design. The computational approach used in this work may be extended to testing different metal ions and different graphene derivatives, or other novel 2D materials, to pursue scenarios under which ssDNA and dsDNA are easy to differentiate due to different behaviour.
3.2. Double-Stranded DNA Adsorption on GO in the Presence of Mg2+ and Cl− Ions
During the equilibration step, the more restrained dsDNA movement allowed the ions to move freely from randomly assigned initial positions to more energetically favourable ones. As was found for the equilibration step for ssDNA simulations, Mg
2+ ions were adsorbed on the surface of the GO near the oxidised functional groups. Moreover, they were also predominantly associated with the negatively charged dsDNA backbone. In effect, Mg
2+ ions formed a thin, positively charged blanket over the negatively charged oxygen atoms present on the GO surface (see
Supplementary Figure S2) and also stabilized the tertiary structure of the DNA molecule. No such specific pattern of distribution was observed in the case of the Cl
− ions.
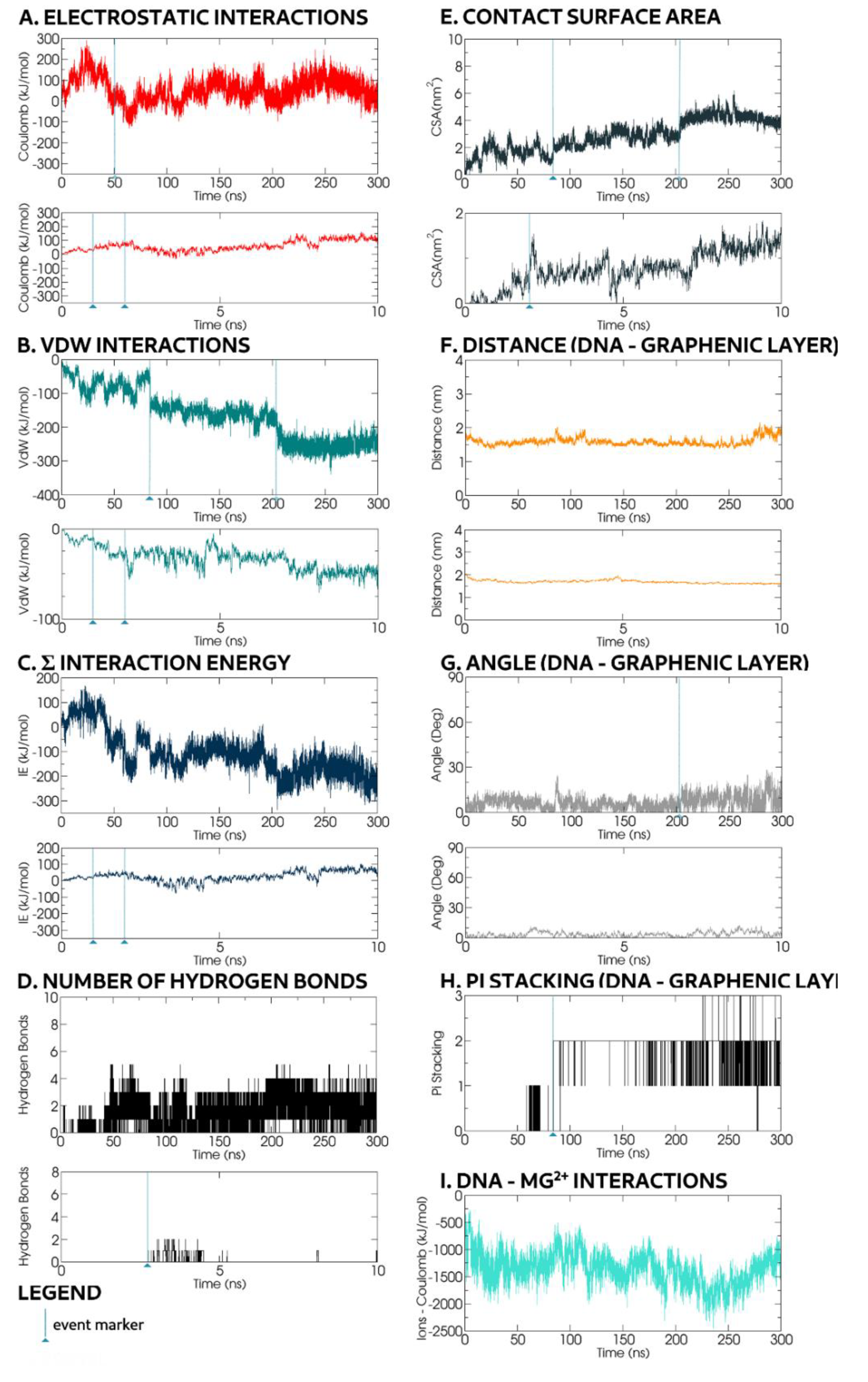
The trajectory snapshots (
Figure 7) for representative dynamic dsDNA adsorption interactions with the GO progressed with the dsDNA backbone parallel to the graphenic surface without significant rotation, regardless of the dsDNA’s initial orientation. During the first 2 ns, the distance between the nucleic backbone and the GO fell from 2 nm to 1.63 nm (
Figure 8F) and remained within a ±0.5 nm range for the rest of the simulation. At the end of the mentioned translocation phase (2 ns), it lay tangentially with only a small angle (0–5°) into the GO plane (
Figure 8G). This finding is in contrast with previous MD studies concerning GO-oligonucleotide without the presence of Mg
2+ or Cl
− ions, where the same dsDNA oligonucleotide axis rotated quickly and landed perpendicularly to the GO surface [
9,
10].
