Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors?


 , and
, and 
Abstract
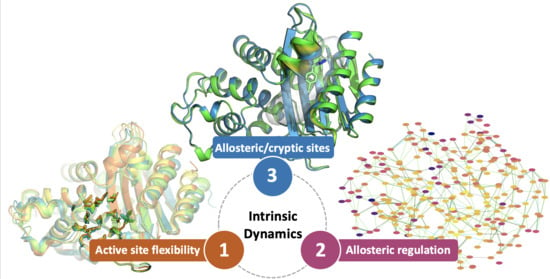
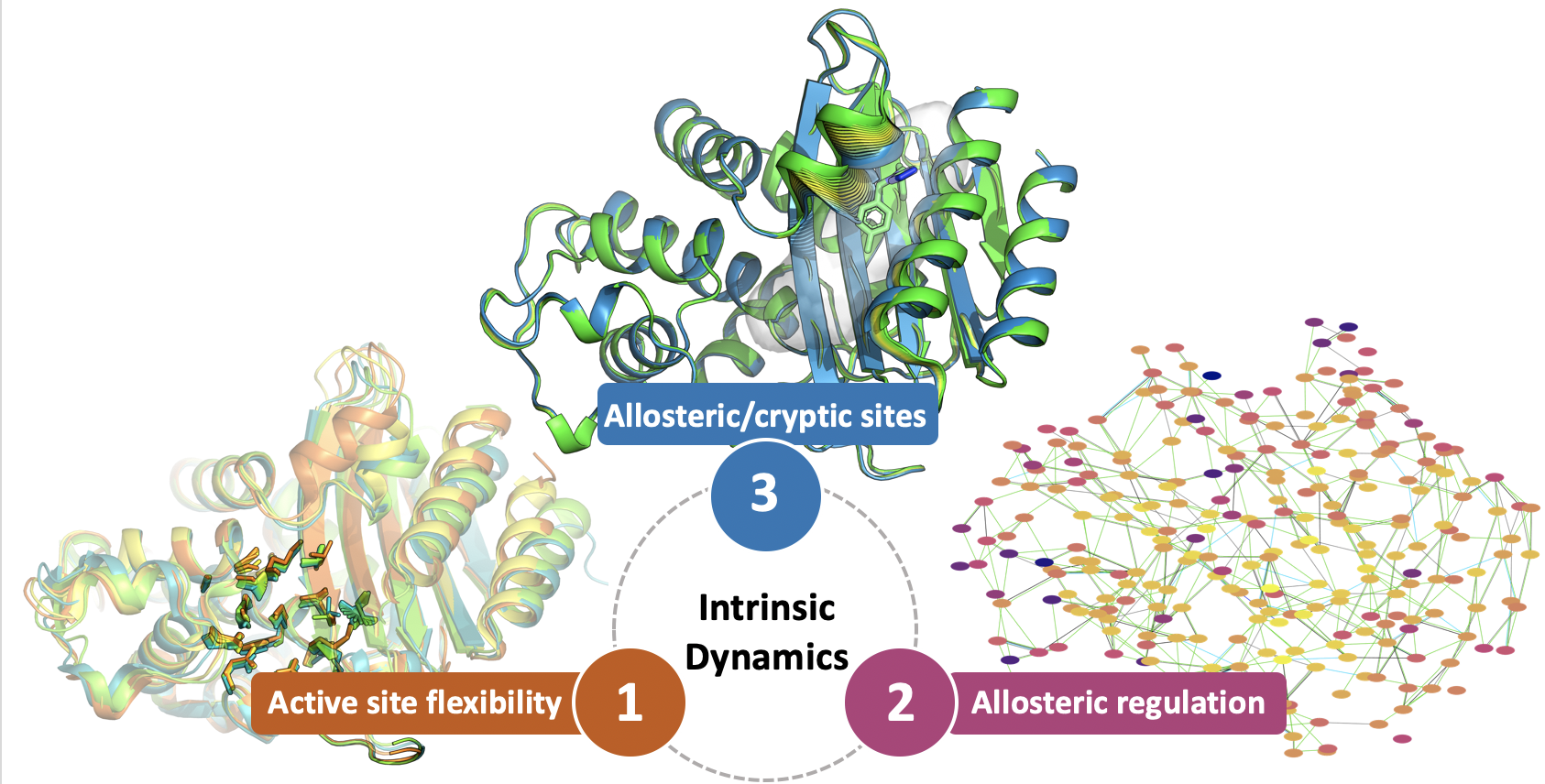
1. Introduction
2. BL Binding Site Flexibility
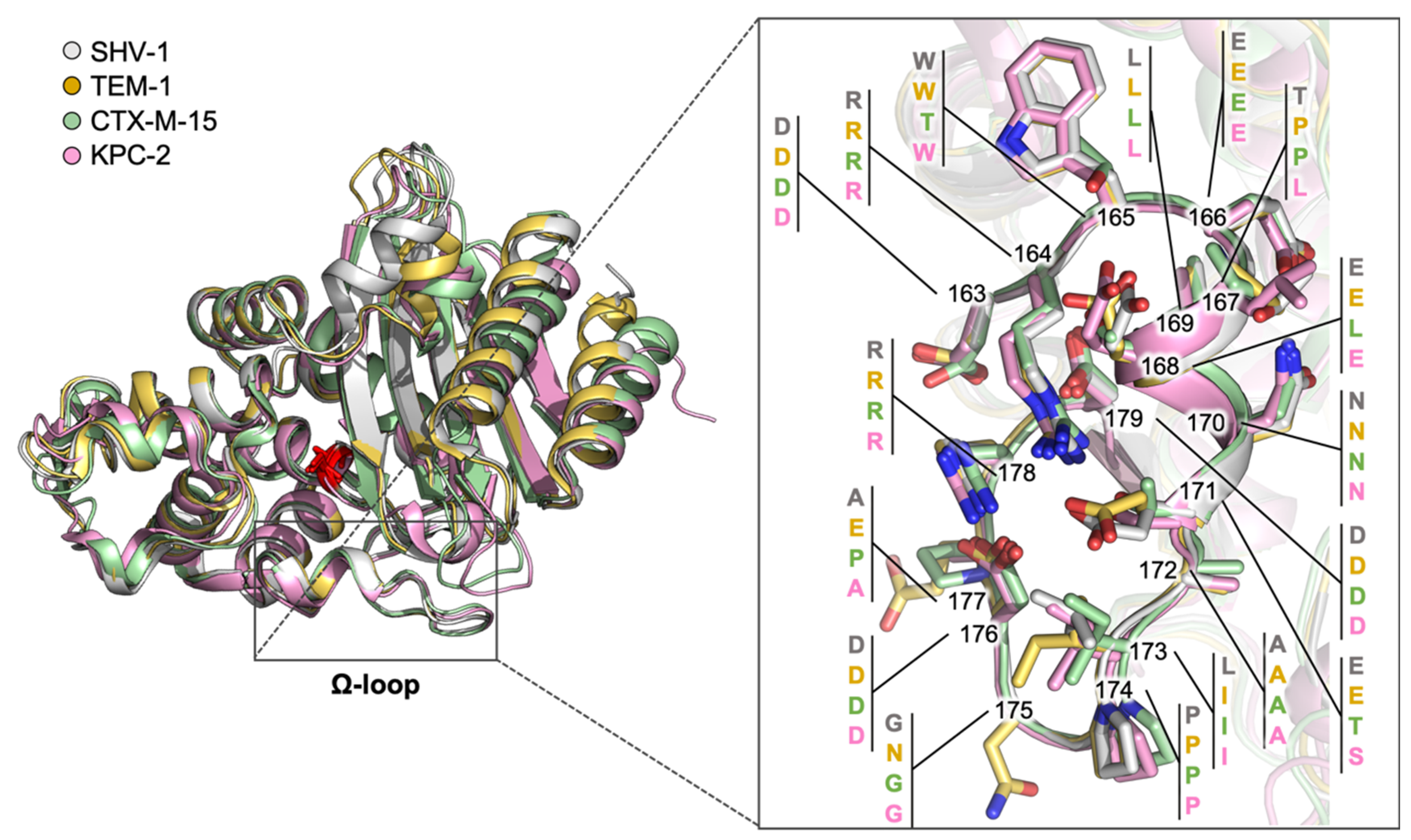
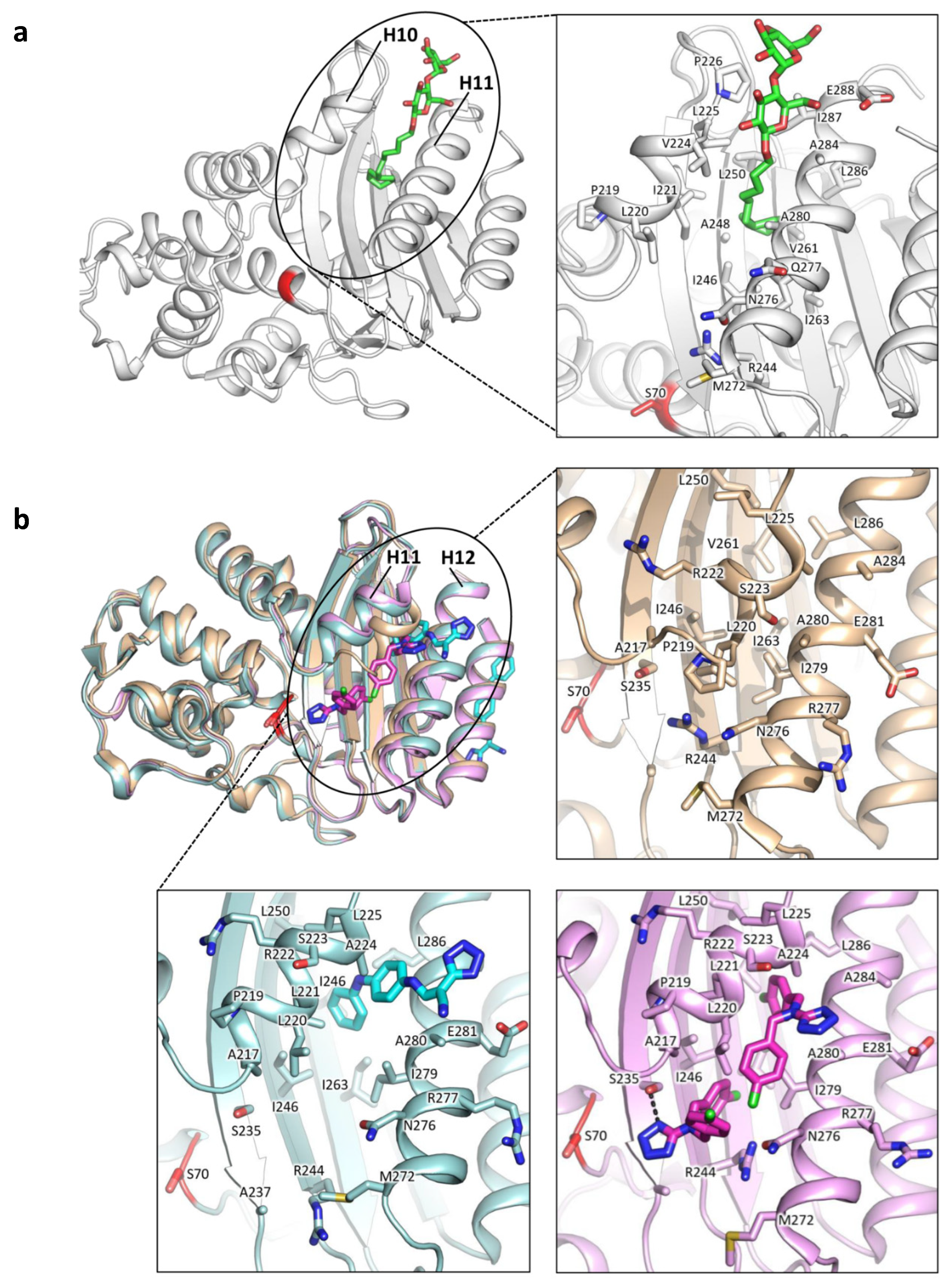
2.1. Class A Flexibility
2.2. Class D Flexibility
2.3. Class B (MBL) Flexibility
3. Allosteric Regulation in BLs: Potential Advantages and Challenges
3.1. Allosteric Effectors in Class A BLs
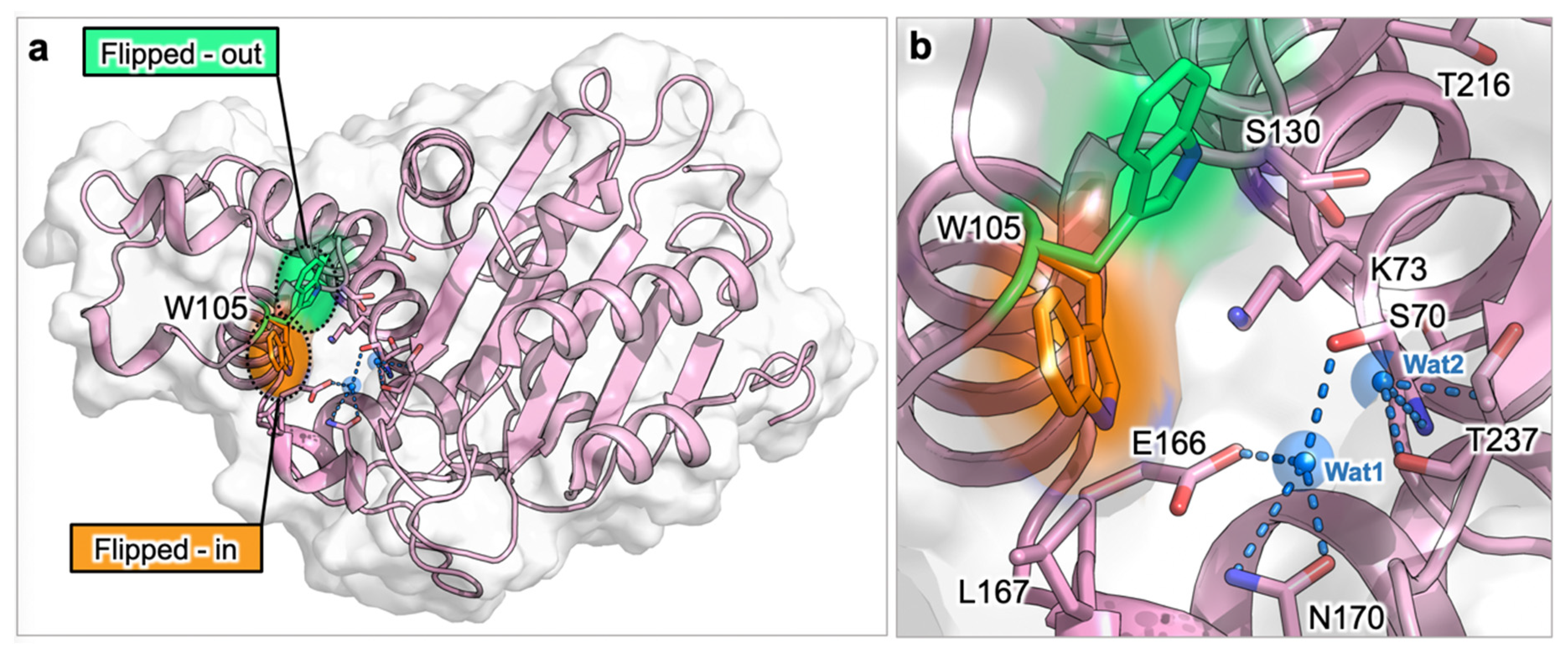
3.1.1. Identification of Cryptic Allosteric Sites in Class A BLs
3.1.2. Relevant Residues Mediating Allosteric Regulation in Class A BLs
3.2. Allosteric Effectors and Mechanisms in Class B BLs
3.3. Allosteric Effectors and Mechanisms in Class C BLs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nathan, C. Resisting antimicrobial resistance. Nat. Rev. Microbiol. 2020, 18, 259–260. [Google Scholar] [CrossRef] [PubMed]
- WHO. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance#:~:text=Antimicrobialresistance(AMR)threatensallgovernmentsectorsandsociety (accessed on 20 November 2020).
- WHO. Global Action Plan on Antimicrobial Resistance. Available online: https://www.who.int/antimicrobial-resistance/global-action-plan/en/ (accessed on 20 November 2020).
- AMR CONTROL. A National Action Plan to Contain Antimicrobial Resistance in China: Contents, Actions and Expectations. Available online: http://resistancecontrol.info/2017/a-national-action-plan-to-contain-antimicrobial-resistance-in-china-contents-actions-and-expectations/ (accessed on 20 November 2020).
- CDC, U.S. Action to Combat Antibiotic Resistance. Available online: https://www.cdc.gov/drugresistance/us-activities.html (accessed on 20 November 2020).
- European Commission. Action on Antimicrobial Resistance. Available online: https://ec.europa.eu/health/antimicrobial-resistance/eu-action-on-antimicrobial-resistance_en (accessed on 20 November 2020).
- Frère, J.-M. Beta-lactamases and bacterial resistance to antibiotics. Mol. Microbiol. 1995, 16, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Taubes, G. The bacteria fight back. Science 2008, 321, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Beta-Lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Ambler, R.P. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. NY. Acad. Sci. 2013, 1227, 91–104. [Google Scholar] [CrossRef]
- Ray, S.; Anand, D.; Purwar, S.; Samanta, A.; Upadhye, K.V.; Gupta, P.; Dhar, D. Association of high mortality with extended–spectrum β-lactamase (ESBL) positive cultures in community acquired infections. J. Crit. Care 2018, 44, 255–260. [Google Scholar] [CrossRef]
