Genomic Characterization of Antimicrobial Resistance, Virulence, and Phylogeny of the Genus Ochrobactrum

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Antimicrobial Resistance Analysis
2.1.1. Phenotypic Data
2.1.2. Resistome Analysis
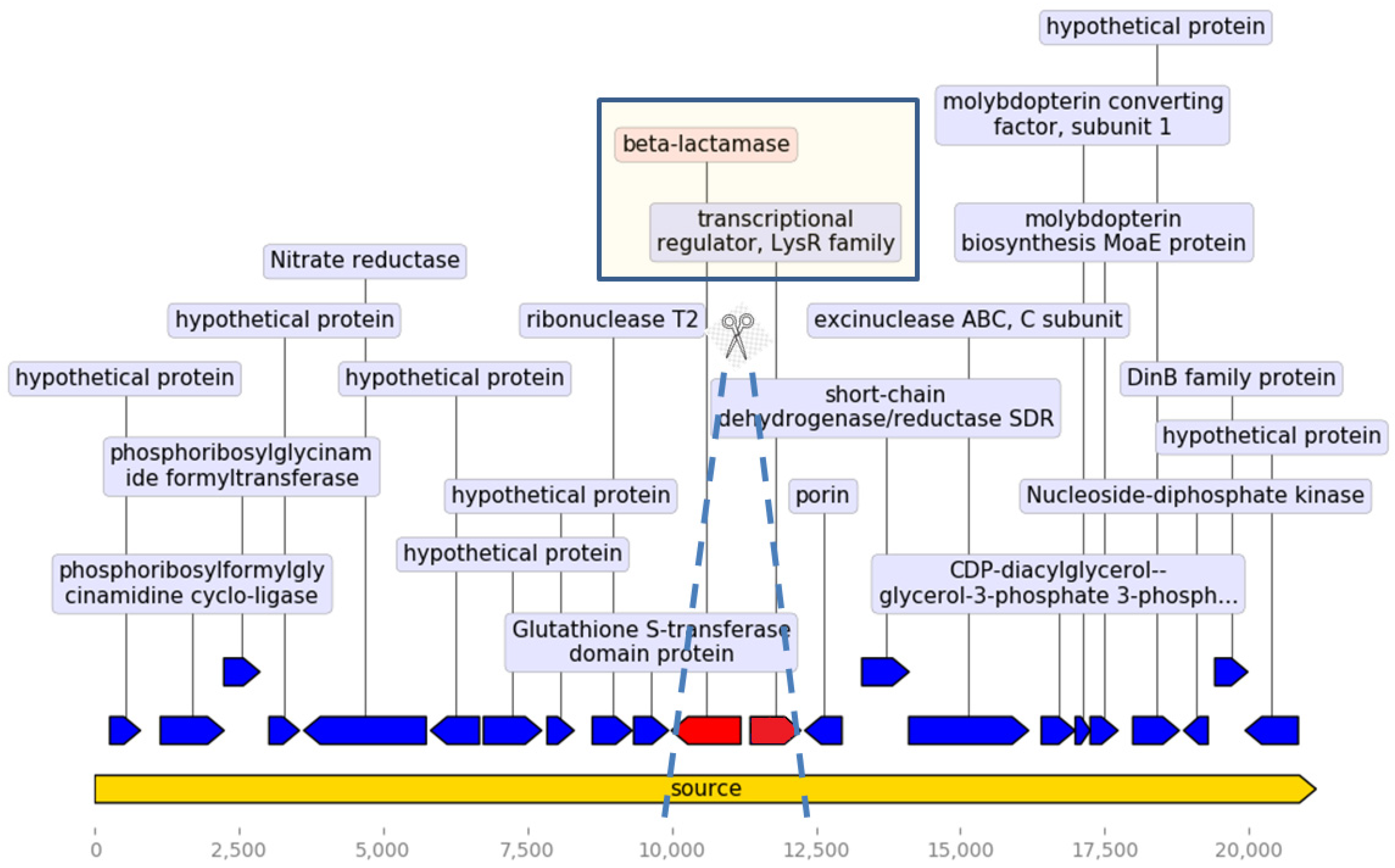
β-Lactam Resistance
Other Resistance Genes
2.2. Virulome Analysis
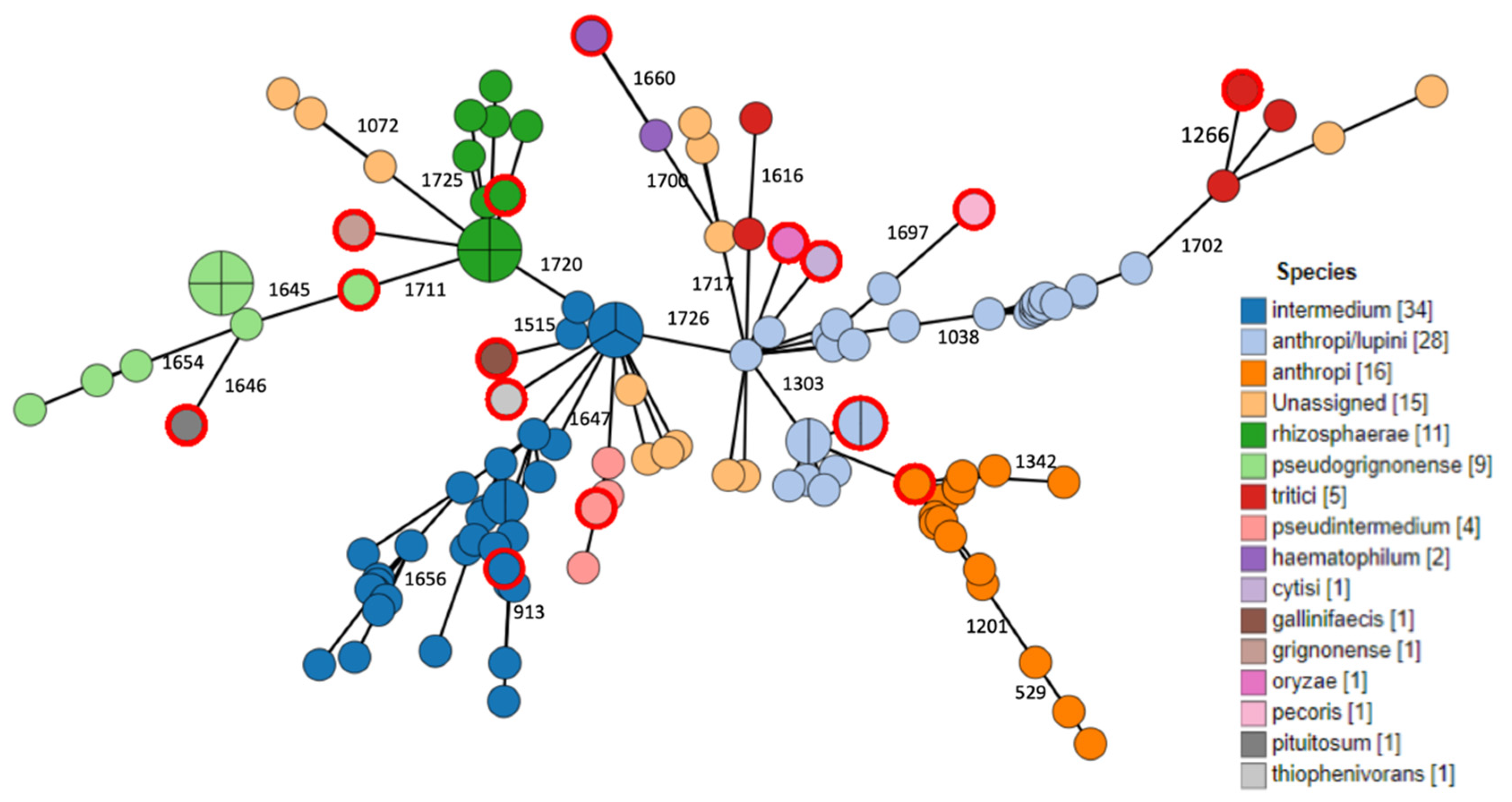
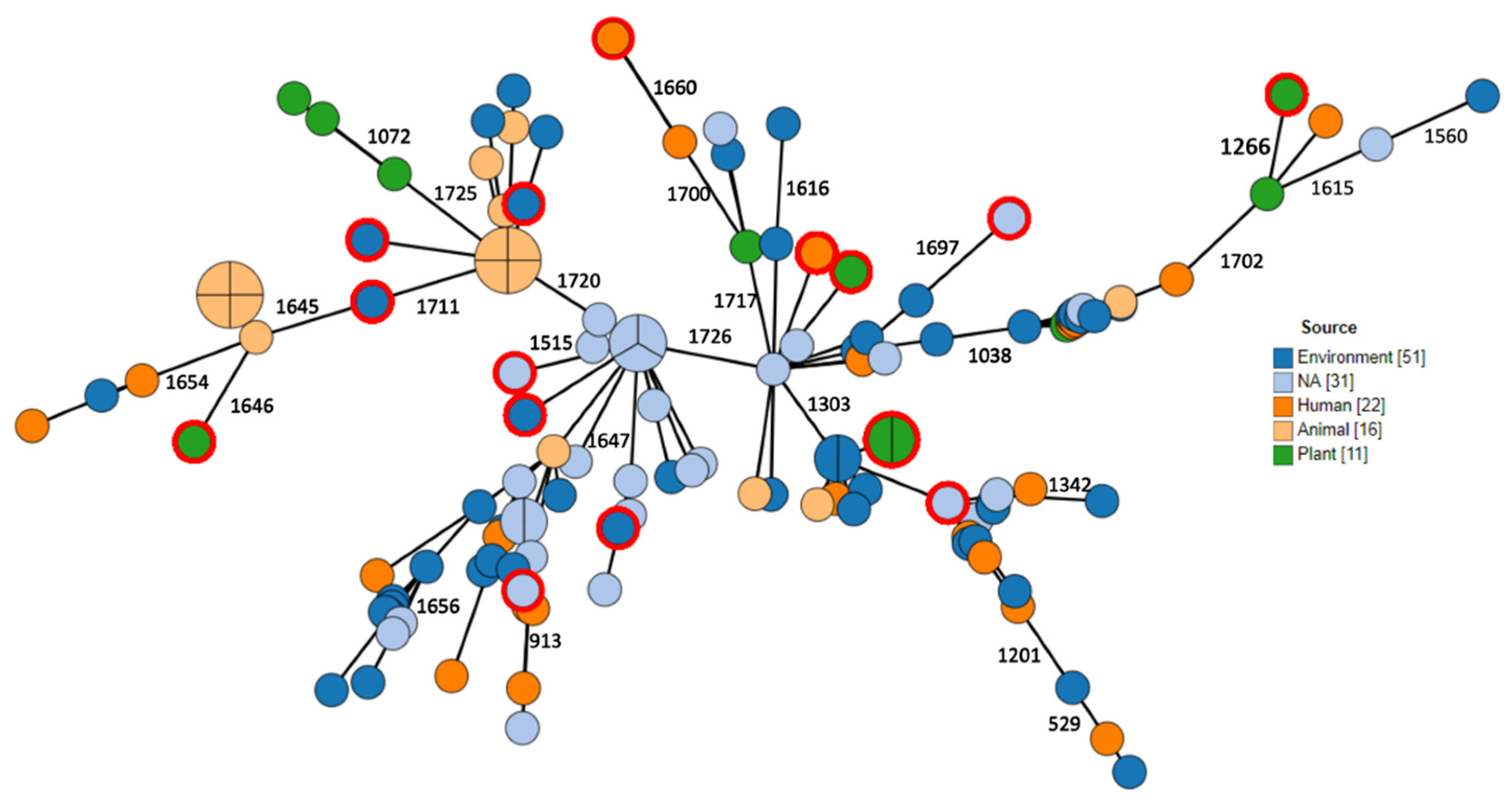
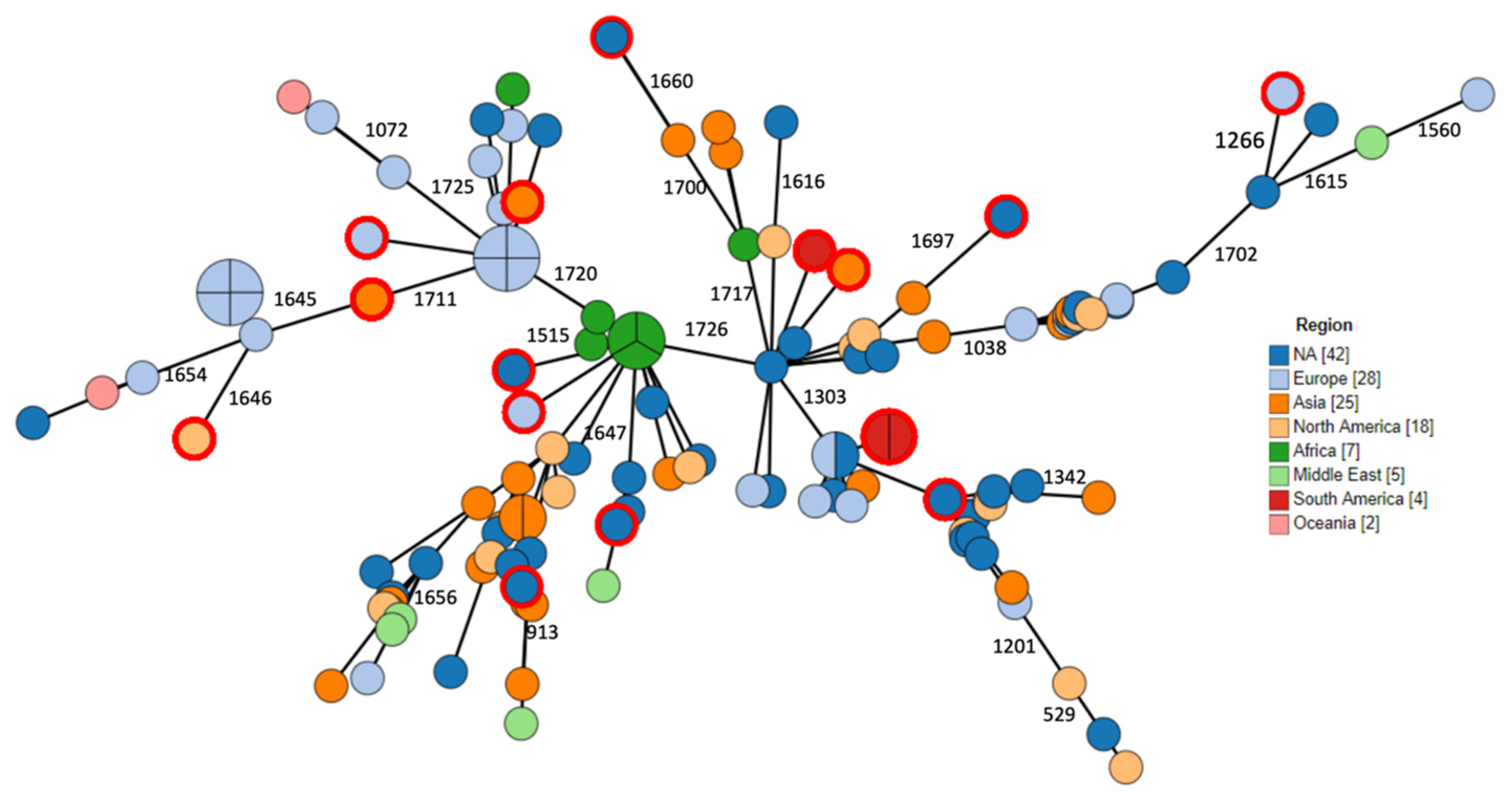
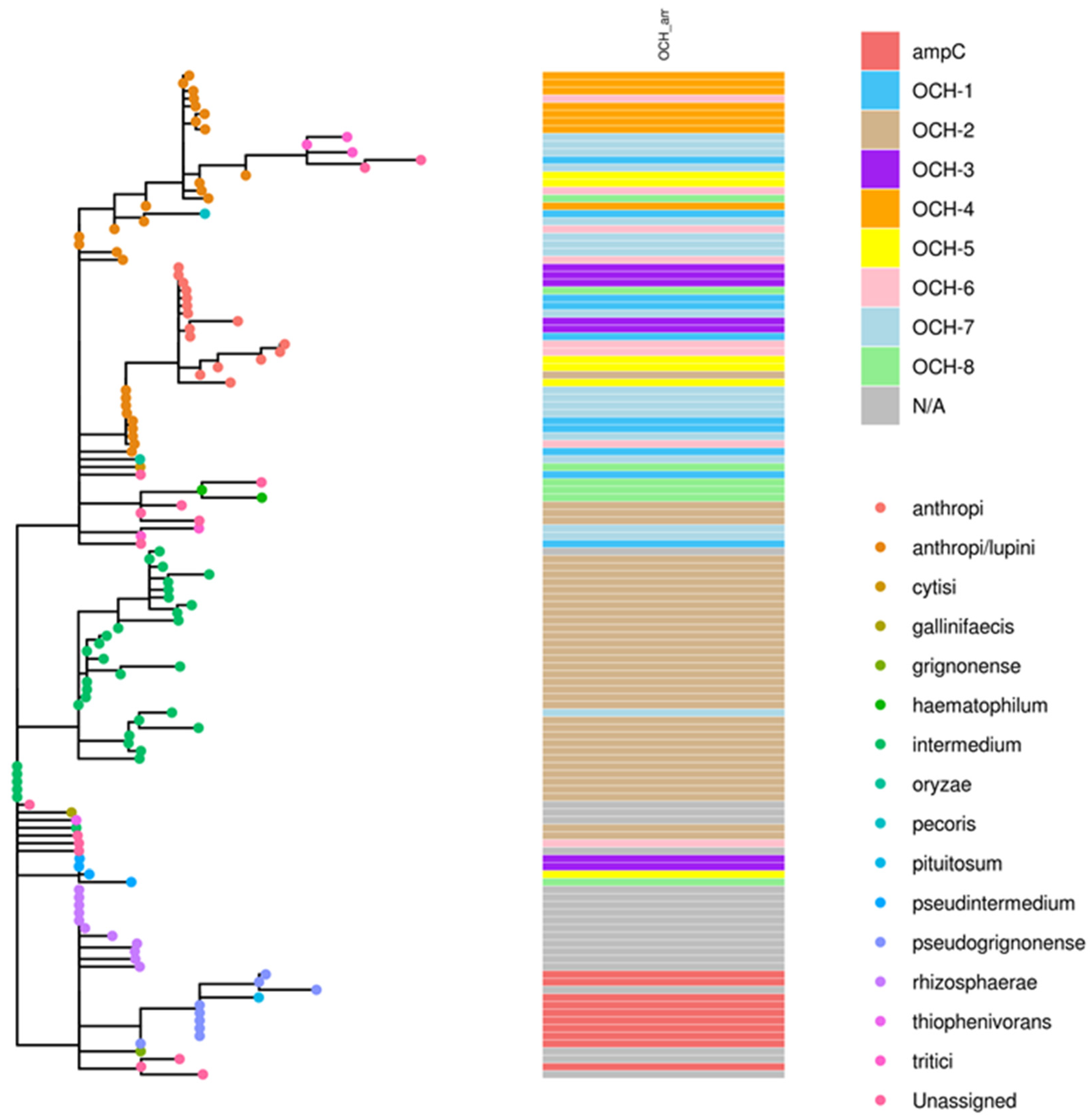
2.3. Phylogenomic Analysis
3. Discussion
4. Materials and Methods
4.1. Collection of Isolates
4.2. DNA Extraction, Library Preparation, and Sequencing
4.3. Collection of Publicly Available Genomes and Bioinformatics Analysis
4.4. Phylogenomic Trees Construction
4.5. Tools and Databases used for Resistance and Virulence Genes Search
4.6. Taxonomic Assignment Using Average Nucleotide Identity (ANI)
4.7. Literature Review for Phenotypic Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Accessibility
References
- Holmes, B.; Popoff, M.; Kiredjian, M.; Kersters, K. Ochrobactrum anthropi gen. nov., sp. nov. from human clinical specimens and previously known as group vd. Int. J. Syst. Bacteriol. 1988, 38, 406–416. [Google Scholar] [CrossRef]
- Lebuhn, M.; Achouak, W.; Schloter, M.; Berge, O.; Meier, H.; Barakat, M.; Hartmann, A.; Heulin, T. Taxonomic characterization of Ochrobactrum sp. isolates from soil samples and wheat roots, and description of Ochrobactrum tritici sp. nov. and Ochrobactrum grignonense sp. nov. Int. J. Syst. Evol. Microbiol. 2000, 50 Pt 6, 2207–2223. [Google Scholar] [CrossRef]
- Kampfer, P.; Scholz, H.C.; Huber, B.; Falsen, E.; Busse, H.-J. Ochrobactrum haematophilum sp. nov. and Ochrobactrum pseudogrignonense sp. nov., isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 2007, 57, 2513–2518. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dirksen, P.; Marsh, S.A.; Braker, I.; Heitland, N.; Wagner, S.; Nakad, R.; Mader, S.; Petersen, C.; Kowallik, V.; Rosenstiel, P.; et al. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 2016, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, G.; Gohil, K.; Misra, V.; Kakrani, A.L.; Misra, S.P.; Patole, M.; Shouche, Y.; Dharne, M. Multilocus sequence typing of Ochrobactrum spp. isolated from gastric niche. J. Infect. Public Health 2017, 10, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Cieslak, T.J.; Robb, M.L.; Drabick, C.J.; Fischer, G.W. Catheter-associated sepsis caused by Ochrobactrum anthropi: Report of a case and review of related nonfermentative bacteria. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1992, 14, 902–907. [Google Scholar] [CrossRef]
