Analogues of a Cyclic Antimicrobial Peptide with a Flexible Linker Show Promising Activity against Pseudomonas aeruginosa and Staphylococcus aureus

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Stage 1. The Impact of Cyclization Point on Antimicrobial Activity—Replacing Lys with Asn.
2.2. Stage 2. The Impact of Manipulating Lipophilicity on Antimicrobial Activity. Replacing Nal (3-(2-naphtyl)-L-alanine) and Bip (L-biphenylananine) with Phe.
2.3. Stage 3. Importance of Amino Acid Sidechain Length on Antimicrobial Activity—Replacing Lys with Dab or Arg.
2.4. Stage 4. Reducing Hydrophobicity of Stage 3 Lead Peptide by Replacing Dab with Arg and Bip with Phe.
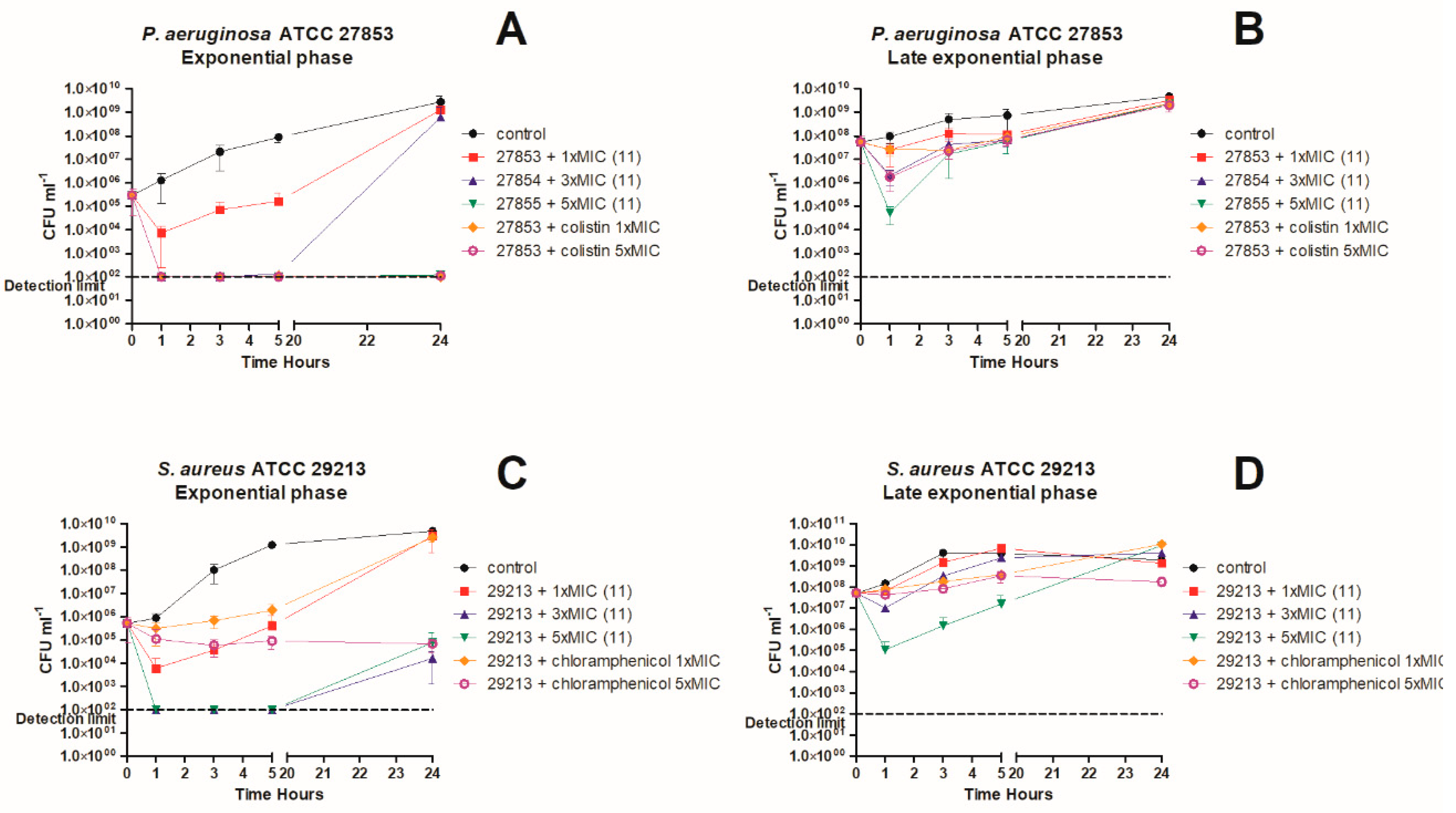
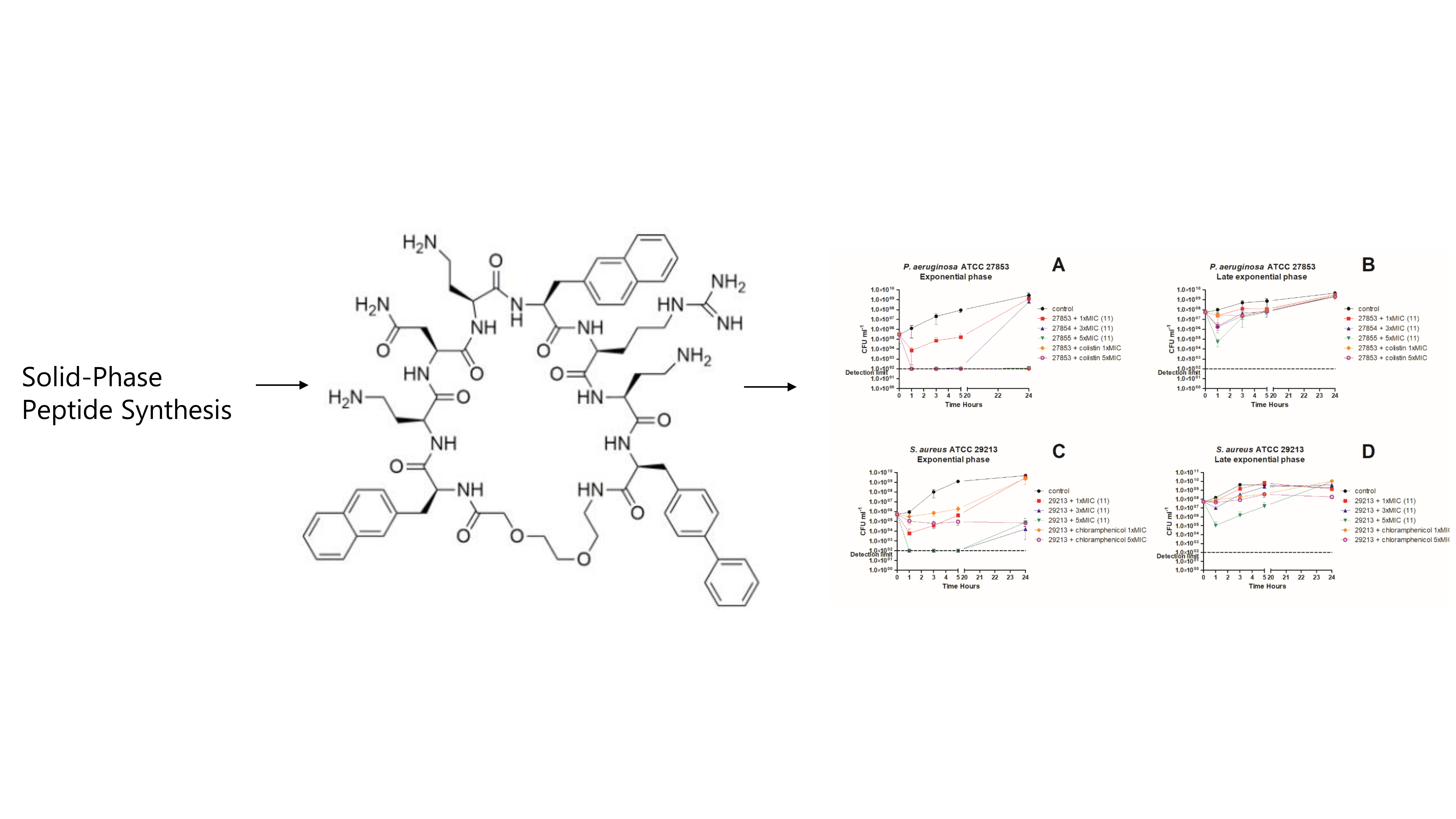
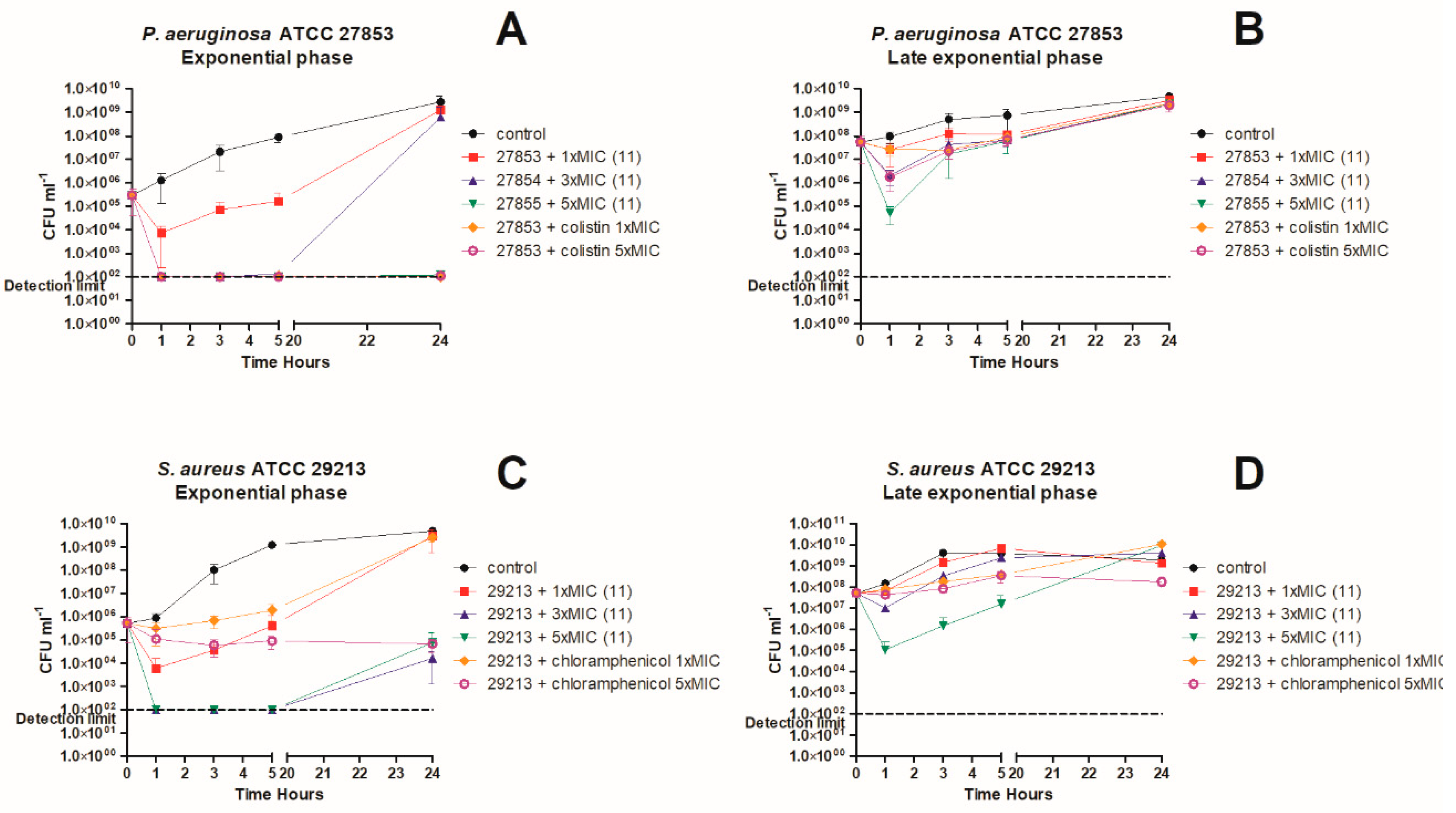
3. In Vitro Killing Kinetics against P. aeruginosa and S. aureus
4. Materials and Methods
4.1. Materials
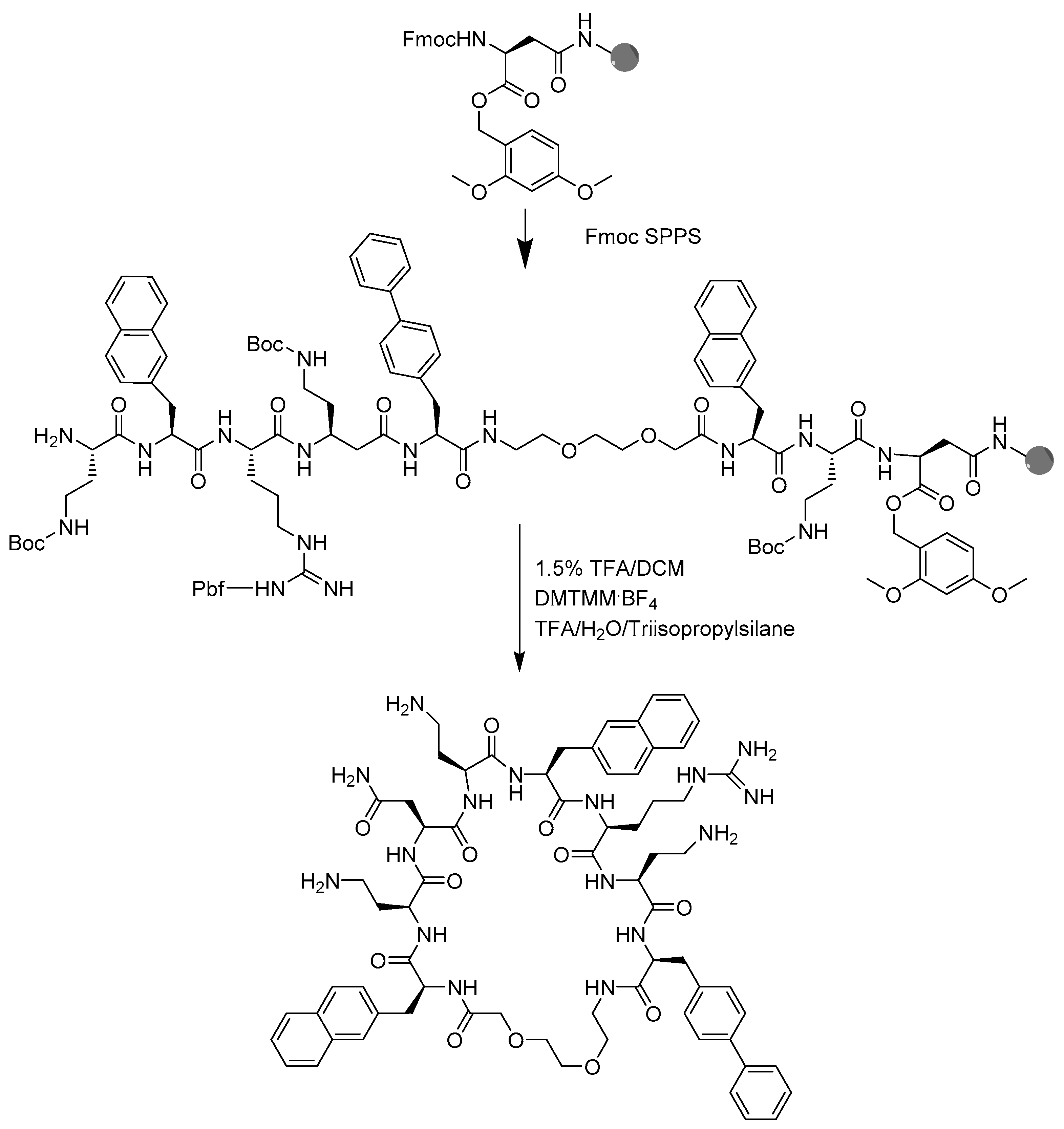
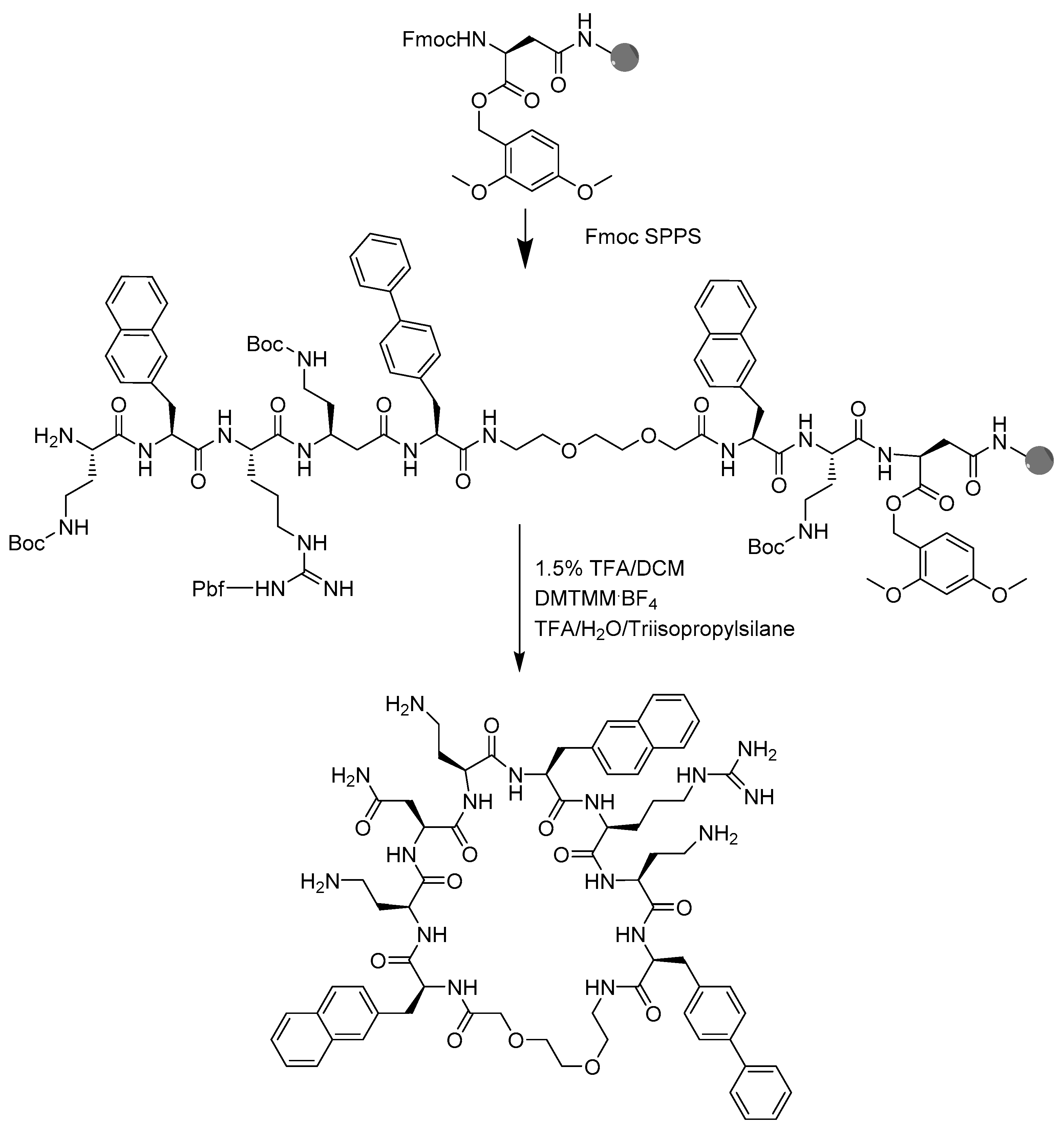
4.2. Peptide Synthesis
4.3. Peptide Macrocyclization
4.4. Peptide Cleavage
4.5. Minimum Inhibitory Concentration Determination
4.6. Hemolysis
4.7. Time-Kill Kinetics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Bip | L-biphenylalanine |
| COMU | 1-Cyano-2-ethoxy-2oxoethylidenaminooxy)-dimethylaminomorpholino-carbenium hexa-fluorophosphate |
| Dab | L-2,4-diaminobutyric acid |
| DCM | Dichloromethane |
| DIEA | Disopropylamine |
| DMF | Dimethylformamide, synthesis grade |
| (DMTMM·BF4) | 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholiniumtetrafluoroborate |
| EUCAST | European Committee on Antimicrobial Susceptibility Testing |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid hexafluoro-phosphate, N-[(Dimethylamino)-1H-1,2,3-triazolo-[4,5-b] pyridin-1-ylmethylene]-N-methylmethan-aminium hexa-fluorophosphate N-oxide |
| HOAt | 1-Hydroxy-7-azabenzotriazole |
| Nal | 3-(2-Naphthyl)-L-alanine |
| O2Oc | 8-amino-3,6-dioxaoctanoic acid |
| Oxyma | Ethyl (hydroxyimino)cyanoacetate |
| PBS | Phosphate-buffered saline |
| RAM | Rink amide Linker |
| RP-HPLC | Reverse Phase Analytical High Performance Liquid Chromatography |
| TFA | trifluoroacetic acid |
| TIS | Triisopropylamine |
References
- Rahbarnia, L.; Farajnia, S.; Naghili, B.; Ahmadzadeh, V.; Veisi, K.; Baghban, R.; Toraby, S. Current trends in targeted therapy for drug-resistant infections. Appl. Microbiol. Biotechnol. 2019, 103, 8301–8314. [Google Scholar] [CrossRef] [Green Version]
- Konaklieva, M.I. Addressing Antimicrobial Resistance through New Medicinal and Synthetic Chemistry Strategies. SLAS Discov. 2018, 24, 419–439. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Brogden, K. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries, R.M.; Pollett, S.; Sakoulas, G. A Current Perspective on Daptomycin for the Clinical Microbiologist. Clin. Microbiol. Rev. 2013, 26, 759–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-Y.; Wang, Y.; Walsh, T.R.; Yi, L.-X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Friedman, L.; Alder, J.D.; Silverman, J.A. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- Trent, M.S.; Ribeiro, A.A.; Doerrler, W.T.; Lin, S.; Cotter, R.J.; Raetz, C.R.H. Accumulation of a Polyisoprene-linked Amino Sugar in Polymyxin-resistant Salmonella typhimurium and Escherichia