Our energy-related graphs indicated a small repulsive net force between 1 and 2 ns in
Figure 8C, as a result of an 80 kJ mol
−1 electrostatic interaction and −30 kJ mol
−1 VdW interaction as shown in
Figure 8A,B. Despite this result, the dsDNA approached the GO because the Mg
2+ ions formed a positively charged blanket over the negatively charged oxygen atoms on the GO surface, thereby bridging the negatively charged dsDNA phosphate backbone with the GO surface. Indeed, within 1.4 nm of both the GO and oligonucleotide, the Mg
2+ ions alone exerted a significant dominant attractive force on the dsDNA (
Figure 8I). The GO–dsDNA intermolecular distance changed minimally over the rest of the simulation time, but yet it was greater than the distances proposed by previous studies of dsDNA adsorption on GO without the Mg
2+ and Cl
− ions, because of the Mg
2+ ions bridged between the dsDNA and GO molecules.
Beyond these ionic interactions, the dsDNA further interacted with the GO through hydrogen bonds, first observed at
t = 2.75 ns (
Figure 7 and
Figure 8D). Correspondingly, the contact surface area reached 0.5 nm
2 at
t = 2 ns (
Figure 8E) and fluctuated between 0.5 and 1.5 nm
2 as the dsDNA conformation oscillated to reach an energetically favourable configuration for the hydrogen bonds to be formed. The initial hydrogen bonds were transient, altered by movement of ions in solution and dsDNA perturbation of ions adsorbed on the GO surface. For the electrostatic interactions that involved the bridging Mg
2+ ions dominating other interactions (
Figure 8I), the dsDNA molecule drifted on the xy-plane with existing hydrogen bonds broken and was recreated in different positions.
Curiously, at the
t = 1 and
t = 2 ns snapshots (
Figure 7), in the presence of Mg
2+, the terminal bases of the dsDNA broke their π–π stacking interactions and hydrogen bonds formed with the other bases, apparently stochastically. This event occurred repeatedly throughout the simulation for all four terminal bases. At
t ≈ 50 ns, one inner base broke its intramolecular interactions to form hydrogen bonds with the nearby GO surface oxidised groups. Concurrently, the dsDNA-GO electrostatic energy dropped below 0 kJ mol
−1 (
Figure 8A). The base continued to move between local functional groups present on the GO surface until, at
t = 72 ns, it reached the edge of an unoxidised region. In the following 12 ns, the two hydrogen bonds formed with persisting nearby epoxy groups. Both Mg
2+ ions and freed dsDNA bases competed for interaction with the oxidised GO surface. Eventually, at
t = 84 ns, the freed base was constrained inside the unoxidised region by Mg
2+ ions, where it quickly formed π–π stacking interactions with the available graphene rings (
Figure 8H). This was marked by a sharp energy drop from −72 kJ mol
−1 to −148 kJ mol
−1 (
Figure 8B) and, for the next 116 ns, only oscillated within ±25 kJ mol
−1. Concurrently, the contact surface area, increased from 1 nm
2 to 2 nm
2 (
Figure 8E). For the next 116 ns, the п-stacked base migrated within the unoxidised region, confining the dsDNA molecule range of movement on the xy-plane.
