A Simple Protocol for the Determination of Lysostaphin Enzymatic Activity

 , , and
, , and 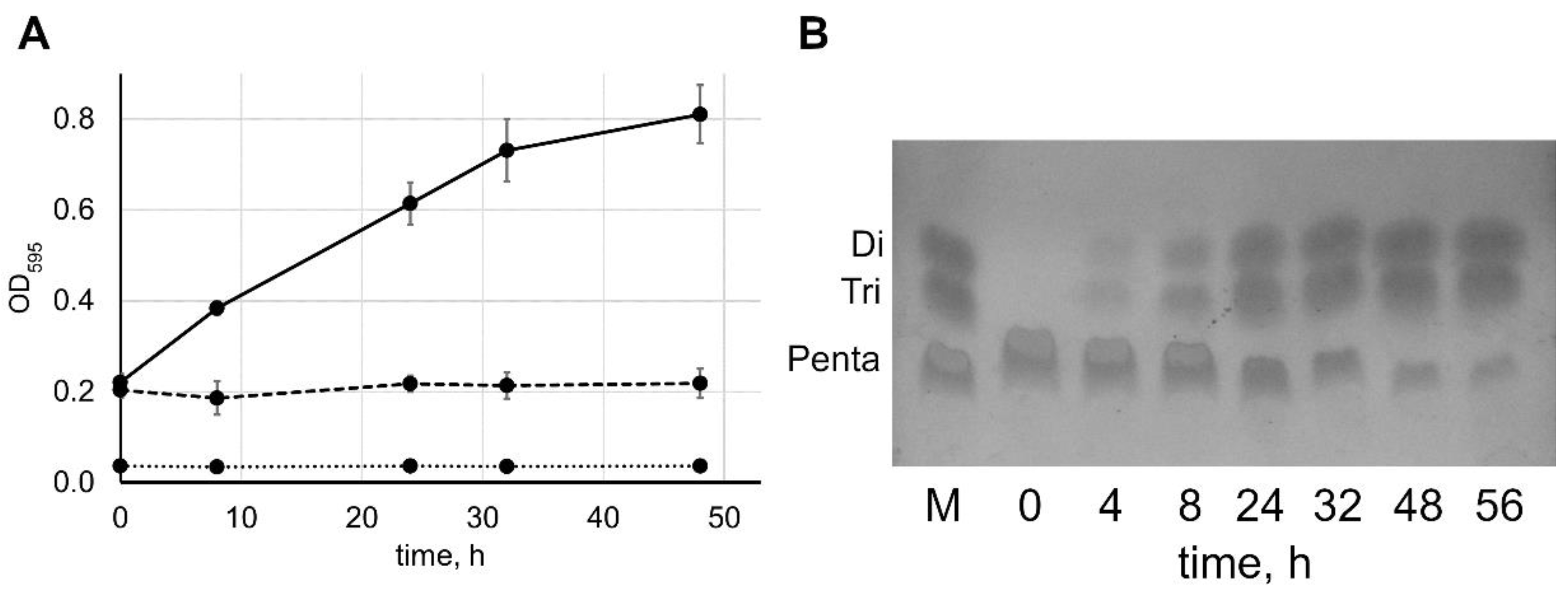{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. General Protocol Description
- Prepare 5 mM pentaglycine stock solution in water by heating the suspension of pentaglycine powder at 99 °C for 20 min.
- Mix pentaglycine stock solution, desired buffer stock solution, and lysostaphin stock solution.
- Aliquot the reaction mixture into 20 μL aliquots in 0.5 mL Eppendorf tubes and incubate the aliquots at the temperature of choice.
- At selected time points, remove the aliquots and place them at −80 °C to stop the reaction and preserve the samples for subsequent analysis.
- Thaw the samples at 99 °C for 10 min, add 100 μL of 0.4% w/v ninhydrin in 80% DMSO/20% water mixture buffered at pH 7.5, and mix thoroughly.