coli: Structural Characterization and Transfer to Lipid A in the Periplasm. J. Biol. Chem. 2001, 276, 43132–43144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2015, 4, 18–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, T.T.; Munita, J.M.; Arias, C.A. Mechanisms of drug resistance: Daptomycin resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 32–53. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxin Derivatives that Sensitize Gram-Negative Bacteria to Other Antibiotics. Molecules 2019, 24, 249. [Google Scholar] [CrossRef] [Green Version]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [Green Version]
- Oddo, A.; Thomsen, T.T.; Britt, H.M.; Løbner-Olesen, A.; Thulstrup, P.W.; Sanderson, J.M.; Hansen, P.R. Modulation of Backbone Flexibility for Effective Dissociation of Antibacterial and Hemolytic Activity in Cyclic Peptides. ACS Med. Chem. Lett. 2016, 7, 741–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, Z.Y.; Wiradharma, N.; Yang, Y.Y. Strategies employed in the design and optimization of synthetic antimicrobial peptide amphiphiles with enhanced therapeutic potentials. Adv. Drug Deliv. Rev. 2014, 78, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, P.; Peschel, A.; Nieuwenhuizen, W.F.; Otto, M.; Götz, F.; Jung, G.; Jack, R.W. Structure-function relationships in the tryptophan-rich, antimicrobial peptide indolicidin. J. Pept. Sci. 2001, 7, 552–564. [Google Scholar] [CrossRef]
- Bluhm, M.E.C.; Knappe, D.; Hoffmann, R. Structure-activity relationship study using peptide arrays to optimize Api137 for an increased antimicrobial activity against Pseudomonas aeruginosa. Eur. J. Med. Chem. 2015, 103, 574–582. [Google Scholar] [CrossRef]
