
Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA)


 and
and 
Abstract
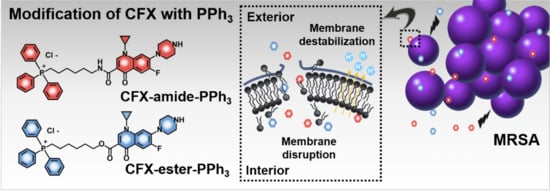
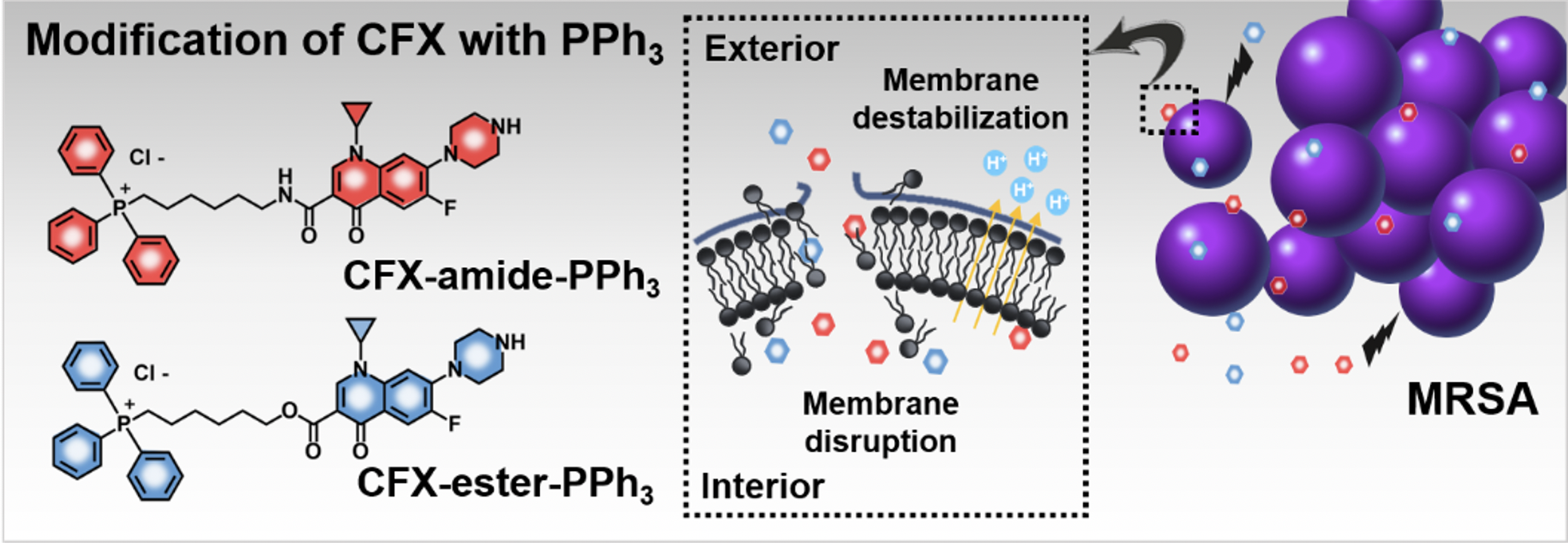
1. Introduction
2. Results and Discussion
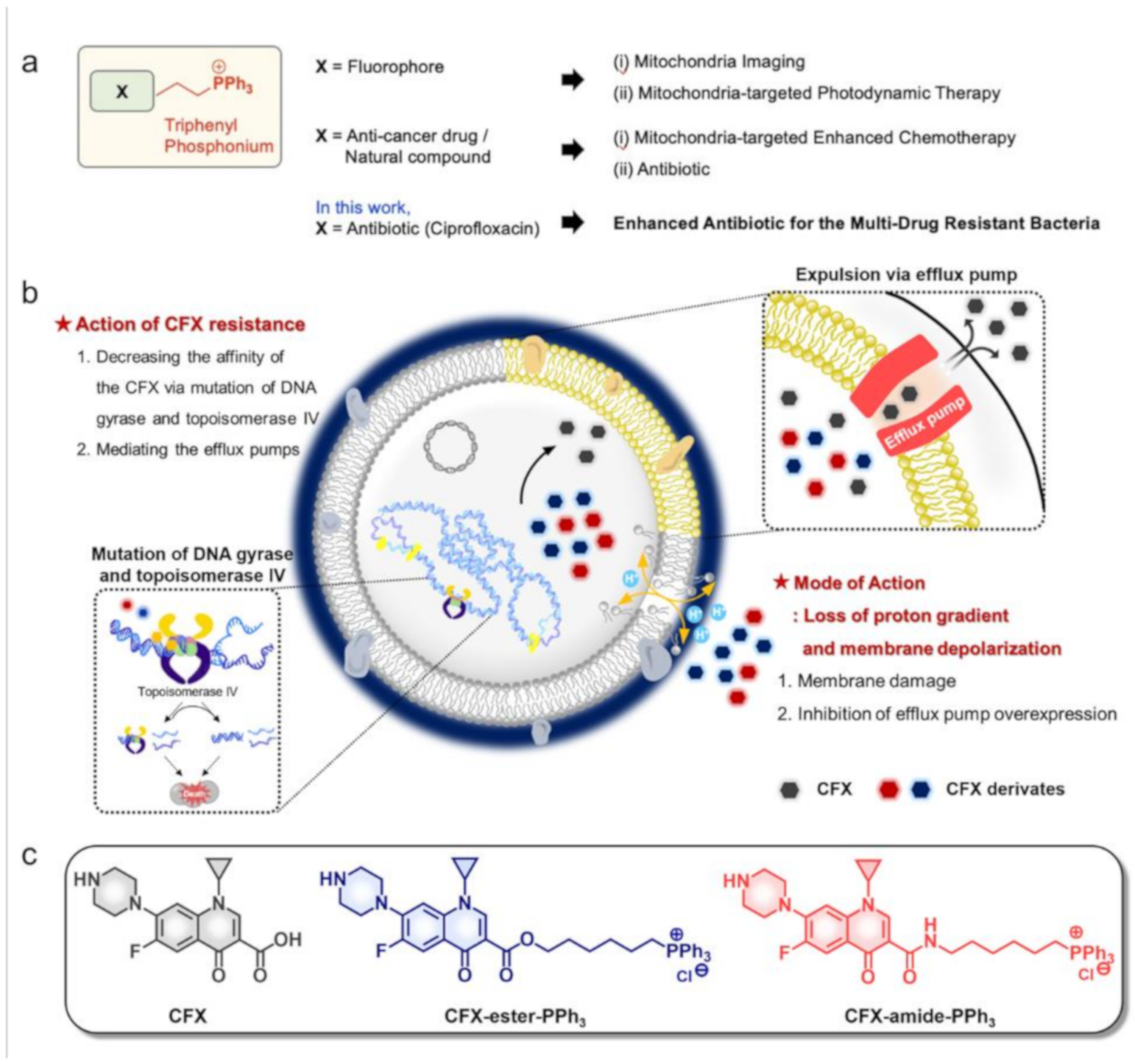
2.1. Rational
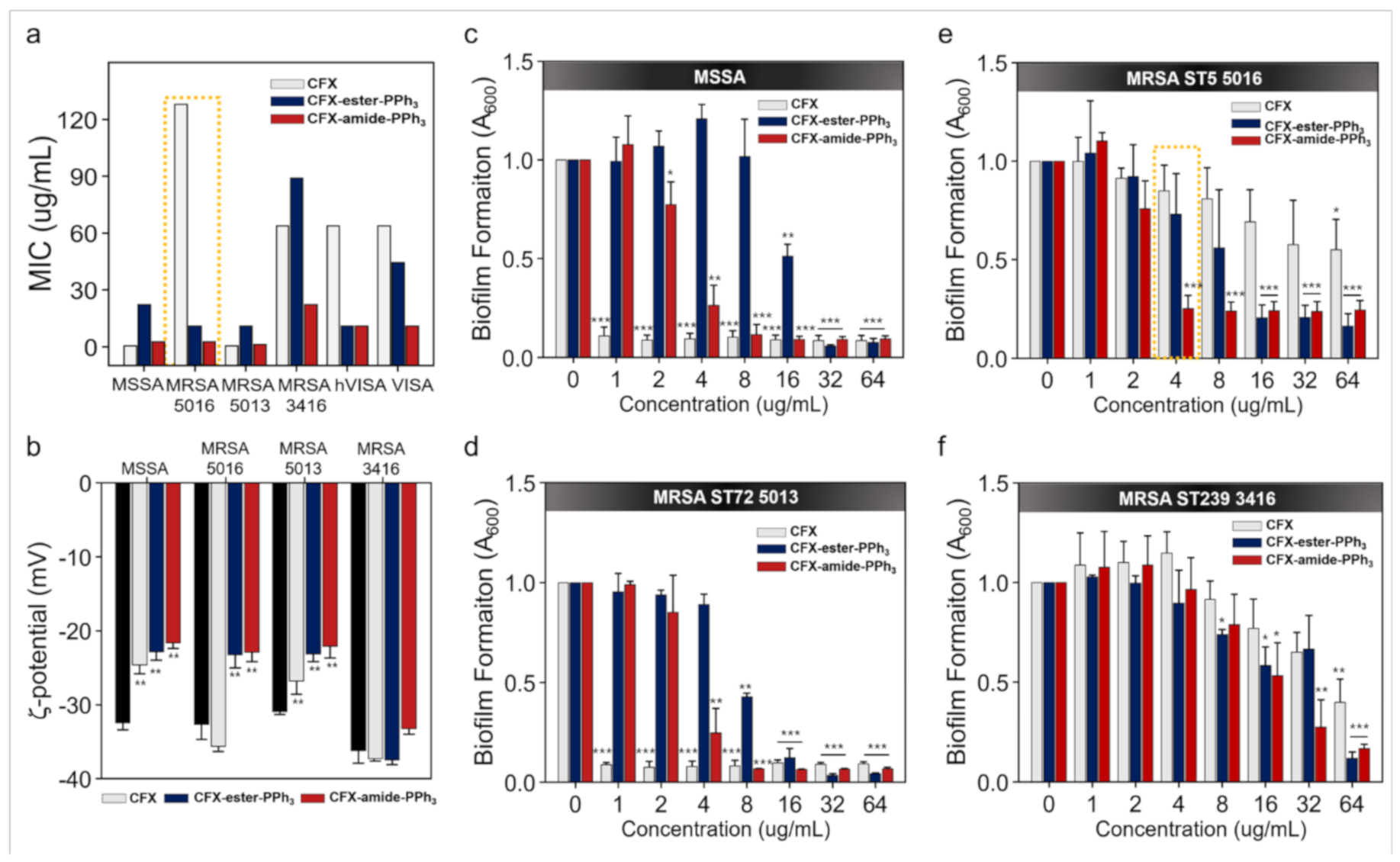
2.2. MIC Assay
2.3. Cytotoxicity Assay
2.4. Membrane-Potential Analysis
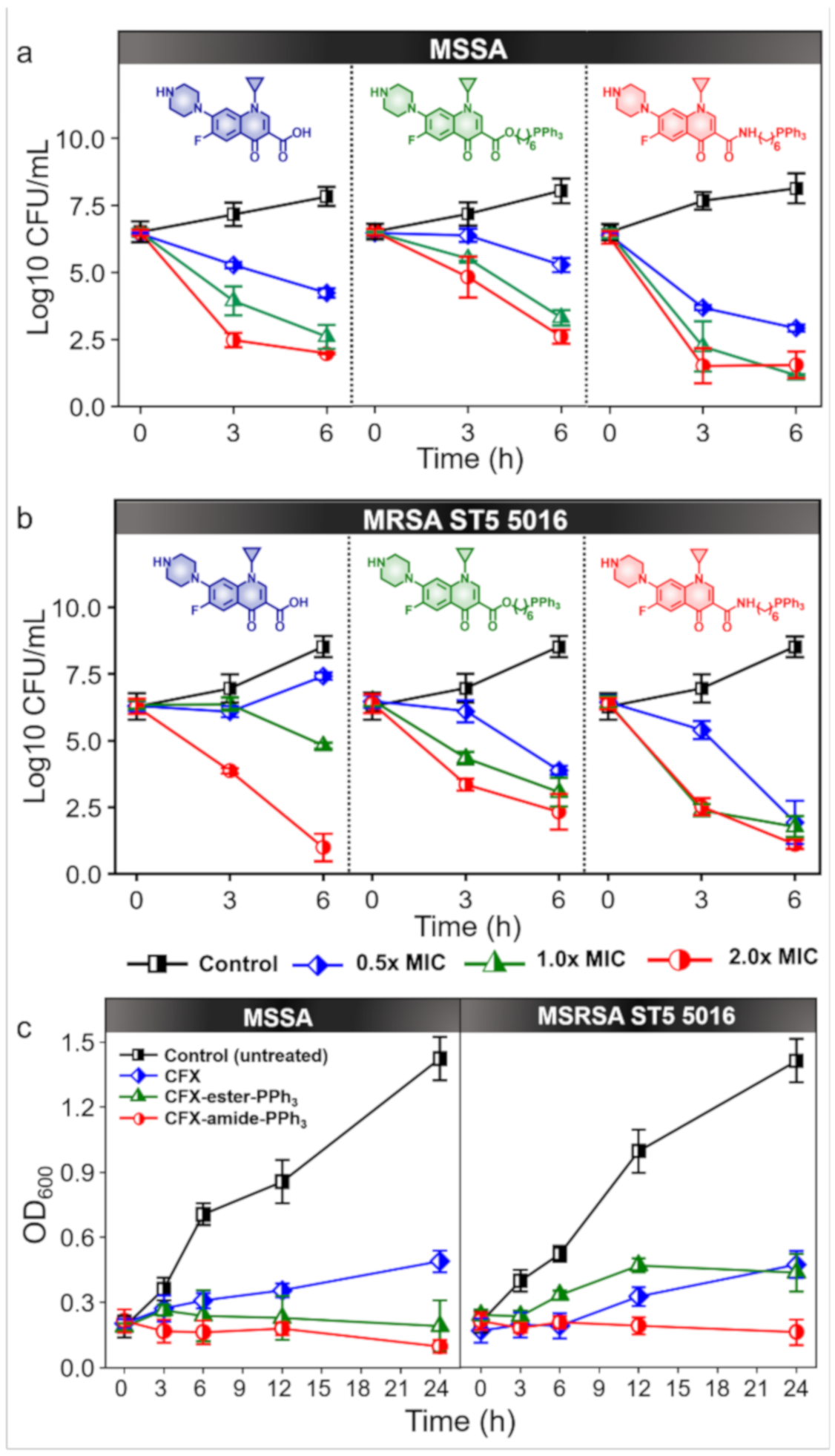
2.5. Crystal Violet Assay
2.6. Time-Kill Assay
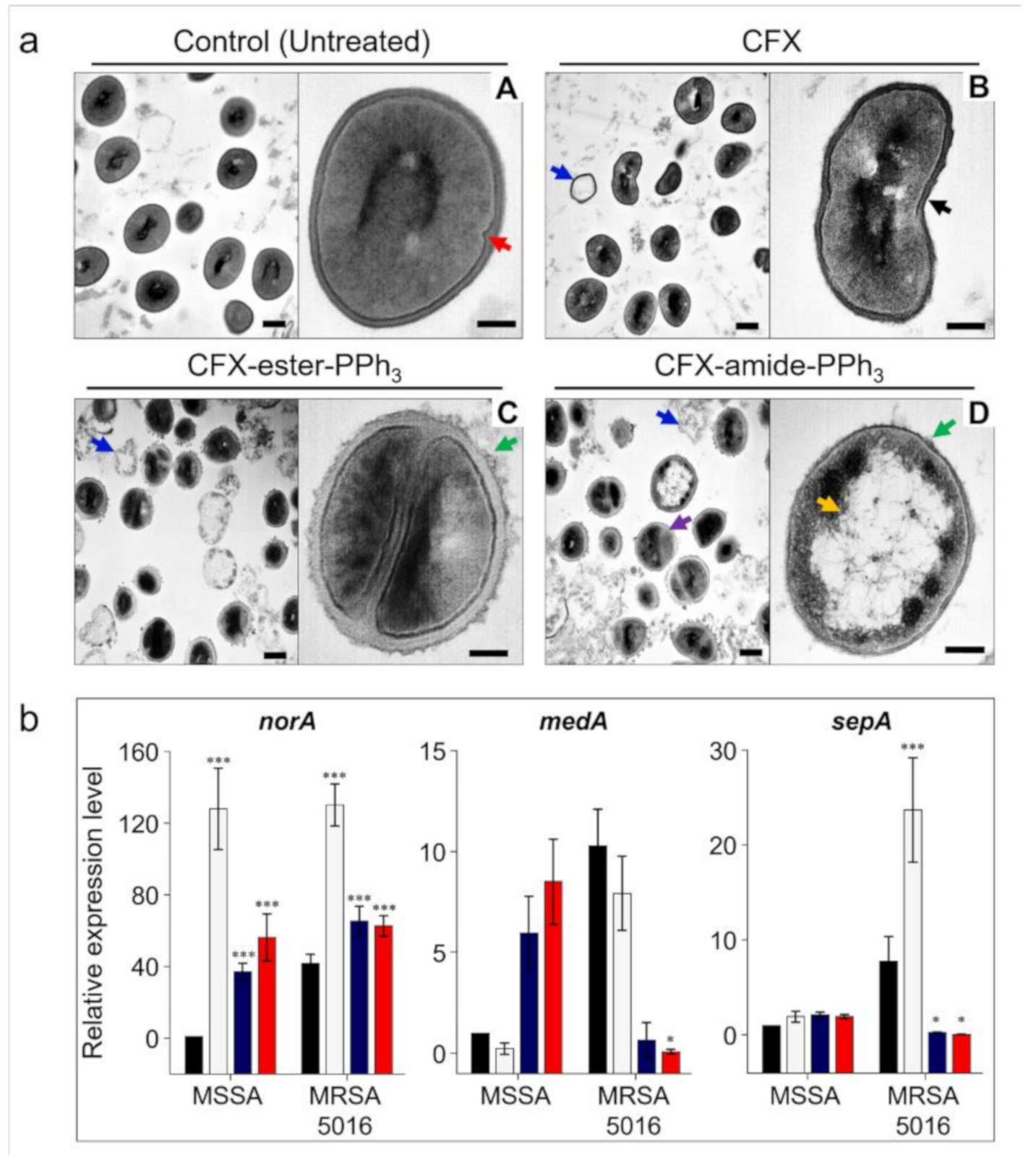
2.7. Morphology Analysis
2.8. Gene Expression Analysis
3. Materials and Methods
3.1. Bacteria Strains and Culture
3.2. Preparation of CFX-PPh3 Derivatives
3.3. MIC Assay
3.4. Membrane-Potential Analysis
3.5. Biofilm Formation Assay
3.6. Time-Kill Assay
3.7. Quantitative Real-Time PCR Analysis
3.8. Transmission Electron Microscopy (TEM) Imaging
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Alam, M.M.; Islam, M.; Wahab, A.; Billah, M. Antimicrobial resistance crisis and combating approaches. J. Med. 2019, 20, 38–45. [Google Scholar] [CrossRef]
- Nicolas, I.; Bordeau, V.; Bondon, A.; Baudy-Floc’h, M.; Felden, B. Novel antibiotics effective against gram-positive and-negative multi-resistant bacteria with limited resistance. PLoS Biol. 2019, 17, e3000337. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, D.; Paterson, D.L. Multidrug-resistant bacteria in the community: Trends and lessons learned. Infect. Dis. Clin. N. Am. 2016, 30, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-resistant bacteria and alternative methods to control them: An overview. Microb. Drug Resist. 2019, 25, 890–908. [Google Scholar] [CrossRef]