- Incubate the samples at 85 °C for 15 min. The color of the samples should become blue.
- Cool the samples to room temperature, add 200 µL of water, and mix. The color of the samples should turn violet.
- Transfer 100 µL of each sample into the wells of a 96-well plate and measure the optical density at 595 nm using a microplate reader.
2.2. Reaction Speed Depends on the Concentrations of Lysostaphin and Pentaglycine
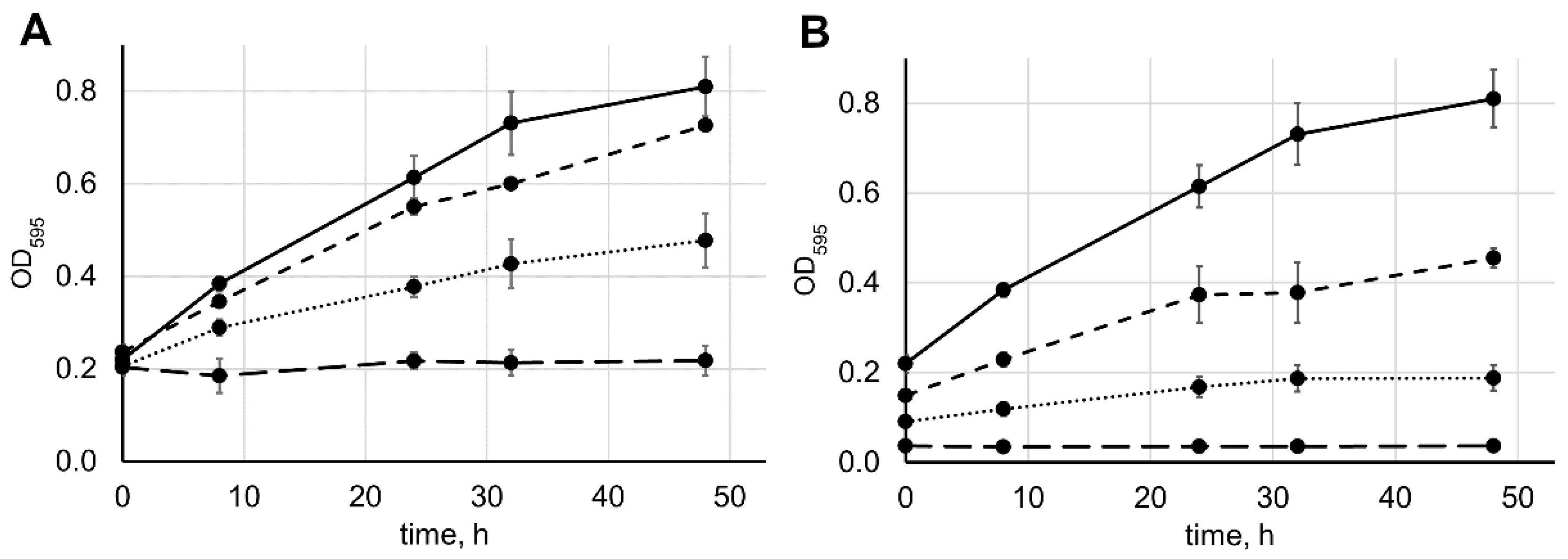
2.3. Comparison of the Enzymatic Activity of Lysostaphin Variants
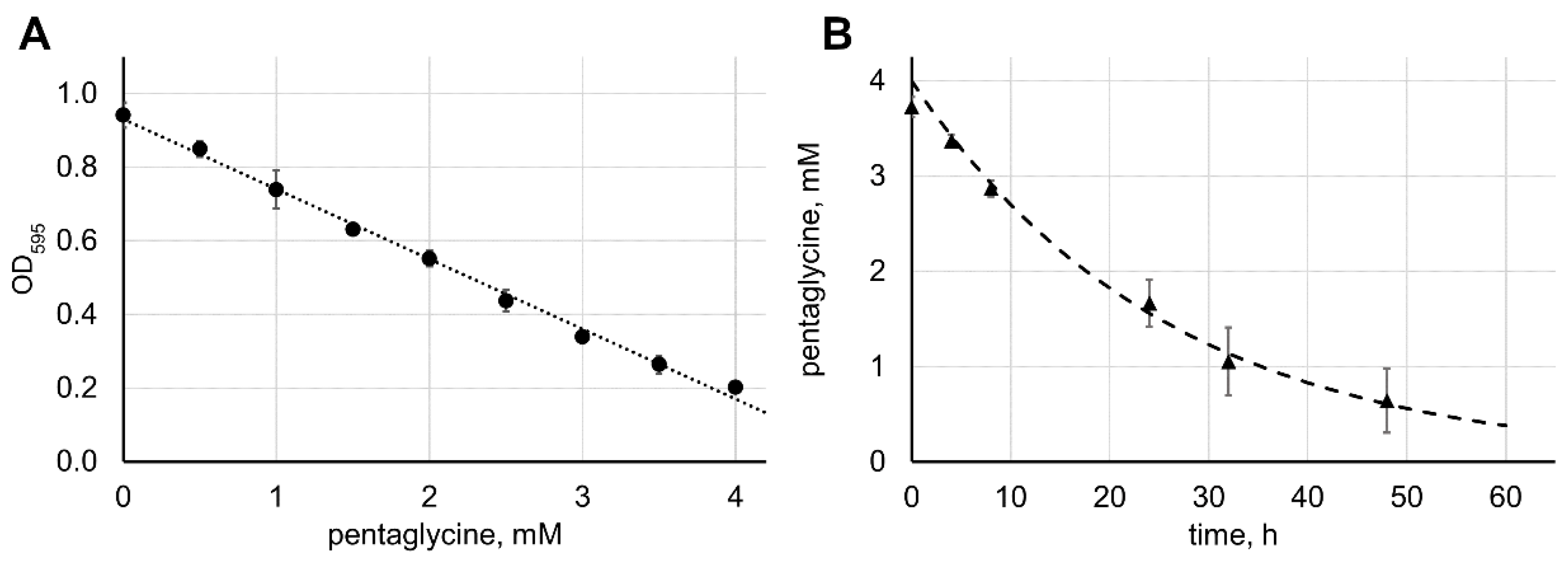
2.4. Calculation of the Lysostaphin Catalytic Parameters
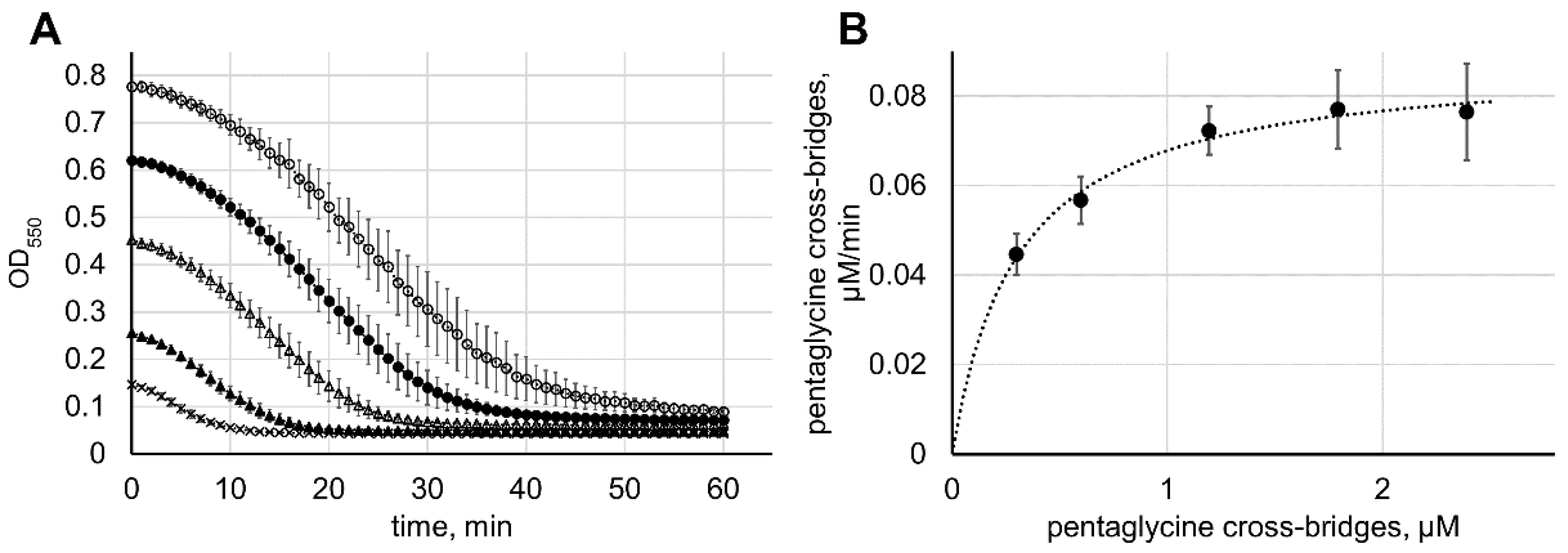
2.5. Comparison of Enzymatic and Bacteriolytic Activities of Lysostaphin
3. Materials and Methods
3.1. Production and Purification of Recombinant Lysostaphin
3.2. Determination of Lysostaphin Enzymatic Activity
3.3. Determination of Lysostaphin Bacteriolytic Activity
3.4. Thin-Layer Chromatography
3.5. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kokai-Kun, J.F. Lysostaphin: A silver bullet for staph. In Antimicrobial Drug Discovery: Emerging Strategies; CABI: Wallingford, CT, USA, 2012; pp. 147–165. ISBN 978-1-84593-943-4. [Google Scholar]
- Pastagia, M.; Schuch, R.; Fischetti, V.A.; Huang, D.B. Lysins: The arrival of pathogen-directed anti-infectives. J. Med. Microbiol. 2013, 62, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Lavigne, R.; Volckaert, G.; Hertveldt, K. A standardized approach for accurate quantification of murein hydrolase activity in high-throughput assays. J. Biochem. Biophys. Methods 2007, 70, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Márová, I.; Kovár, J. Spectrophotometric detection of bacteriolytic activity of diluted lysostaphin solutions. Folia Microbiol. 