- Wheen, L.; Taylor, S.; Godfrey, K. Vertebral osteomyelitis due to Ochrobactrum anthropi. Intern. Med. J. 2002, 32, 426–428. [Google Scholar] [CrossRef]
- Ozdemir, D.; Soypacacı, Z.; Sahin, I.; Bicik, Z.; Sencan, I. Ochrobactrum anthropi endocarditis and septic shock in a patient with no prosthetic valve or rheumatic heart disease: Case report and review of the literature. Jpn. J. Infect. Dis. 2006, 59, 264–265. [Google Scholar]
- Alparslan, C.; Yavascan, O.; Kose, E.; Sanlioglu, P.; Aksu, N. An opportunistic pathogen in a peritoneal dialysis patient: Ochrobactrum anthropi. Indian J. Pediatr. 2013, 80, 72–74. [Google Scholar] [CrossRef]
- Hagiya, H.; Ohnishi, K.; Maki, M.; Watanabe, N.; Murase, T. Clinical characteristics of Ochrobactrum anthropi bacteremia. J. Clin. Microbiol. 2013, 51, 1330–1333. [Google Scholar] [CrossRef]
- Mattos, F.B.; Saraiva, F.P.; Angotti-Neto, H.; Passos, A.F. Outbreak of Ochrobactrum anthropi endophthalmitis following cataract surgery. J. Hosp. Infect. 2013, 83, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Khasawneh, W.; Yusef, D. Ochrobactrum anthropi fulminant early-onset neonatal sepsis: A case report and review of literature. Pediatr. Infect. Dis. J. 2017, 36, 1167–1168. [Google Scholar] [CrossRef]
- Gigi, R.; Flusser, G.; Kadar, A.; Salai, M.; Elias, S. Ochrobactrum anthropi-caused osteomyelitis in the foot mimicking a bone tumor: Case report and review of the literature. J. Foot Ankle Surg. 2017, 56, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Galanakis, E.; Bitsori, M.; Samonis, G.; Christidou, A.; Georgiladakis, A.; Sbyrakis, S.; Tselentis, Y. Ochrobactrum anthropi bacteraemia in immunocompetent children. Scand. J. Infect. Dis. 2002, 34, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, T.C. Ochrobactrum anthropi septic arthritis of the acromioclavicular joint in an immunocompetent 17 year old. Orthopedics 2008, 31, 606. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, N.; Mathur, P. Ochrobactrum anthropi: An emerging pathogen causing meningitis with sepsis in a neurotrauma patient. J. Infect. Dev. Ctries. 2017, 11, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.S.; Murtough, S.M.; Williamson, E.; Hiom, S.J.; Payne, D.J.; Russell, A.D.; Walsh, T.R. Resistance to antibiotics and biocides among non-fermenting Gram-negative bacteria. Clin. Microbiol. Infect. 2001, 7, 308–315. [Google Scholar] [CrossRef]