- Tay, S.B.; Yew, W.S. Development of quorum-based anti-virulence therapeutics targeting Gram-negative bacterial pathogens. Int. J. Mol. Sci. 2013, 14, 16570–16599. [Google Scholar] [CrossRef]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.; Silva-Pereira, I.; Kyaw, C. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353–365. [Google Scholar] [CrossRef]
- Ghannad, M.S.; Mohammadi, A. Bacteriophage: Time to re-evaluate the potential of phage therapy as a promising agent to control multidrug-resistant bacteria. Iran. J. Basic Med. Sci. 2012, 15, 693. [Google Scholar]
- Bassegoda, A.; Ivanova, K.; Ramon, E.; Tzanov, T. Strategies to prevent the occurrence of resistance against antibiotics by using advanced materials. Appl. Microbiol. Biot. 2018, 102, 2075–2089. [Google Scholar] [CrossRef]
- Baker, S.J.; Payne, D.J.; Rappuoli, R.; De Gregorio, E. Technologies to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12887–12895. [Google Scholar] [CrossRef]
- Bresolí-Obach, R.; Gispert, I.; Peña, D.G.; Boga, S.; Gulias, Ó.; Agut, M.; Vázquez, M.E.; Nonell, S. Triphenylphosphonium cation: A valuable functional group for antimicrobial photodynamic therapy. J. Biophotonics 2018, 11, e201800054. [Google Scholar]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Khasiyatullina, N.R.; Mironov, V.F.; Voloshina, A.D.; Sapunova, A.S. Synthesis and antimicrobial properties of novel phosphonium salts bearing 1, 4-dihydroxyaryl fragment. Chem. Biodivers. 2019, 16, e1900039. [Google Scholar] [CrossRef] [PubMed]