- Bush, K. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr. Opin. Microbiol. 2010, 13, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Bonomo, R.A. Status report on carbapenemases: Challenges and prospects. Expert Rev. Anti. Infect. Ther. 2011, 9, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Halat, D.H.; Moubareck, C.A. The current burden of carbapenemases: Review of significant properties and dissemination among gram-negative bacteria. Antibiotics 2020, 9, 186. [Google Scholar] [CrossRef] [PubMed]
- Torelli, N.J.; Akhtar, A.; Defrees, K.; Jaishankar, P.; Pemberton, O.A.; Zhang, X.; Johnson, C.; Renslo, A.R.; Chen, Y. Active-Site Druggability of Carbapenemases and Broad-Spectrum Inhibitor Discovery. ACS Infect. Dis. 2019, 5, 1013–1021. [Google Scholar] [CrossRef]
- King, D.T.; Worrall, L.J.; Gruninger, R.; Strynadka, N.C.J. New delhi metallo β-lactamase: Structural insights into β-lactam recognition and inhibition. J. Am. Chem. Soc. 2012, 134, 11362–11365. [Google Scholar] [CrossRef]
- Lobkovsky, E.; Moews, P.C.; Liu, H.; Zhao, H.; Frere, J.M.; Knox, J.R. Evolution of an enzyme activity: Crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class a penicillinase. Proc. Natl. Acad. Sci. USA. 1993, 90, 11257–11261. [Google Scholar] [CrossRef]
- Roccatano, D.; Sbardella, G.; Aschi, M.; Amicosante, G.; Bossa, C.; Di Nola, A.; Mazza, F. Dynamical aspects of TEM-1 β-Lactamase probed by molecular dynamics. J. Comput. Aided. Mol. Des. 2005, 19. [Google Scholar] [CrossRef]
- Egorov, A.; Rubtsova, M.; Grigorenko, V.; Uporov, I.; Veselovsky, A. The role of the Ω-loop in regulation of the catalytic activity of TEM-type β-lactamases. Biomolecules 2019, 9, 854. [Google Scholar] [CrossRef]
- Dellus-Gur, E.; Elias, M.; Caselli, E.; Prati, F.; Salverda, M.L.M.; De Visser, J.A.G.M.; Fraser, J.S.; Tawfik, D.S. Negative epistasis and evolvability in TEM-1 β-lactamase—The thin line between an enzyme’s conformational freedom and disorder. J. Mol. Biol. 2015, 427, 2396–2409. [Google Scholar] [CrossRef]
- Wang, X.; Minasov, G.; Shoichet, B.K. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 2002, 320, 85–95. [Google Scholar] [CrossRef]
- Bös, F.; Pleiss, J. Multiple molecular dynamics simulations of TEM β-lactamase: Dynamics and water binding of the Ω-loop. Biophys. J. 2009, 97, 2550–2558. [Google Scholar] [CrossRef] [PubMed]