- Thoma, B.; Straube, E.; Scholz, H.C.; Al Dahouk, S.; Zöller, L.; Pfeffer, M.; Neubauer, H.; Tomaso, H. Identification and antimicrobial susceptibilities of Ochrobactrum spp. Int. J. Med. Microbiol. 2009, 299, 209–220. [Google Scholar] [CrossRef]
- Chmelař, D.; Holý, O.; Kasáková, I.; Hájek, M.; Lazarová, A.; Gonzalez-Rey, C.; Lasák, J.; Raclavský, V.; Čižnár, I. Antibiotic susceptibility and production of endotoxin by Ochrobactrum anthropi isolated from environment and from patients with cystic fibrosis. Folia Microbiol. (Praha) 2019, 64, 861–865. [Google Scholar] [CrossRef]
- Montaña, S.; Fernandez, J.S.; Barenboim, M.; Hernandez, M.; Kayriyama, C.; Carulla, M.; Iriarte, A.; Ramirez, M.S.; Almuzara, M. Whole-genome analysis and description of an outbreak due to carbapenem-resistant Ochrobactrum anthropi causing pseudo-bacteraemias. New Microbes New Infect. 2018, 26, 100–106. [Google Scholar] [CrossRef]
- Teyssier, C. Molecular and phenotypic features for identification of the opportunistic pathogens Ochrobactrum spp. J. Med. Microbiol. 2005, 54, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, D.; Labia, R.; Cerceau, C.; Bizet, C.; Philippon, A.; Arlet, G. Molecular characterization of chromosomal class C β-Lactamase and its regulatory gene in Ochrobactrum anthropi. Antimicrob. Agents Chemother. 2001, 45, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.S.; Avison, M.B.; Jamieson, L.; Simm, A.M.; Bennett, P.M.; Walsh, T.R. Characterization, cloning and sequence analysis of the inducible Ochrobactrum anthropi AmpC beta-lactamase. J. Antimicrob. Chemother. 2001, 47, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.A.; Kwabugge, Y.A.; Anyanwu, M.U.; Torres, C.; Chah, K.F. Diversity of Ochrobactrum species in food animals, antibiotic resistance phenotypes and polymorphisms in the blaOCH gene. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef]
- Poonawala, H.; Marrs Conner, T.; Peaper, D.R. The brief case: Misidentification of Brucella melitensis as Ochrobactrum anthropi by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry (MALDI-TOF MS). J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef]
- Trêpa, J.; Mendes, P.; Gonçalves, R.; Chaves, C.; Brás, A.M.; Mesa, A.; Ramos, I.; Sá, R.; da Cunha, J.G.S. Brucella vertebral osteomyelitis misidentified as an Ochrobactrum anthropi infection. IDCases 2018, 11, 74–76. [Google Scholar] [CrossRef]