- Castro, W.; Navarro, M.; Biot, C. Medicinal potential of ciprofloxacin and its derivatives. Future Med. Chem. 2013, 5, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.M.; Rimland, D.; Carroll, D.J.; Terry, P.; Wachsmuth, I.K. Rapid development of ciprofloxacin resistance in methicillin-susceptible and-resistant Staphylococcus aureus. J. Infect. Dis. 1991, 163, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.S.; Cho, S.Y.; Yang, H.Y.; Park, K.S.; Jang, J.-H.; Kim, Y.-T.; Jeong, J.-w.; Suh, J.-T.; Lee, H.J. Investigation of mutation distribution in DNA gyrase and topoisomerase IV genes in ciprofloxacin-non-susceptible Enterobacteriaceae isolated from blood cultures in a tertiary care university hospital in South Korea, 2005–2010. Int. J. Antimicrob. Agents 2013, 41, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.; Sanchez, M.B.; Martínez, J.L. Quinolone resistance: Much more than predicted. Front. Microbiol. 2011, 2, 22. [Google Scholar]
- Pinto, T.; Banerjee, A.; Nazarov, P.A. Triphenyl phosphonium-based substances are alternatives to common antibiotics. Bull. Russ. State Med. Univ. 2018, 7, 16–25. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Guo, Z.; Zhang, S.; Yan, C.; Tian, H.; Zhu, W.H. High-Fidelity Trapping of Spatial–Temporal Mitochondria with Rational Design of Aggregation-Induced Emission Probes. Adv. Funct. Mater. 2019, 29, 1808153. [Google Scholar] [CrossRef]