- Ifrah, D.; Doisy, X.; Ryge, T.; Hansen, P. Structure-activity relationship study of anoplin. J. Pept. Sci. 2005, 11, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Zhu, N.; Zhu, Y.; Liu, T.; Gou, S.; Xie, J.; Yao, J.; Ni, J. Antimicrobial peptides conjugated with fatty acids on the side chain of D-amino acid promises antimicrobial potency against multidrug-resistant bacteria. Eur. J. Pharm. Sci. 2020, 141, 105123. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Wang, J.; Thompson, P.E.; Li, J. Teaching ‘Old’ Polymyxins New Tricks: New-Generation Lipopeptides Targeting Gram-Negative ‘Superbugs’. ACS Chem. Biol. 2014, 9, 1172–1177. [Google Scholar] [CrossRef]
- Kondejewski, L.H.; Lee, D.L.; Jelokhani-Niaraki, M.; Farmer, S.W.; Hancock, R.E.W.; Hodges, R.S. Optimization of Microbial Specificity in Cyclic Peptides by Modulation of Hydrophobicity within a Defined Structural Framework. J. Biol. Chem. 2002, 277, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Oddo, A.; Münzker, L.; Hansen, P.R. Peptide Macrocycles Featuring a Backbone Secondary Amine: A Convenient Strategy for the Synthesis of Lipidated Cyclic and Bicyclic Peptides on Solid Support. Org. Lett. 2015, 17, 2502–2505. [Google Scholar] [CrossRef]
- Oddo, A.; Nyberg, N.T.; Frimodt-Moller, N.; Thulstrup, P.W.; Hansen, P.R. The effect of glycine replacement with flexible omega-amino acids on the antimicrobial and haemolytic activity of an amphipathic cyclic heptapeptide. Eur. J. Med. Chem. 2015, 102, 574–581. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipatic, a-helical anticrobial peptides. Pept. Sci. 