- Hujer, A.M.; Bethel, C.R.; Bonomo, R.A. Antibody mapping of the linear epitopes of CMY-2 and SHV-1 β-lactamases. Antimicrob. Agents Chemother. 2004, 48, 3980–3988. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambler, R.P.; Coulson, A.F.W.; Frère, J.M.; Ghuysen, J.M.; Joris, B.; Forsman, M.; Levesque, R.C.; Tiraby, G.; Waley, S.G. A standard numbering scheme for the class A β-lactamases. Biochem. J. 1991, 276. Pt 1, 269. [Google Scholar] [CrossRef]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Poirel, L. Emerging carbapenemases in Gram-negative aerobes. Clin. Microbiol. Infect. 2002, 8, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.S.; Ward, S.; Kania, M.; Page, M.G.P.; Wharton, C.W. Multiple conformations of the acylenzyme formed in the hydrolysis of methicillin by Citrobacter freundii β-lactamase: A time-resolved FTIR spectroscopic study. Biochemistry 1999, 38, 3851–3856. [Google Scholar] [CrossRef] [PubMed]
- Maveyraud, L.; Pratt, R.F.; Samama, J.P. Crystal structure of an acylation transition-state analog of the TEM-1 β-lactamase. Mechanistic implications for class A β-lactamases. Biochemistry 1998, 37, 2622–2628. [Google Scholar] [CrossRef]
- Fonseca, F.; Chudyk, E.I.; Van Der Kamp, M.W.; Correia, A.; Mulholland, A.J.; Spencer, J. The basis for carbapenem hydrolysis by class a beta-lactamases: A combined investigation using crystallography and simulations. J. Am. Chem. Soc. 2012, 134, 18275–18285. [Google Scholar] [CrossRef]
- Cortina, G.A.; Hays, J.M.; Kasson, P.M. Conformational Intermediate That Controls KPC-2 Catalysis and Beta-Lactam Drug Resistance. ACS Catal. 2018, 8, 2741–2747. [Google Scholar] [CrossRef]
- Galdadas, I.; Lovera, S.; Pérez-Hernández, G.; Barnes, M.D.; Healy, J.; Afsharikho, H.; Woodford, N.; Bonomo, R.A.; Gervasio, F.L.; Haider, S. Defining the architecture of KPC-2 Carbapenemase: Identifying allosteric networks to fight antibiotics resistance. Sci. Rep. 2018, 8, 12916. [Google Scholar] [CrossRef]
- Petit, A.; Maveyraud, L.; Lenfant, F.; Samama, J.P.; Labia, R.; Masson, J.M. Multiple substitutions at position 104 of β-lactamase TEM-1: Assessing the role of this residue in substrate specificity. Biochem. J. 1995, 305, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, R.; Recule, C.; Baraduc, R.; Chanal, C.; Sirot, D.; De Champs, C.; Sirot, J. Effect of D240G substitution in a novel ESBL CTX-M-27. J. Antimicrob. Chemother. 2003, 52, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.C.; Rice, K.; Palzkill, T. Natural Variants of the KPC-2 Carbapenemase have Evolved Increased Catalytic Efficiency for Ceftazidime Hydrolysis at the Cost of Enzyme Stability. PLoS Pathog. 2015, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Arnold, F.H. In the light of directed evolution: Pathways of adaptive protein evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 9995–10000. [Google Scholar] [CrossRef]