- Sunwoo, K.; Won, M.; Ko, K.-P.; Choi, M.; Arambula, J.F.; Chi, S.-G.; Sessler, J.L.; Verwilst, P.; Kim, J.S. Mitochondrial Relocation of a Common Synthetic Antibiotic: A Non-genotoxic Approach to Cancer Therapy. Chem 2020, 6, 1408–1419. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing; M100-S26; Clinical Lab. Standards Institute: Wayne, PA, USA, 2016. [Google Scholar]
- Matsuzaki, K.; Sugishita, K.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with lipopolysaccharide-containing liposomes as a model for outer membranes of gram-negative bacteria. FEBS Lett. 1999, 449, 221–224. [Google Scholar] [CrossRef]
- Madak, J.T.; Neamati, N. Membrane permeable lipophilic cations as mitochondrial directing groups. Curr. Top. Med. Chem. 2015, 15, 745–766. [Google Scholar] [CrossRef]
- O’Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef]
- Choi, U.; Lee, C.R. Antimicrobial agents that inhibit the outer membrane assembly machines of gram-negative bacteria. J. Microbiol. Biotechnol. 2019, 29, 1–10. [Google Scholar] [CrossRef]
- Wu, S.-C.; Yang, Z.-Q.; Liu, F.; Peng, W.-J.; Qu, S.-Q.; Li, Q.; Song, X.-B.; Zhu, K.; Shen, J.-Z. Antibacterial effect and mode of action of flavonoids from licorice against methicillin-resistant Staphylococcus aureus. Front. Microbiol. 2019, 10, 2489. [Google Scholar] [CrossRef] [PubMed]
- Arakha, M.; Saleem, M.; Mallick, B.C.; Jha, S. The effects of interfacial potential on antimicrobial propensity of ZnO nanoparticle. Sci. Rep. 2015, 5, 9578. [Google Scholar] [CrossRef] [PubMed]