- Horvat, R.T.; El Atrouni, W.; Hammoud, K.; Hawkinson, D.; Cowden, S. Ribosomal RNA sequence analysis of Brucella infection misidentified as Ochrobactrum anthropi infection. J. Clin. Microbiol. 2011, 49, 1165–1168. [Google Scholar] [CrossRef]
- Vila, A. Brucella suis bacteremia misidentified as Ochrobactrum anthropi by the VITEK 2 system. J. Infect. Dev. Ctries. 2016, 10, 432. [Google Scholar] [CrossRef]
- Scholz, H.C.; Al Dahouk, S.; Tomaso, H.; Neubauer, H.; Witte, A.; Schloter, M.; Kämpfer, P.; Falsen, E.; Pfeffer, M.; Engel, M. Genetic diversity and phylogenetic relationships of bacteria belonging to the Ochrobactrum–Brucella group by recA and 16S rRNA gene-based comparative sequence analysis. Syst. Appl. Microbiol. 2008, 31, 1–16. [Google Scholar] [CrossRef]
- Kulkarni, G.; Dhotre, D.; Dharne, M.; Shetty, S.; Chowdhury, S.; Misra, V.; Misra, S.; Patole, M.; Shouche, Y. Draft genome of Ochrobactrum intermedium strain M86 isolated from non-ulcer dyspeptic individual from India. Gut Pathog. 2013, 5, 7. [Google Scholar] [CrossRef]
- Liang, Z.; Li, G.; Mai, B.; Ma, H.; An, T. Application of a novel gene encoding bromophenol dehalogenase from Ochrobactrum sp. T in TBBPA degradation. Chemosphere 2019, 217, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.; Jiang, X.; Zhang, F.; Zheng, B.; Shu, F.; Wang, Z.; Cui, Q.; Dong, H.; Zhang, Z.; Hou, D.; et al. Isolation and characterization of a crude oil degrading bacteria from formation water: Comparative genomic analysis of environmental Ochrobactrum intermedium isolate versus clinical strains. J. Zhejiang Univ. Sci. B 2015, 16, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Poszytek, K.; Karczewska-Golec, J.; Ciok, A.; Decewicz, P.; Dziurzynski, M.; Gorecki, A.; Jakusz, G.; Krucon, T.; Lomza, P.; Romaniuk, K.; et al. Genome-guided characterization of Ochrobactrum sp. POC9 enhancing sewage sludge utilization—Biotechnological potential and biosafety considerations. Int. J. Environ. Res. Public. Health 2018, 15, 1501. [Google Scholar] [CrossRef] [PubMed]
- Gazolla Volpiano, C.; Hayashi Sant’Anna, F.; Ambrosini, A.; Brito Lisboa, B.; Kayser Vargas, L.; Passaglia, L.M.P. Reclassification of Ochrobactrum lupini as a later heterotypic synonym of Ochrobactrum anthropi based on whole-genome sequence analysis. Int. J. Syst. Evol. Microbiol. 2019, 69, 2312–2314. [Google Scholar] [CrossRef]