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Uggerhøj, L.E.; Poulsen, T.J.; Munk, J.K.; Fredborg, M.; Sondergaard, T.E.; Frimodt-Moller, N.; Hansen, P.R.; Wimmer, R. Rational Design of Alpha-Helical Antimicrobial Peptides: Do’s and Don’ts. ChemBioChem 2015, 16, 242–253. [Google Scholar]
- Almaaytah, A.; Qaoud, M.T.; Khalil Mohammed, G.; Abualhaijaa, A.; Knappe, D.; Hoffmann, R.; Al-Balas, Q. Antimicrobial and Antibiofilm Activity of UP-5, an Ultrashort Antimicrobial Peptide Designed Using Only Arginine and Biphenylalanine. Pharmaceuticals 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- McCoy, L.S.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Velkov, T.; Li, J.; Tor, Y. Polymyxins and Analogues Bind to Ribosomal RNA and Interfere with Eukaryotic Translation in Vitro. ChemBioChem 2013, 14, 2083–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondelle, S.E.; Simpkins, L.R.; Pérez-Payá, E.; Houghten, R.A. Influence of tryptophan residues on melittin’s hemolytic activity. Biochim. Biophys. Acta (BBA) 1993, 1202, 331–336. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Yang, J.C.; Yohonn, L.; Ly, N.S.; Brown, S.V.; Hondt, R.E.; Jusko, W.J.; Forrest, A.; Tsuji, B.T. Attenuation of Colistin Bactericidal Activity by High Inoculum of Pseudomonas Aeruginosa Characterized by a New Mechanism-Based Population Pharmacodynamic Model. Antimicrob. Agents Chemother. 2010, 54, 2051–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savini, F.; Luca, V.; Bocedi, A.; Massoud, R.; Park, Y.; Mangoni, M.L.; Stella, L. Cell-Density Dependence of Host-Defense Peptide Activity and Selectivity in the Presence of Host Cells. ACS Chem. Biol. 2017, 12, 52–56. [Google Scholar] [CrossRef] [Green Version]