- Knies, J.L.; Cai, F.; Weinreich, D.M. Enzyme Efficiency but Not Thermostability Drives Cefotaxime Resistance Evolution in TEM-1 β-Lactamase. Mol. Biol. Evol. 2017, 34, 1040–1054. [Google Scholar] [CrossRef]
- Modi, T.; Banu Ozkan, S. Mutations utilize dynamic allostery to confer resistance in TEM-1 β-lactamase. Int. J. Mol. Sci. 2018, 19, 3808. [Google Scholar] [CrossRef]
- Horn, J.R.; Shoichet, B.K. Allosteric Inhibition Through Core Disruption. J. Mol. Biol. 2004, 336, 1283–1291. [Google Scholar] [CrossRef]
- Tokuriki, N.; Stricher, F.; Serrano, L.; Tawfik, D.S. How protein stability and new functions trade off. PLoS Comput. Biol. 2008, 4, e1000002. [Google Scholar] [CrossRef]
- Fisher, J.F.; Meroueh, S.O.; Mobashery, S. Bacterial resistance to β-lactam antibiotics: Compelling opportunism, compelling opportunity. Chem. Rev. 2005, 105, 395–424. [Google Scholar] [CrossRef]
- Kaitany, K.C.J.; Klinger, N.V.; June, C.M.; Ramey, M.E.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Structures of the class D carbapenemases OXA-23 and OXA-146: Mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob. Agents Chemother. 2013, 57, 4848–4855. [Google Scholar] [CrossRef]
- Mitchell, J.M.; Clasman, J.R.; June, C.M.; Kaitany, K.C.J.; LaFleur, J.R.; Taracila, M.A.; Klinger, N.V.; Bonomo, R.A.; Wymore, T.; Szarecka, A.; et al. Structural Basis of Activity against Aztreonam and Extended Spectrum Cephalosporins for Two Carbapenem-Hydrolyzing Class D β-Lactamases from Acinetobacter baumannii. Biochemistry 2015, 54, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- Maveyraud, L.; Golemi, D.; Kotra, L.P.; Tranier, S.; Vakulenko, S.; Mobashery, S.; Samama, J.P. Insights into class D β-lactamases are revealed by the crystal structure of the OXA10 enzyme from Pseudomonas aeruginosa. Structure 2000, 8. [Google Scholar] [CrossRef]
- Szarecka, A.; Lesnock, K.R.; Ramirez-Mondragon, C.A.; Nicholas, H.B.; Wymore, T. The Class D β-lactamase family: Residues governing the maintenance and diversity of function. Protein Eng. Des. Sel. 2011, 24, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Antunes, N.T.; Stewart, N.K.; Toth, M.; Kumarasiri, M.; Chang, M.; Mobashery, S.; Vakulenko, S.B. Structural basis for carbapenemase activity of the OXA-23 β-Lactamase from acinetobacter baumannii. Chem. Biol. 2013, 20, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Legorreta, E.M.; Garza-Ramos, U.; Barrios-Camacho, H.; Sanchez-Perez, A.; Galicia-Paredes, A.; Meza-Chavez, A.; Silva-Sanchez, J. Identification of OXA-23 carbapenemases: Novel variant OXA-239 in Acinetobacter baumannii ST758 clinical isolates in Mexico. New Microbes New Infect. 2014, 2, 173–174. [Google Scholar] [CrossRef]