- Trnka, J.; Elkalaf, M.; Andel, M. Lipophilic triphenylphosphonium cations inhibit mitochondrial electron transport chain and induce mitochondrial proton leak. PLoS ONE 2015, 10, e0121837. [Google Scholar] [CrossRef]
- Sani, M.A.; Henriques, S.T.; Weber, D.; Separovic, F. Bacteria May cope differently from similar membrane damage caused by the australian tree frog antimicrobial peptide maculatin 1.1. J. Biol. Chem. 2015, 290, 19853–19862. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Liu, J.; Zhang, M.; Shi, H.; Zheng, S.; Jin, X.; Gao, Y.; Wang, S.; Ji, A.; Lou, H. Efflux pump-mediated resistance to antifungal compounds can be prevented by conjugation with triphenylphosphonium cation. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Carvalho, A.R., Jr.; Martins, A.L.d.B.; Cutrim, B.d.S.; Santos, D.M.; Maia, H.S.; da Silva, M.S.M.; Zagmignan, A.; Silva, M.R.C.; Monteiro, C.d.A.; Guilhon, G.M.S.P.; et al. Betulinic acid prevents the acquisition of ciprofloxacin-mediated mutagenesis in Staphylococcus aureus. Molecules 2019, 24, 1757. [Google Scholar] [CrossRef]
- Dong, G.; Li, J.; Chen, L.; Bi, W.; Zhang, X.; Liu, H.; Zhi, X.; Zhou, T.; Cao, J. Effects of sub-minimum inhibitory concentrations of ciprofloxacin on biofilm formation and virulence factors of Escherichia coli. Braz. J. Infect. Dis. 2019, 23, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Yadav, K.K.; Sarkar, R.; Mukherjee, S.; Saha, P.; Haldar, S.; Karmakar, S.; Sen, T. Alteration of Zeta potential and membrane permeability in bacteria: A study with cationic agents. SpringerPlus 2015, 4, 1–14. [Google Scholar] [CrossRef]