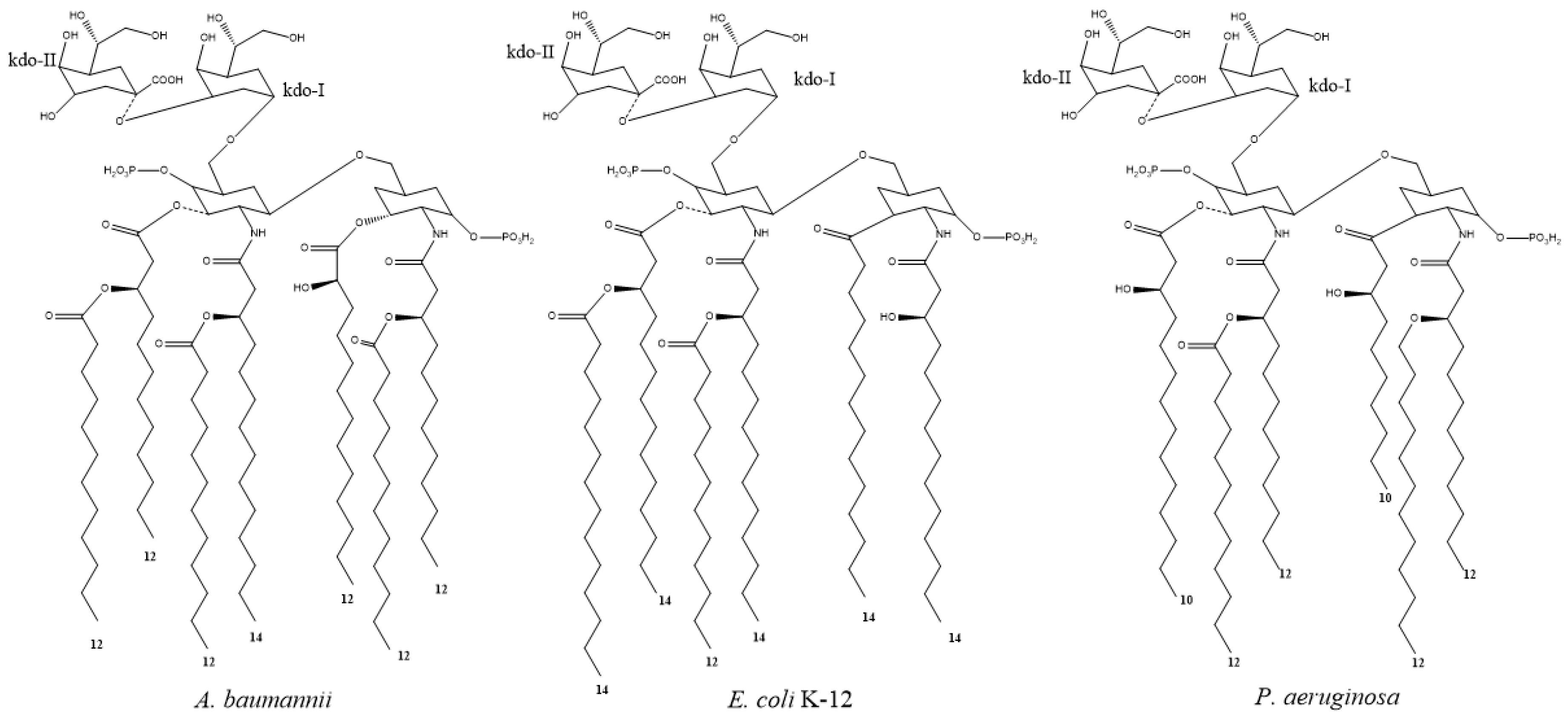
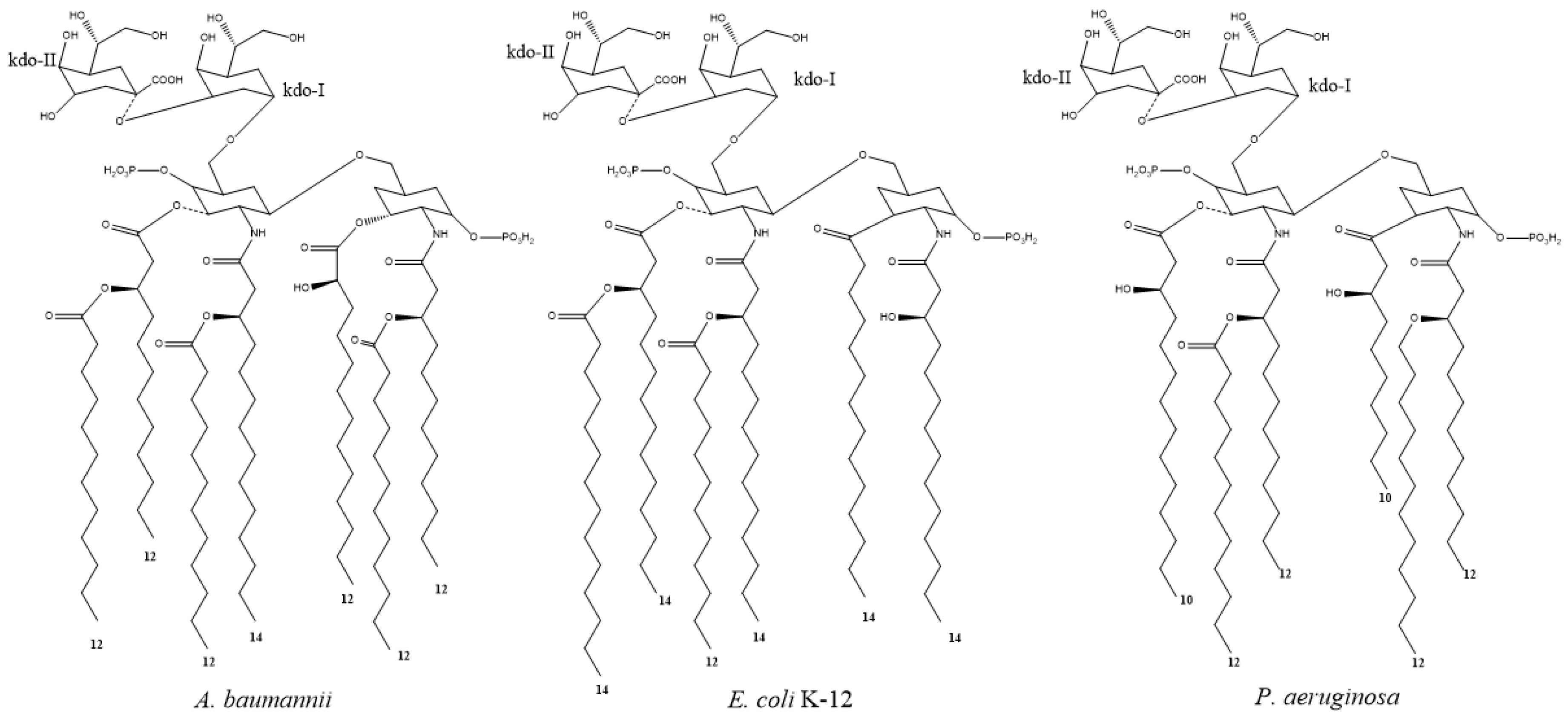
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A Modification Systems in Gram-Negative Bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Boll, J.M.; Tucker, A.T.; Klein, D.R.; Beltran, A.M.; Brodbelt, J.S.; Davies, B.W.; Trent, M.S. Reinforcing Lipid A Acylation on the Cell Surface of Acinetobacter baumannii Promotes Cationic Antimicrobial Peptide Resistance and Desiccation Survival. mBio 2015, 6, e00478-15. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yun, J.; Liu, L.; Li, Y.; Wang, X. Identification of Two Genes Encoding for the Late Acyltransferases of Lipid A in Klebsiella pneumoniae. Curr. Microbiol. 2016, 73, 732–738. [Google Scholar] [CrossRef]
- Kim, S.; Patel, D.S.; Park, S.; Slusky, J.; Klauda, J.B.; Widmalm, G.; Im, W. Bilayer Properties of Lipid A from Various Gram-Negative Bacteria. Biophys. J. 2016, 111, 1750–1760. [Google Scholar] [CrossRef]
- Ofek, I.; Cohen, S.; Rahmani, R.; Kabha, K.; Tamarkin, D.; Herzig, Y.; Rubinstein, E. Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 1994, 38, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nature 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Hansen, P.R. Hemolytic Activity of Antimicrobial Peptides. Methods Mol. Biol. 2017, 1548, 427–435. [Google Scholar]