- Harper, T.M.; June, C.M.; Taracila, M.A.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Multiple substitutions lead to increased loop flexibility and expanded specificity in Acinetobacter baumannii carbapenemase OXA-239. Biochem. J. 2018, 475, 273–288. [Google Scholar] [CrossRef]
- Beadle, B.M.; Shoichet, B.K. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 2002, 321, 285–296. [Google Scholar] [CrossRef]
- Marciano, D.C.; Pennington, J.M.; Wang, X.; Wang, J.; Chen, Y.; Thomas, V.L.; Shoichet, B.K.; Palzkill, T. Genetic and Structural Characterization of an L201P Global Suppressor Substitution in TEM-1 β-Lactamase. J. Mol. Biol. 2008, 384. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Feng, H.; Ding, J.; Zhu, D.; Liu, X.; Xu, X.; Zhang, Y.; Zang, S.; Wang, D.C.; Liu, W. Structural and mechanistic insights into NDM-1 catalyzed hydrolysis of cephalosporins. J. Am. Chem. Soc. 2014, 136, 14694–14697. [Google Scholar] [CrossRef]
- Feng, H.; Liu, X.; Wang, S.; Fleming, J.; Wang, D.C.; Liu, W. The mechanism of NDM-1-catalyzed carbapenem hydrolysis is distinct from that of penicillin or cephalosporin hydrolysis. Nat. Commun. 2017, 8, 2242. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Kupper, M.B.; Herzog, K.; Bennink, S.; Schlömer, P.; Bogaerts, P.; Glupczynski, Y.; Fischer, R.; Bebrone, C.; Hoffmann, K.M. The three-dimensional structure of VIM-31—A metallo-β-lactamase from Enterobacter cloacae in its native and oxidized form. FEBS J. 2015, 282, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Matsueda, S.; Matsunaga, K.; Takashio, N.; Toma-Fukai, S.; Yamagata, Y.; Shibata, N.; Wachino, J.I.; Shibayama, K.; Arakawa, Y.; et al. Crystal structure of IMP-2 metallo-β-lactamase from Acinetobacter spp.: Comparison of active-site loop structures between IMP-1 and IMP-2. Biol. Pharm. Bull. 2015, 38, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Borra, P.S.; Leiros, H.K.S.; Ahmad, R.; Spencer, J.; Leiros, I.; Walsh, T.R.; Sundsfjord, A.; Samuelsen, E. Structural and computational investigations of VIM-7: Insights into the substrate specificity of VIM metallo-β-lactamases. J. Mol. Biol. 2011, 411, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Palacios, A.R.; Mojica, M.F.; Giannini, E.; Taracila, M.A.; Bethel, C.R.; Alzari, P.M.; Otero, L.H.; Klinke, S.; Llarrull, L.I.; Bonomo, R.A.; et al. The reaction mechanism of metallo-lactamases is tuned by the conformation of an active-site mobile loop. Antimicrob. Agents Chemother. 2018, 63, e01754-18. [Google Scholar] [CrossRef] [PubMed]