At
t ≈ 200 ns, a second inner dsDNA base, complementary to the base within an unoxidised region, broke away from the intramolecular π–π stacking interactions with neighbouring bases, moved chaotically and soon formed hydrogen bonds with nearby oxidised groups on the GO surface. Thus, the CSA increase from 3 to 4 nm
2 (
Figure 8E) and a two-step increase of VdW attractive forces from −187 to −240 kJ mol
−1 (
Figure 8B) corresponded to the formation of two hydrogen bonds. Subsequently, the VdW energy value remained constant at −250 kJ mol
−1 throughout the rest of the simulation. The CSA value remained constant at 4 nm
2 with only small fluctuations of ±0.25 nm
2 (
Figure 8E). These values indicated that the dsDNA molecule was adsorbed on the surface of the GO molecule.
Notably, when situated in an oxidised area, the newly freed base’s inclination changed according to the influence of nearby ions, but its x–y plane position remained fixed by hydrogen bonds between its corresponding dsDNA backbone segment and the nearby oxidised groups. Furthermore, when the dsDNA was anchored at one end by its two freed bases, the dsDNA backbone angle with the GO plane oscillated between 5 and 30°. During the final 100 ns of the simulation, the dsDNA moved away from the GO before returning to an almost parallel position, while anchored.
In summary, the dsDNA was adsorbed on GO in the presence of Mg2+ and Cl− and first lay axially flat, its negatively charged phosphate backbone bridged by Mg2+ ions to oxidised groups on the GO surface. Given that the electrostatic attractive forces between the dsDNA and Mg2+ ions dominated the interactions of the dsDNA with the GO, we expect the distribution of the ions adsorbed on the GO surface to be the most influential factor for successful adsorption. However, solute ions could disrupt intramolecular π–π stacking interactions, allowing the oligonucleotide bases to instead associate via hydrogen bonds to oxidised GO surface regions. In adjacent unoxidised GO regions, the oligonucleotide base could form π–π stacking interactions that anchored it to the graphene rings of the graphene molecule. The unravelling of the terminal bases of the dsDNA at different times throughout the simulations could ultimately invoke stronger π–π interaction-mediated oligonucleotide anchoring on the GO surface. This phenomenon may be dependent on ionic concentration. Future studies should address whether the predicted oscillatory pattern from the data for 300ns simulations for the dsDNA backbone may persist over longer simulation times. Generally, the energy and CSA values were greater for dsDNA compared with ssDNA, and thus in the presence of Mg2+ ions we would expect dsDNA to have a greater binding affinity for GO than ssDNA. For building a broader overview of dsDNA adsorption in the presence of Mg2+ and Cl− ions, different ionic concentrations and dsDNA molecules of varying lengths should be simulated.
3.3. Single-Stranded DNA Adsorption on rGO-PEG-NH2 in the Presence of Mg2+ and Cl− Ions
After the NVT equilibration step, Cl
− ions positioned themselves between and around the PEG–NH
2 chains, whereas Mg
2+ ions were primarily located further away from the rGO surface. However, some Mg
2+ ions were found around the negatively charged oligonucleotide backbone. Through a series of cooperative non-bonding interactions, the ssDNA molecule was adsorbed on the rGO-PEG-NH
2 species in the presence of the Mg
2+ and Cl
− ions. Two main simulation outcomes were revealed: The ssDNA backbone would either lie entirely on top of the PEG-NH
2 chains, or one of its ends would reach through the PEG-NH
2 chains and contact the rGO surface, eventually forming п–п interactions with one of its graphene rings. The adsorbed ssDNA conformation was not necessarily stretched, but could form loops, hairpins and pseudoknots [
29] due to the torsion of its backbone. The main forces involved were electrostatic interactions, hydrogen bonds and eventually π–π stacking interactions.