- Peeters, E.; Nelis, H.J.; Coenye, T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Berditsch, M.; Hawecker, J.; Ardakani, M.F.; Gerthsen, D.; Ulrich, A.S. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef]
- Law, B.; Bunn, W.; Hesterberg, T. Dissolution of natural mineral and man-made vitreous fibers in Karnovsky’s and formalin fixatives. Inhal. Toxicol. 1991, 3, 309–321. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strains | Drug Sensitive or Resistant Bacteria |
|---|---|---|
| Gram-negative | ||
| Escherichia coli | ATCC® 25922 | Carbapenem-susceptible E. coli |
| AMCEC 22365 | Carbapenem-resistant E. coli (NDM-1 type) | |
| Klebsiella pneumoniae | ATCC® 13883 | Carbapenem-susceptible K. pneumoniae |
| AMCKP 24272 | Carbapenem-resistant K. pneumoniae (KPC type) | |
| Pseudomonas aeruginosa | ATCC® 27853 | Carbapenem-susceptible P. aeruginosa (CSPA) |
| CCARM 2321 | Carbapenem-resistant P. aeruginosa (CRPA) | |
| Acinetobacter baumannii | ATCC® 19606 | Carbapenem-susceptible A. baumannii (CSAB) |
| AMCAB 643 | Carbapenem-resistant A. baumannii (CRAB) | |
| Gram-positive | ||
| Enterococcus faecium | ATCC® 29212 | Vancomycin-susceptible E. faecium (VSE) |
| CCARM 5024 | Vancomycin-resistant E. faecium (VRE) | |
| Staphylococcus aureus | ATCC® 29213 | Methicillin-susceptible S. aureus (MSSA) |
| AMCSA 5016 | Methicillin-resistant S. aureus (MRSA, ST5) | |
| AMCSA 5013 | MRSA (ST72) | |