- Makena, A.; Düzgün, A.; Brem, J.; McDonough, M.A.; Rydzik, A.M.; Abboud, M.I.; Saral, A.; Çiçek, A.; Sandalli, C.; Schofield, C.J. Comparison of verona integron-borne metallo-β-lactamase (VIM) variants reveals differences in stability and inhibition profiles. Antimicrob. Agents Chemother. 2016, 60, 1377–1384. [Google Scholar] [CrossRef]
- Linciano, P.; Gianquinto, E.; Montanari, M.; Maso, L.; Bellio, P.; Cebrian-Sastre, E.; Celenza, G.; Blazquez, J.; Cendron, L.; Spyrakis, F.; et al. 4-amino-1,2,4-triazole-3-thione as promising scaffold for the inhibition of serine and metallo beta-lactamases. Pharmaceuticals 2020, 13, 52. [Google Scholar] [CrossRef]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.D.; Cendron, L.; et al. Virtual screening identifies broad-spectrum β-lactamase inhibitors with activity on clinically relevant serine- and metallo-carbapenemases. Sci. Rep. 2020, 10, 12763. [Google Scholar] [CrossRef]
- Kuzin, A.P.; Nukaga, M.; Nukaga, Y.; Hujer, A.M.; Bonomo, R.A.; Knox, J.R. Structure of the SHV-1 β-lactamase. Biochemistry 1999, 38, 5720–5727. [Google Scholar] [CrossRef]
- Grigorenko, V.G.; Andreeva, I.P.; Rubtsova, M.Y.; Deygen, I.M.; Antipin, R.L.; Majouga, A.G.; Egorov, A.M.; Beshnova, D.A.; Kallio, J.; Hackenberg, C.; et al. Novel non-β-lactam inhibitor of β-lactamase TEM-171 based on acylated phenoxyaniline. Biochimie 2017, 132, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Moeder, K.E.; Ho, C.M.W.; Zimmerman, M.I.; Frederick, T.E.; Bowman, G.R. Designing small molecules to target cryptic pockets yields both positive and negative allosteric modulators. PLoS ONE 2017, 12, e0178678. [Google Scholar] [CrossRef] [PubMed]
- Avci, F.G.; Altinisik, F.E.; Karacan, I.; Senturk Karagoz, D.; Ersahin, S.; Eren, A.; Sayar, N.A.; Vardar Ulu, D.; Ozkirimli, E.; Sariyar Akbulut, B. Targeting a hidden site on class A beta-lactamases. J. Mol. Graph. Model. 2018, 84, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.N.; Barrios, H.; Mathonet, P.; Evrard, C.; Ubbink, M.; Declercq, J.P.; Soumillion, P.; Fastrez, J. Engineering an Allosteric Binding Site for Aminoglycosides into TEM1-β-Lactamase. ChemBioChem 2011, 12, 904–913. [Google Scholar] [CrossRef]