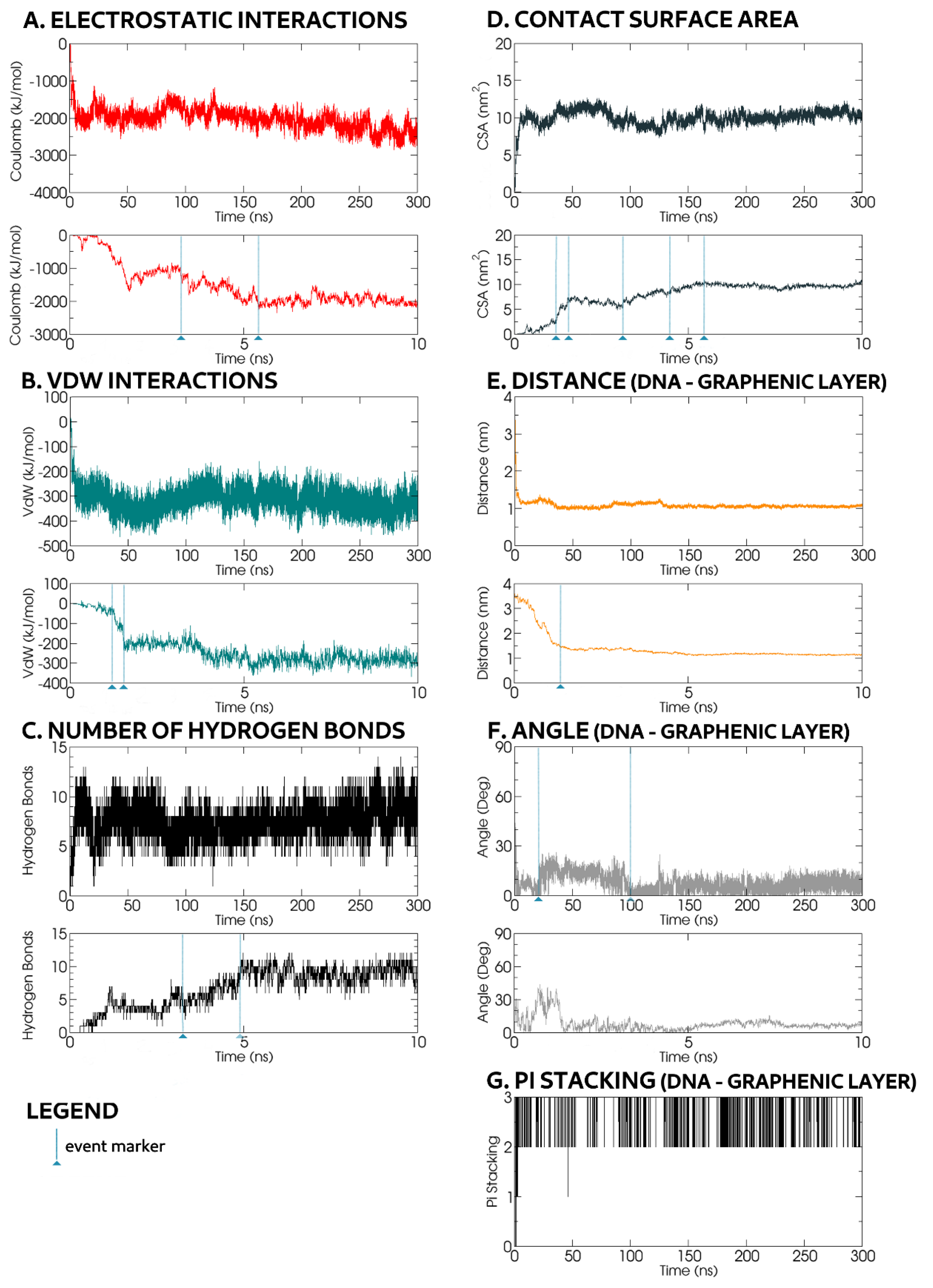
In a typical example, most dynamic adsorption events took place within the first 2 ns. Depending on the initial ssDNA orientation, one end approached the rGO-PEG-NH
2, with its phosphate backbone facing the GO surface, achieving contact in ~0.5 ns (
Figure 9). Concurrently, the CSA value rose (
Figure 10D), while transient hydrogen bonds were reformed (
Figure 10C).
The opposite ssDNA end moved towards the PEG-NH
2 chains, while the one already in contact with rGO sank deeper into the PEG chain layer according to chain orientation. Within
t = 1.33 ns, the intermolecular distance decreased from 4 nm to 1.33 nm, reaching 1.07 nm at 300 ns (
Figure 10E). At
t = 1.21 ns, during intermolecular proximation, CSA (
Figure 10D) and VdW (
Figure 10B) values suddenly changed: CSA increased from 2.19 nm
2 at
t = 1.21 ns to 7.2 nm
2 at
t = 1.58 ns and VdW energy decreased from −40 kJ mol
−1 at
t = 1.21 ns to −220 kJ mol
−1 at
t = 1.58 ns (
Figure 10B). These events corresponded to the formation of a π–π stacking structure (
Figure 10G). Thus, although one ssDNA end was attracted to the top of the PEG-NH
2 chains, the oligonucleotide bases at the other end were anchored the ssDNA to the rGO surface via π–π interactions. This behaviour was more likely when ssDNA was situated in a region with a lower density of PEG-NH
2 chains, where more space was available for its bases to form π–π stacking structures with the graphene rings.