1993, 38, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.M.; Hardman, J.K.; Sloan, G.L. Relationship between lysostaphin endopeptidase production and cell wall composition in Staphylococcus staphylolyticus. J. Bacteriol. 1979, 137, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.A.; Schuhardt, V.T. Lysostaphin: A new bacteriolytic agent for the staphylococcus. Proc. Natl. Acad. Sci. USA 1964, 51, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Sugai, M.; Fujiwara, T.; Akiyama, T.; Ohara, M.; Komatsuzawa, H.; Inoue, S.; Suginaka, H. Purification and molecular characterization of glycylglycine endopeptidase produced by Staphylococcus capitis EPK1. J. Bacteriol. 1997, 179, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- DeHart, H.P.; Heath, H.E.; Heath, L.S.; LeBlanc, P.A.; Sloan, G.L. The lysostaphin endopeptidase resistance gene (epr) specifies modification of peptidoglycan cross bridges in Staphylococcus simulans and Staphylococcus aureus. Appl. Environ. Microbiol. 1995, 61, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Warfield, R.; Bardelang, P.; Saunders, H.; Chan, W.C.; Penfold, C.; James, R.; Thomas, N.R. Internally quenched peptides for the study of lysostaphin: An antimicrobial protease that kills Staphylococcus aureus. Org. Biomol. Chem. 2006, 4, 3626–3638. [Google Scholar] [CrossRef] [PubMed]