- Oddo, A.; Thomsen, T.T.; Kjelstrup, S.; Gorey, C.; Franzyk, H.; Frimodt-Møller, N.; Løbner-Olesen, A.; Hansen, P.R. An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter baumannii and a dual mode of action. Antimicrob. Agents Chemother. 2016, 60, 592–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | SA [a] | PA [b] | EC [c] | AB [d] | %H [e] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BSI-9 | c(Lys | Nal | Lys | Lys | Bip | O2Oc | Nal | Lys | Asn) [f] | 32 | 16 | 64 | 32 | 33 | |
| Stage 1 | 1 | c(Lys | Nal | Lys | Lys | Bip | O2Oc | Nal | Asn | Lys) | >64 | 32 | >64 | 32 | ND |
| 2 | c(Lys | Nal | Lys | Asn | Bip | O2Oc | Nal | Lys | Lys) | >64 | 64 | >64 | >64 | ND | |
| 3 | c(Lys | Nal | Asn | Lys | Bip | O2Oc | Nal | Lys | Lys) | 64 | 64 | >64 | 64 | ND | |
| 4 | c(Asn | Nal | Lys | Lys | Bip | O2Oc | Nal | Lys | Lys) | >64 | 64 | >64 | 64 | ND | |
| Stage 2 | 5 | c(Lys | Phe | Lys | Lys | Bip | O2Oc | Nal | Lys | Asn) | >64 | 32 | >64 | >64 | 4 |
| 6 | c(Lys | Nal | Lys | Lys | Phe | O2Oc | Nal | Lys | Asn) | >64 | 32 | >64 | >64 | 6 | |
| 7 | c(Lys | Nal | Lys | Lys | Bip | O2Oc | Phe | Lys | Asn) | >64 | 64 | >64 | 64 | 7 | |
| 8 | c(Lys | Phe | Lys | Lys | Phe | O2Oc | Phe | Lys | Asn) | >64 | >64 | >64 | >64 | 3 | |
| 9 | c(Dab | Nal | Dab | Dab | Bip | O2Oc | Nal | Dab | Asn) | 8 | 4 | >64 | 32 | 76 | |
| Stage 3 | 10 | c(Arg | Nal | Dab | Dab | Bip | O2Oc | Nal | Dab | Asn) | 2 | 2 | 32 | 32 | 66 |
| 11 | c(Dab | Nal | Arg | Dab | Bip | O2Oc | Nal | Dab | Asn) | 8 | 4 | 32 | 32 | 36 | |
| 12 | c(Dab | Nal | Dab | Arg | Bip | O2Oc | Nal | Dab | Asn) | 1 | 4 | 32 | 32 | 98 | |
| 13 | c(Dab | Nal | Dab | Dab | Bip | O2Oc | Nal | Arg | Asn) | 1 | 4 | 32 | 64 | 80 | |
| 14 | c(Arg | Nal | Arg | Arg | Bip | O2Oc | Nal | Arg | Asn) | 32 | 32 | >64 | >64 | 68 | |
| Stage 4 | 15 | c(Arg | Nal | Arg | Dap | Bip | O2Oc | Nal | Dab | Asn) | 8 | 16 | 64 | >64 | 100 |