The shape of the ssDNA molecule after
t = 2 ns remained largely similar until the end of the simulation at
t = 300 ns (
Figure 9). At
t = 22 ns, the angle between the oligonuncleotide backbone and the rGO surface increased to 15–20°, but decreased to 5–10° at
t = 100 ns, remaining so until the end of the simulation (
Figure 10F).
Events occurring after
t = 2 ns included CSA incrementation, from 5 nm
2 at
t = 3.18 ns to 8 nm
2 at
t = 4.47 ns and 10 nm
2 at
t = 5.46 ns (
Figure 10D), associated with ssDNA shape rearrangement as the number of hydrogen bonds with the rGO-PEG-NH
2 molecule grew from 4–5 at
t = 3.18 ns to 10 at
t = 4.9 ns (
Figure 10C), while Coulomb interactions grew from −1000 kJ mol
−1 at
t = 3.18 ns to −2200 kJ mol
−1 at
t = 5.4 ns (
Figure 10A).
3.4. Double-Stranded DNA Adsorption on rGO-PEG-NH2 in the Presence of Mg2+ and Cl− Ions
During the NVT equilibration step, Cl
− ions positioned themselves between and around the PEG-NH
2 chains, whereas Mg
2+ ions were primarily located further away from the graphene. However, some Mg
2+ ions were found around the negatively charged phosphate backbone of the dsDNA. Snapshots of simulation events are displayed in
Figure 11, while the graphs of specific parameters are shown in
Figure 12.
For rGO-PEG-NH2 in the presence of Mg2+ and Cl− ions, dsDNA was adsorbed with its backbone orientation parallel to the graphene layer. The main forces involved were electrostatic interactions and to a lesser extent hydrogen bonds. In contrast with GO simulations, π–π stacking interactions did not play a role in the dsDNA adsorption process.
Initially, the dsDNA molecule moved towards the rGO, swaying with loss of some associated Mg
2+ ions. Within
t = 2 ns, the oligonucleotide backbone to rGO surface distance fell from 4 to 2 nm (
Figure 12E), and CSA increased sharply to 5 nm
2 (
Figure 12D) as the two molecules made contact with the dsDNA backbone captured by the PEG-NH
2 chains (
Figure 11). The predominant electrostatic Coulomb interactions reached −2000 kJ mol
−1 (
Figure 12A). Approximately 7–8 hydrogen bonds were formed early in the reaction (
Figure 12C); however, the VdW interaction value was relatively weak at −100 kJ mol
−1 (
Figure 12B).
From this point onward, the position of the dsDNA persisted throughout the rest of the simulation. We noted only modest trends, e.g., the value of the inter-distance graph (
Figure 12E) fell from 2 nm at
t = 2 ns to 1.65 nm at
t = 300 ns as the number of hydrogen bonds increased to between 10 and 12. The energy of the VdW interactions increased correspondingly from the aforementioned −100 kJ mol
−1 to −200 kJ mol
−1 at
t = 300 ns. We could therefore assume that the dsDNA became further associated with the PEG chains following their initial interaction.
Of note, the adsorbed dsDNA was influenced by both the Mg
2+ ions and PEG chains. As the dsDNA sank among the PEG chains, the associated segments of the backbone became stretched out. In contrast, for dsDNA grooves facing away from the rGO layer, Mg
2+ ions maintained a more contracted oligonucleotide conformation. Overall, the dsDNA became arched, changing its orientation angle (
Figure 11). The angle between the oligonucleotide axis and the graphenic surface fluctuated slightly (±5°) over time (
Figure 12F).
Thus, we propose that dsDNA molecule was adsorbed on the surface of the rGO-PEG-NH2 species primarily due to electrostatic interactions with the oligonucleotide backbone in an almost parallel orientation to the plane of the graphenic surface. Future studies may seek to optimize the charge derivation method for new computational models of graphene derivatives by making use of ab initio techniques, such as Density Functional Theory.
To aid with following the events described in this section, please see
Table 1.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