- Baumann, M.; Simon, H.; Schneider, K.H.; Danneel, H.J.; Küster, U.; Giffhorn, F. Susceptibility of Rhodobacter sphaeroides to beta-lactam antibiotics: Isolation and characterization of a periplasmic beta-lactamase (cephalosporinase). J. Bacteriol. 1989, 171, 308–313. [Google Scholar] [CrossRef]
- Ntreh, A.T.; Weeks, J.W.; Nickels, L.M.; Zgurskaya, H.I. Opening the channel: The two functional interfaces of Pseudomonas aeruginosa opmH with the triclosan efflux pump triABC. J. Bacteriol. 2016, 198, 3176–3185. [Google Scholar] [CrossRef]
- Guglierame, P.; Pasca, M.R.; De Rossi, E.; Buroni, S.; Arrigo, P.; Manina, G.; Riccardi, G. Efflux pump genes of the resistance-nodulation-division family in Burkholderia cenocepacia genome. BMC Microbiol. 2006, 6, 66. [Google Scholar] [CrossRef]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [CrossRef]
- Johnning, A.; Moore, E.R.B.; Svensson-Stadler, L.; Shouche, Y.S.; Larsson, D.G.J.; Kristiansson, E. The acquired genetic mechanisms of a multi-resistant bacterium isolated from a treatment plant receiving wastewater from antibiotic production. Appl. Environ. Microbiol. 2013, 79, 7256–7263. [Google Scholar] [CrossRef]
- Henderson, B.; Fares, M.A.; Lund, P.A. Chaperonin 60: A paradoxical, evolutionarily conserved protein family with multiple moonlighting functions. Biol. Rev. Camb. Philos. Soc. 2013, 88, 955–987. [Google Scholar] [CrossRef]
- Chong, A.; Lima, C.A.; Allan, D.S.; Nasrallah, G.K.; Garduño, R.A. The purified and recombinant Legionella pneumophila chaperonin alters mitochondrial trafficking and microfilament organization. Infect. Immun. 2009, 77, 4724–4739. [Google Scholar] [CrossRef]
- Noah, C.E.; Malik, M.; Bublitz, D.C.; Camenares, D.; Sellati, T.J.; Benach, J.L.; Furie, M.B. GroEL and lipopolysaccharide from Francisella tularensis live vaccine strain synergistically activate human macrophages. Infect. Immun. 2010, 78, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Oeda, K.; Watanabe, E.; Mikami, T.; Fukita, Y.; Nishimura, K.; Komai, K.; Matsuda, K. Protein function. Chaperonin turned insect toxin. Nature 2001, 411, 44. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, Y.; Li, W.; Chen, Z. Type IV secretion system of Brucella spp. and its effectors. Front. Cell. Infect. Microbiol. 2015, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 March 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Machado, M.P.; Silva, D.N.; Rossi, M.; Moran-Gilad, J.; Santos, S.; Ramirez, M.; Carriço, J.A. chewBBACA: A complete suite for gene-by-gene schema creation and strain identification. Microb. Genomics 2018, 4, e000166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Alikhan, N.