- Lood, R.; Molina, H.; Fischetti, V.A. Determining bacteriophage endopeptidase activity using either fluorophore-quencher labeled peptides combined with liquid chromatography-mass spectrometry (LC-MS) or Förster resonance energy transfer (FRET) assays. PLoS ONE 2017, 12, e0173919. [Google Scholar] [CrossRef] [PubMed]
- Bardelang, P.; Vankemmelbeke, M.; Zhang, Y.; Jarvis, H.; Antoniadou, E.; Rochette, S.; Thomas, N.R.; Penfold, C.N.; James, R. Design of a polypeptide FRET substrate that facilitates study of the antimicrobial protease lysostaphin. Biochem. J. 2009, 418, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kline, S.A.; Delaharpe, J.; Blackburn, P. A Colorimetric Microtiter Plate Assay for Lysostaphin Using a Hexaglycine Substrate. Anal. Biochem. 1994, 217, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Raulinaitis, V.; Tossavainen, H.; Aitio, O.; Juuti, J.T.; Hiramatsu, K.; Kontinen, V.; Permi, P. Identification and structural characterization of LytU, a unique peptidoglycan endopeptidase from the lysostaphin family. Sci. Rep. 2017, 7, 6020. [Google Scholar] [CrossRef] [PubMed]
- Tossavainen, H.; Raulinaitis, V.; Kauppinen, L.; Pentikäinen, U.; Maaheimo, H.; Permi, P. Structural and Functional Insights into Lysostaphin–Substrate Interaction. Front. Mol. Biosci. 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Mitkowski, P.; Jagielska, E.; Nowak, E.; Bujnicki, J.M.; Stefaniak, F.; Niedziałek, D.; Bochtler, M.; Sabała, I. Structural bases of peptidoglycan recognition by lysostaphin SH3b domain. Sci. Rep. 2019, 9, 5965. [Google Scholar] [CrossRef] [PubMed]