| AMCSA 3416 | MRSA (ST239) | |
| ATCC® 700698 | Heterogeneous VISA (hVISA) | |
| ATCC® 700699 | Vancomycin intermediate-resistant S. aureus (VISA) | |
| Type or Strain | CLSI Breakpoint (μg/mL) | QC Range | MIC (μg/mL) | Biofilm Inhibition Rate (%) * | ||||
|---|---|---|---|---|---|---|---|---|
| CFX | CFX- Ester-PPh3 | CFX- Amide-PPh3 | CFX | CFX- Ester-PPh3 | CFX- Amide-PPh3 | |||
| Gram-negative Bacteria | ||||||||
| E. coli | ≤1 | 0.004–0.015 | 0.004 | 5.56 | 5.56 | <0.5 (96.49%) | 32 (76.0%) | 8 (85.4%) |
| NDM-1 type | ≤1 | - | 16 | >178.05 | 89.02 | - | - | - |
| K. pneumoniae | ≤1 | - | 0.031 | 22.25 | 44.51 | - | - | - |
| KPC type | ≤1 | - | 32 | >178.05 | 178.05 | 128 (74.8%) | >512 (28.5%) | 256 (51.2%) |
| P. aeruginosa | ≤1 | - | 0.25–0.125 | 89.02 | 89.02 | 2 (73.2%) | 64 (50.1%) | 256 (54.0%) |
| CRPA | ≤1 | 0.25–2.0 | 32 | >178.05 | 178.05 | <0.5 (80.3%) | 64 (74.4%) | 32 (56.5%) |
| A. baumannii | ≤1 | 0.5 | 89.02 | 89.02 | 256 (66.0%) | >512 (48.2%) | 256 (53.4%) | |
| CRAB | ≤1 | 64 | 89.02 | 89.02 | - | - | - | |
| Gram-positive Bacteria | ||||||||
| E. faecium | ≤1 | 0.25–2.0 | 1.0-0.5 | 178.05 | 44.51 | <0.5 (65.0%) | 64 (60.0%) | 64 (91.5%) |
| VRE | ≤1 | - | 256 | 89.02 | 22.25 | - | - | - |
| S. aureus | ≤1 | 0.12–0.5 | 0.5 | 22.25 | 2.78 | <0.5 (89.49%) | 16 (48.9%) | 4 (73.6%) |
| MRSA 5016 | ≤1 | 128 | 11.12 | 2.78 | 128 (54.1%) | 8 (43.9%) | 4 (74.9%) | |
| MRSA 5013 | ≤1 | 0.5 | 11.12 | 1.39 | <0.5 (88.1%) | 8 (57.2%) | 4 (75.2%) | |