-F.; Sergeant, M.J.; Luhmann, N.; Vaz, C.; Francisco, A.P.; Carriço, J.A.; Achtman, M. GrapeTree: Visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 2018, 28, 1395–1404. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genomics 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate No. | OCH-ISR1 | OCH-ISR2 | OCH-ISR3 | OCH-ISR4 | OCH-ISR5 |
|---|---|---|---|---|---|
| MALDI TOF MS identification—Best match | O. intermedium | O. triciti | O. intermedium | O. intermedium | O. intermedium |
| Origin | Horse, on admission to veterinary hospital | Horse, on admission to veterinary hospital | Healthy horse 1, community collection | Healthy horse 1, community collection | Fat sand rat (Psammomys obesus), Zoo collection |
| Comments | 12-year-old gelding, gastrointestinal pathology | 1-month-old filly, respiratory pathology | 16-year-old healthy mare, gut colonization | 9-year-old healthy mare, gut colonization | − |
| WGS data: | |||||
| Total reads | 1,984,624 | 1,137,438 | 925,646 | 1,497,780 | 1,693,904 |
| Total bases | 276,000,000 | 160,000,000 | 131,000,000 | 203,000,000 | 241,000,000 |
| % GC | 57.4 | 55.6 | 57.2 | 57 | 57.3 |
| Minimum read length | 35 | 35 | 35 | 35 | 35 |
| Average read length | 139 | 140 | 141 | 135 | 142 |
| Maximum read length | 151 | 151 | 151 | 151 | 151 |
| Mode read length | 151 | 151 | 151 | 151 | 151 |
| Average quality 2 | 35.5 | 35.3 | 35 | 34.9 | 35.6 |
| Calculating depth | 5,000,000 | 5,000,000 | 5,000,000 | 5,000,000 | 5,000,000 |
| Sequencing depth of coverage 3 | 55x | 31x | 26x | 40x | 48x |
| Number of contigs | 53 | 115 | 171 | 71 | 52 |
| Total Base Pairs | 3,813,327 | 4,758,714 | 4,270,779 | 4,807,944 | 4,904,698 |
| Minimum | 2.05 × 102 | 2.00 × 102 | 2.01 × 102 | 200 | 2.03 × 102 |
| Average | 86,928 | 41,380 | 249,75 | 67,717 | 94321 |
| Maximum | 912,538 | 500,181 | 476,636 | 874,803 | 830,669 |
| N50 | 427,705 | 308,273 | 240,761 | 363,912 | 397,644 |
| Antimicrobial Agent | Measured MIC Values (µg/mL) | Reported MIC Values (µg/mL) | |||||
|---|---|---|---|---|---|---|---|
| OCH-ISR1 | OCH-ISR2 | OCH-ISR3 | OCH-ISR4 | OCH-ISR5 | Median, (MIC Range) | Number of Isolates | |
| Ampicillin | ≥32 | ≥32 | ≥32 | ≥32 | ≥32 | 256, (32–256) | 101 |
| Ampicillin–Sulbactam | ≥32 | ≥32 | ≥32 | ≥32 | ≥32 | 32, (32–32) | 5 |
| Amoxicillin–Clavulanic Acid | ≥32 | ≥32 | ≥32 | ≥32 | ≥32 | 256, (32–256) | 101 |
| Ticaricillin | ≥128 | ≥128 | ≥64 | ≥128 | ≥128 | 128, (64–128) | 12 |