- Van De Water, K.; Soror, S.H.; Wohlkonig, A.; Van Nuland, N.A.J.; Volkov, A.N. Crystallization and preliminary X-ray diffraction analysis of kanamycin-binding β-lactamase in complex with its ligand. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, F67, 703–706. [Google Scholar] [CrossRef]
- Vajda, S.; Beglov, D.; Wakefield, A.E.; Egbert, M.; Whitty, A. Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. [Google Scholar] [CrossRef]
- Cimermancic, P.; Weinkam, P.; Rettenmaier, T.J.; Bichmann, L.; Keedy, D.A.; Woldeyes, R.A.; Schneidman-Duhovny, D.; Demerdash, O.N.; Mitchell, J.C.; Wells, J.A.; et al. CryptoSite: Expanding the Druggable Proteome by Characterization and Prediction of Cryptic Binding Sites. J. Mol. Biol. 2016, 428, 709–719. [Google Scholar] [CrossRef]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef]
- Bowman, G.R.; Geissler, P.L. Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. USA 2012, 109, 11681–11686. [Google Scholar] [CrossRef]
- Bowman, G.R.; Bolin, E.R.; Hart, K.M.; Maguire, B.C.; Marqusee, S. Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. USA 2015, 112, 2734–2739. [Google Scholar] [CrossRef]
- Porter, J.R.; Moeder, K.E.; Sibbald, C.A.; Zimmerman, M.I.; Hart, K.M.; Greenberg, M.J.; Bowman, G.R. Cooperative Changes in Solvent Exposure Identify Cryptic Pockets, Switches, and Allosteric Coupling. Biophys. J. 2019, 116, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Meneksedag, D.; Dogan, A.; Kanlikilicer, P.; Ozkirimli, E. Communication between the active site and the allosteric site in class A beta-lactamases. Comput. Biol. Chem. 2013, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Avci, F.G.; Altinisik, F.E.; Vardar Ulu, D.; Ozkirimli Olmez, E.; Sariyar Akbulut, B. An evolutionarily conserved allosteric site modulates beta-lactamase activity. J. Enzyme Inhib. Med. Chem. 2016, 33–40. [Google Scholar] [CrossRef]
- Huang, L.; So, P.K.; Chen, Y.W.; Leung, Y.C.; Yao, Z.P. Conformational Dynamics of the Helix 10 Region as an Allosteric Site in Class A β-Lactamase Inhibitory Binding. J. Am. Chem. Soc. 2020, 142, 13756–13767. [Google Scholar] [CrossRef] [PubMed]
- Latallo, M.J.; Cortina, G.A.; Faham, S.; Nakamoto, R.K.; Kasson, P.M. Predicting allosteric mutants that increase activity of a major antibiotic resistance enzyme. Chem. Sci. 2017, 8, 6484–6492. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Chang, Y.N.; Ge, Y.; Kang, J.S.; Zhang, Y.L.; Liu, X.L.; Oelschlaeger, P.; Yang, K.W. Azolylthioacetamides as a potent scaffold for the development of metallo-β-lactamase inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 5225–5229. [Google Scholar] [CrossRef]