| MRSA 3416 | ≤1 | 64 | 89.02 | 22.25 | 64 (60.2%) | 64 (88.2%) | 32 (72.5%) | |
| hVISA | ≤1 | 64 | 11.12 | 11.12 | - | - | - | |
| VISA | ≤1 | 64 | 44.51 | 11.12 | - | - | - | |
| MIC | MSSA | MRSA ST5 5016 | ||||
|---|---|---|---|---|---|---|
| CFX | CFX-ester-PPh3 | CFX-amide-PPh3 | CFX | CFX-ester-PPh3 | CFX-amide-PPh3 | |
| 0.5× | 0.25 | 11.12 | 1.39 | 64 | 5.56 | 1.39 |
| 1.0× | 0.5 | 22.24 | 2.78 | 128 | 11.12 | 2.78 |
| 2.0× | 1.0 | 44.48 | 5.56 | 256 | 22.24 | 5.56 |
| 4.0× | 2.0 | 89.96 | 11.12 | 512 | 44.48 | 11.12 |
| 8.0× | 4.0 | 178 | 22.25 | 1,024 | 88.96 | 22.24 |
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.; Sunwoo, K.; Jung, Y.; Hur, J.K.; Park, K.-H.; Kim, J.S.; Kim, D. Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA). Antibiotics 2020, 9, 758. https://doi.org/10.3390/antibiotics9110758
Kang S, Sunwoo K, Jung Y, Hur JK, Park K-H, Kim JS, Kim D. Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA). Antibiotics. 2020; 9(11):758. https://doi.org/10.3390/antibiotics9110758
Chicago/Turabian StyleKang, Sangrim, Kyoung Sunwoo, Yuna Jung, Junho K. Hur, Ki-Ho Park, Jong Seung Kim, and Dokyoung Kim. 2020. "Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA)" Antibiotics 9, no. 11: 758. https://doi.org/10.3390/antibiotics9110758
APA StyleKang, S., Sunwoo, K., Jung, Y., Hur, J. K., Park, K.-H., Kim, J. S., & Kim, D. (2020). Membrane-Targeting Triphenylphosphonium Functionalized Ciprofloxacin for Methicillin-Resistant Staphylococcus aureus (MRSA). Antibiotics, 9(11), 758. https://doi.org/10.3390/antibiotics9110758