| Piperacillin | ≥128 | ≥128 | ≥128 | ≥128 | ≥128 | 256, (128–256) | 101 |
| Piperacillin–tazobactam | ≥128 | ≥128 | ≥128 | ≥128 | ≥1228 | 128, (16–256) | 22 |
| Cefalexin | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 | 64, (64–64) | 5 |
| Cefuroxime | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 | 64, (32–128) | 19 |
| Cefoxitin | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 | 96, (64–128) | 12 |
| Ceftazidime | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 | 256, (8–256) | 111 |
| Ceftriaxone | ≥64 | ≥64 | ≥64 | ≥64 | ≥64 | 256, (4–256) | 93 |
| Ertapenem | ≤0.5 | ≤0.5 | ≤0.5 | ≤0.5 | ≤0.5 | 0.25, (0.064–0.5) | 12 |
| Imipenem | 1 | 0.5 | 0.5 | 1 | 1 | 2, (0.128–256) | 114 |
| Meropenem | 1 | 0.5 | 0.5 | 1 | 1 | 0.5, (0.128–16) | 21 |
| Amikacin | ≥64 | 4 | 16 | 16 | ≥64 | 3, (1–64) | 12 |
| Gentamicin | 8 | ≤1 | ≤1 | 8 | 8 | 4, (0.256–256) | 104 |
| Tobramycin | ≥16 | ≤1 | ≤1 | ≥16 | ≥16 | 16, (1–16) | 5 |
| Ciprofloxacin | ≤0.25 | ≤0.25 | 0.5 | ≤0.25 | ≤0.25 | 0.25, (0.25–0.5) | 12 |
| Levofloxacin | 0.25 | 0.25 | 0.25 | 0.25 | 0.25 | 0.25, (0.25–0.25) | 5 |
| Minocycline | ≤1 | ≤1 | ≤1 | ≤1 | ≤1 | 1, (1–1) | 5 |
| Tigecycline | 4 | 2 | 1 | 4 | 4 | 2, (0.28–4) | 12 |
| Fosfomycin | ≥256 | ≥256 | ≥256 | ≥256 | ≥256 | 256, (256–256) | 5 |
| Nitrofurantoin | 256 | 256 | 256 | 256 | 256 | 256, (256–256) | 5 |
| Trimethoprim/ sulfamethoxazole (TMP–SMX) | ≤20 | ≤20 | ≤20 | ≤20 | ≤20 | 0.064, (0.064–256) | 104 |
| Gene Name | AMR Gene Family | Drug Target | Resistance Mechanism | % of Total Sequences (n = 130) |
|---|---|---|---|---|
| β-lactamase genes | ||||
| OCH-1 | OCH β-lactamase | Cephalosporin, cephamycin, monobactam | Antibiotic inactivation | 7.7 (10) |
| OCH-2 | 27.7 (36) | |||
| OCH-3 | 4.6 (6) | |||
| OCH-4 | 6.2 (8) | |||
| OCH-5 | 4.6 (6) | |||
| OCH-6 | 7 (9) | |||
| OCH-7 | 13.1 (17) | |||
| OCH-8 | 5.4 (7) | |||
| Any OCH | 76.2 (99) | |||
| AmpC | AmpC-type β-lactamase | Cephalosporin, penicillin | − | 7 1 (9) |
| Any β-lactamase | − | − | − | 83.1 (108) |
| Other resistance genes | ||||
| triC | Resistance-nodulation-cell division (RND) efflux pump system and related genes | Triclosan | Antibiotic efflux complex | 97.7 (127) |
| ceoB | Aminoglycosides, fluoroquinolones | 96.2 (125) | ||
| mdsB | Phenicol antibiotics, β-lactams | 42.3 (55) | ||
| aac, ant and aph variants | Aminoglycoside-modifying enzymes | Aminoglycosides | Antibiotic inactivation | 10.8 2 (14) |