- Huang, P.J.J.; Pautler, R.; Shanmugaraj, J.; Labbé, G.; Liu, J. Inhibiting the VIM-2 metallo-β-lactamase by graphene oxide and carbon nanotubes. ACS Appl. Mater. Interfaces 2015, 7, 9898–9903. [Google Scholar] [CrossRef]
- Sohier, J.S.; Laurent, C.; Chevigné, A.; Pardon, E.; Srinivasan, V.; Wernery, U.; Lassaux, P.; Steyaert, J.; Galleni, M. Allosteric inhibition of VIM metallo-β-lactamases by a camelid nanobody. Biochem. J. 2013, 450, 477–486. [Google Scholar] [CrossRef]
- Ouyang, X.; Chang, Y.N.; Yang, K.W.; Wang, W.M.; Bai, J.J.; Wang, J.W.; Zhang, Y.J.; Wang, S.Y.; Xie, B.B.; Wang, L.L. A DNA nanoribbon as a potent inhibitor of metallo-β-lactamases. Chem. Commun. 2017, 53, 8878–8881. [Google Scholar] [CrossRef]
- Khan, N.H.; Bui, A.A.; Xiao, Y.; Sutton, R.B.; Shaw, R.W.; Wylie, B.J.; Latham, M.P. A DNA aptamer reveals an allosteric site for inhibition in metallo-β-lactamases. PLoS ONE 2019, 14, e0214440. [Google Scholar] [CrossRef]
- Brown, J.R.; Livesay, D.R. Flexibility correlation between active site regions is conserved across four AmpC β-lactamase enzymes. PLoS ONE 2015, 10, e0125832. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Compound Structure | BL Inhibition (µM) | References | PDB ID |
|---|---|---|---|---|
| Cymal-6 |  | IC50 = 100 [68] vs. TEM-1 and 20–25% reaction rate decrease vs. SHV-1 [65] | [65] | 1shv |
| FTA |  | Ki = 490 ± 40 vs. TEM-1 | [42] | 1pzp |
| CBT |  | Ki = 480 ± 20 vs. TEM-1 | [42] | 1pzo |
| NSC 341597 |  | EC50 = 57 ± 3 vs. TEM-1 | [67] | / |
| Name | Compound structure | BL activation (µM) | References | PDB ID |
| NSC 333009 |  | EC50 = 63 ± 9 vs TEM-1 | [67] | / |
| NSC 350086 |  | EC50 = 162 ± 15 vs TEM-1 | [67] | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianquinto, E.; Tondi, D.; D'Arrigo, G.; Lazzarato, L.; Spyrakis, F. Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors? Antibiotics 2020, 9, 833. https://doi.org/10.3390/antibiotics9110833
Gianquinto E, Tondi D, D'Arrigo G, Lazzarato L, Spyrakis F. Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors? Antibiotics. 2020; 9(11):833. https://doi.org/10.3390/antibiotics9110833
Chicago/Turabian StyleGianquinto, Eleonora, Donatella Tondi, Giulia D'Arrigo, Loretta Lazzarato, and Francesca Spyrakis. 2020. "Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors?" Antibiotics 9, no. 11: 833. https://doi.org/10.3390/antibiotics9110833
APA StyleGianquinto, E., Tondi, D., D'Arrigo, G., Lazzarato, L., & Spyrakis, F. (2020). Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors? Antibiotics, 9(11), 833. https://doi.org/10.3390/antibiotics9110833