| 16 | c(Arg | Nal | Arg | Dab | Phe | O2Oc | Nal | Dab | Asn) | 32 | 16 | 64 | 64 | 16 | |
| 17 | c(Arg | Nal | Dab | Dab | Phe | O2Oc | Nal | Dab | Asn) | 64 | 16 | 64 | >64 | 3 | |
| 18 | c(Dab | Nal | Arg | Dab | Phe | O2Oc | Nal | Dab | Asn) | 64 | 16 | 64 | >64 | 30 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomsen, T.T.; Mendel, H.C.; Al-Mansour, W.; Oddo, A.; Løbner-Olesen, A.; Hansen, P.R. Analogues of a Cyclic Antimicrobial Peptide with a Flexible Linker Show Promising Activity against Pseudomonas aeruginosa and Staphylococcus aureus. Antibiotics 2020, 9, 366. https://doi.org/10.3390/antibiotics9070366
Thomsen TT, Mendel HC, Al-Mansour W, Oddo A, Løbner-Olesen A, Hansen PR. Analogues of a Cyclic Antimicrobial Peptide with a Flexible Linker Show Promising Activity against Pseudomonas aeruginosa and Staphylococcus aureus. Antibiotics. 2020; 9(7):366. https://doi.org/10.3390/antibiotics9070366
Chicago/Turabian StyleThomsen, Thomas T., Helen C. Mendel, Wafaa Al-Mansour, Alberto Oddo, Anders Løbner-Olesen, and Paul R. Hansen. 2020. "Analogues of a Cyclic Antimicrobial Peptide with a Flexible Linker Show Promising Activity against Pseudomonas aeruginosa and Staphylococcus aureus" Antibiotics 9, no. 7: 366. https://doi.org/10.3390/antibiotics9070366
APA StyleThomsen, T. T., Mendel, H. C., Al-Mansour, W., Oddo, A., Løbner-Olesen, A., & Hansen, P. R. (2020). Analogues of a Cyclic Antimicrobial Peptide with a Flexible Linker Show Promising Activity against Pseudomonas aeruginosa and Staphylococcus aureus. Antibiotics, 9(7), 366. https://doi.org/10.3390/antibiotics9070366