| Gene Name | Protein | Function | % of Human Isolates (n = 22) | % of Other Isolates (n = 82) | % of Total Isolates (n = 130) |
|---|---|---|---|---|---|
| Percent (n) | |||||
| acpXL | Acyl carrier protein | Lipid A biosynthesis | 100 (22) | 100 (82) | 100 (130) |
| htrB | Lauroyltransferase | 100 (22) | 98 (80) | 98 (127) | |
| ipx variants | Multiple | 100 1 (22) | 1001 (82) | 99 1 (129) | |
| fabZ | Acyl carrier protein | Fatty acid biosynthesis | 100 (22) | 98 (80) | 98 (127) |
| pgm | Phosphoglucomutase-1 | Carbohydrate metabolism | 95 (21) | 98 (80) | 97 (126) |
| cgs | Glucan synthesis | 95 (21) | 96 (79) | 95 (123) | |
| wbpL | Glucosyltransferase | Cell wall synthesis and organization | 100 (22) | 96 (79) | 96 (125) |
| ricA | Regulator protein | Biofilm formation | 100 (22) | 93 (76) | 95 (123) |
| htpB | Chaperone | Folding, adhesion, invasion factor | 36 (8) | 48 (39) | 40 (52) |
| man variants | Multiple | Polysaccharide synthesis | 92 (2) | 8.52 (7) | 9.22 (12) |
| wbpZ | Glycosyltransferase | 9 (2) | 8.5 (7) | 9.2 (12) | |
| mgtB | Magnesium-transporting ATPase | Mediates magnesium influx | 4.5 (1) | 3.7 (3) | 4.6 (6) |
| ureB | Urease subunit | Urea degradation | 4.5 (1) | 2.4 (2) | 2 (3) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yagel, Y.; Sestito, S.; Motro, Y.; Shnaiderman-Torban, A.; Khalfin, B.; Sagi, O.; Navon-Venezia, S.; Steinman, A.; Moran-Gilad, J. Genomic Characterization of Antimicrobial Resistance, Virulence, and Phylogeny of the Genus Ochrobactrum. Antibiotics 2020, 9, 177. https://doi.org/10.3390/antibiotics9040177
Yagel Y, Sestito S, Motro Y, Shnaiderman-Torban A, Khalfin B, Sagi O, Navon-Venezia S, Steinman A, Moran-Gilad J. Genomic Characterization of Antimicrobial Resistance, Virulence, and Phylogeny of the Genus Ochrobactrum. Antibiotics. 2020; 9(4):177. https://doi.org/10.3390/antibiotics9040177
Chicago/Turabian StyleYagel, Yael, Stephanie Sestito, Yair Motro, Anat Shnaiderman-Torban, Boris Khalfin, Orly Sagi, Shiri Navon-Venezia, Amir Steinman, and Jacob Moran-Gilad. 2020. "Genomic Characterization of Antimicrobial Resistance, Virulence, and Phylogeny of the Genus Ochrobactrum" Antibiotics 9, no. 4: 177. https://doi.org/10.3390/antibiotics9040177
APA StyleYagel, Y., Sestito, S., Motro, Y., Shnaiderman-Torban, A., Khalfin, B., Sagi, O., Navon-Venezia, S., Steinman, A., & Moran-Gilad, J. (2020). Genomic Characterization of Antimicrobial Resistance, Virulence, and Phylogeny of the Genus Ochrobactrum. Antibiotics, 9(4), 177. https://doi.org/10.3390/antibiotics9040177