- Boksha, I.S.; Lavrova, N.V.; Grishin, A.V.; Demidenko, A.V.; Lyashchuk, A.M.; Galushkina, Z.M.; Ovchinnikov, R.S.; Umyarov, A.M.; Avetisian, L.R.; Chernukha, M.I.; et al. Staphylococcus simulans recombinant lysostaphin: Production, purification, and determination of antistaphylococcal activity. Biochemistry 2016, 81, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.V.; Lavrova, N.V.; Lyashchuk, A.M.; Strukova, N.V.; Generalova, M.S.; Ryazanova, A.V.; Shestak, N.V.; Boksha, I.S.; Polyakov, N.B.; Galushkina, Z.M.; et al. The Influence of Dimerization on the Pharmacokinetics and Activity of an Antibacterial Enzyme Lysostaphin. Molecules 2019, 24, 1879. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, A.V.; Konstantinova, S.V.; Vasina, I.V.; Shestak, N.V.; Karyagina, A.S.; Lunin, V.G. A Simple Protocol for the Determination of Lysostaphin Enzymatic Activity. Antibiotics 2020, 9, 917. https://doi.org/10.3390/antibiotics9120917
Grishin AV, Konstantinova SV, Vasina IV, Shestak NV, Karyagina AS, Lunin VG. A Simple Protocol for the Determination of Lysostaphin Enzymatic Activity. Antibiotics. 2020; 9(12):917. https://doi.org/10.3390/antibiotics9120917
Chicago/Turabian StyleGrishin, Alexander V., Svetlana V. Konstantinova, Irina V. Vasina, Nikita V. Shestak, Anna S. Karyagina, and Vladimir G. Lunin. 2020. "A Simple Protocol for the Determination of Lysostaphin Enzymatic Activity" Antibiotics 9, no. 12: 917. https://doi.org/10.3390/antibiotics9120917
APA StyleGrishin, A. V., Konstantinova, S. V., Vasina, I. V., Shestak, N. V., Karyagina, A. S., & Lunin, V. G. (2020). A Simple Protocol for the Determination of Lysostaphin Enzymatic Activity. Antibiotics, 9(12), 917. https://doi.org/10.3390/antibiotics9120917