Inhibitory Activity of a Scorpion Defensin BmKDfsin3 against Hepatitis C Virus

 ,
,  ,
, _Kwok.png) ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
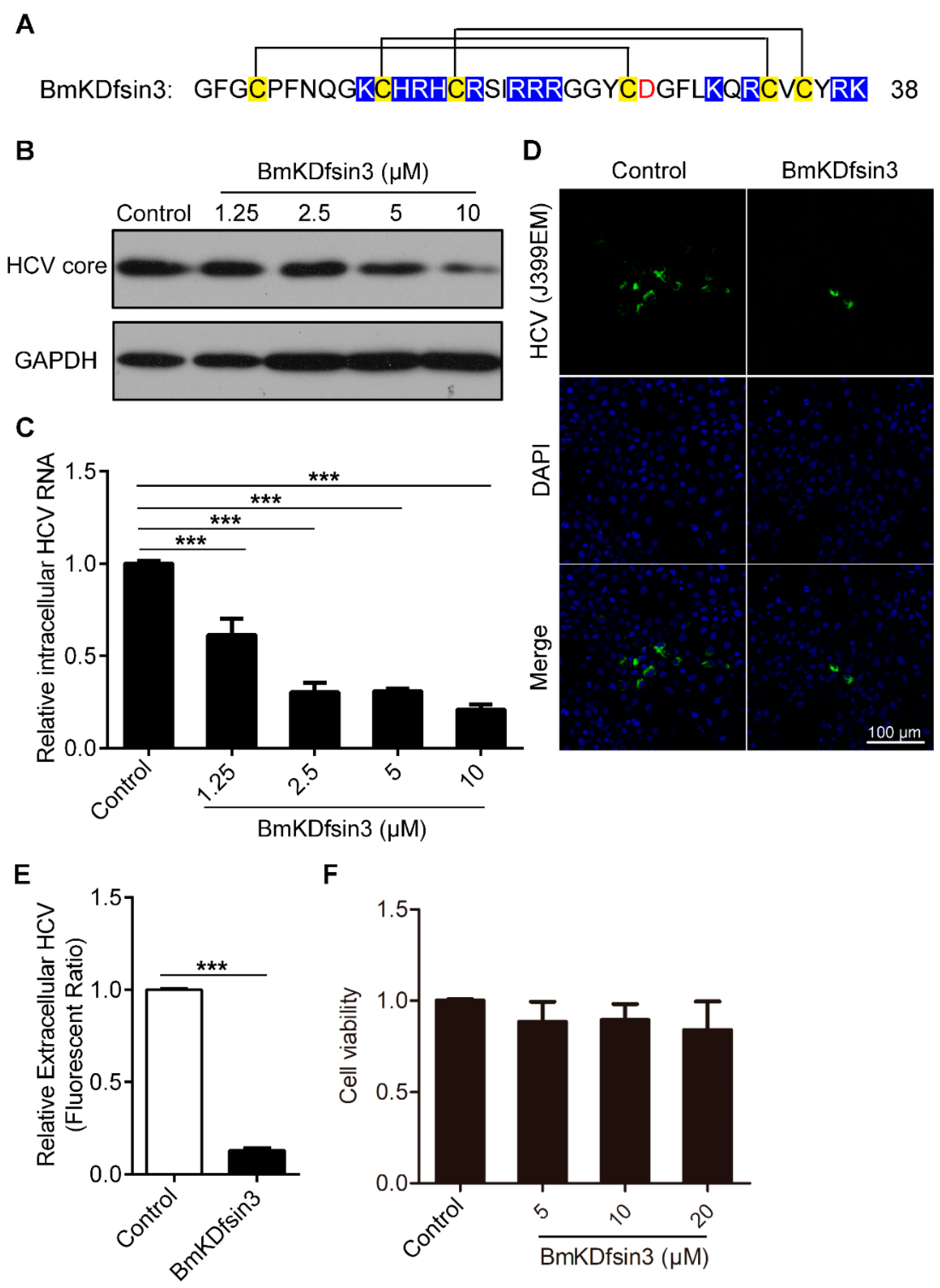
2.1. BmKDfsin3 Inhibits HCV Replication In Vitro at Noncytotoxic Concentrations
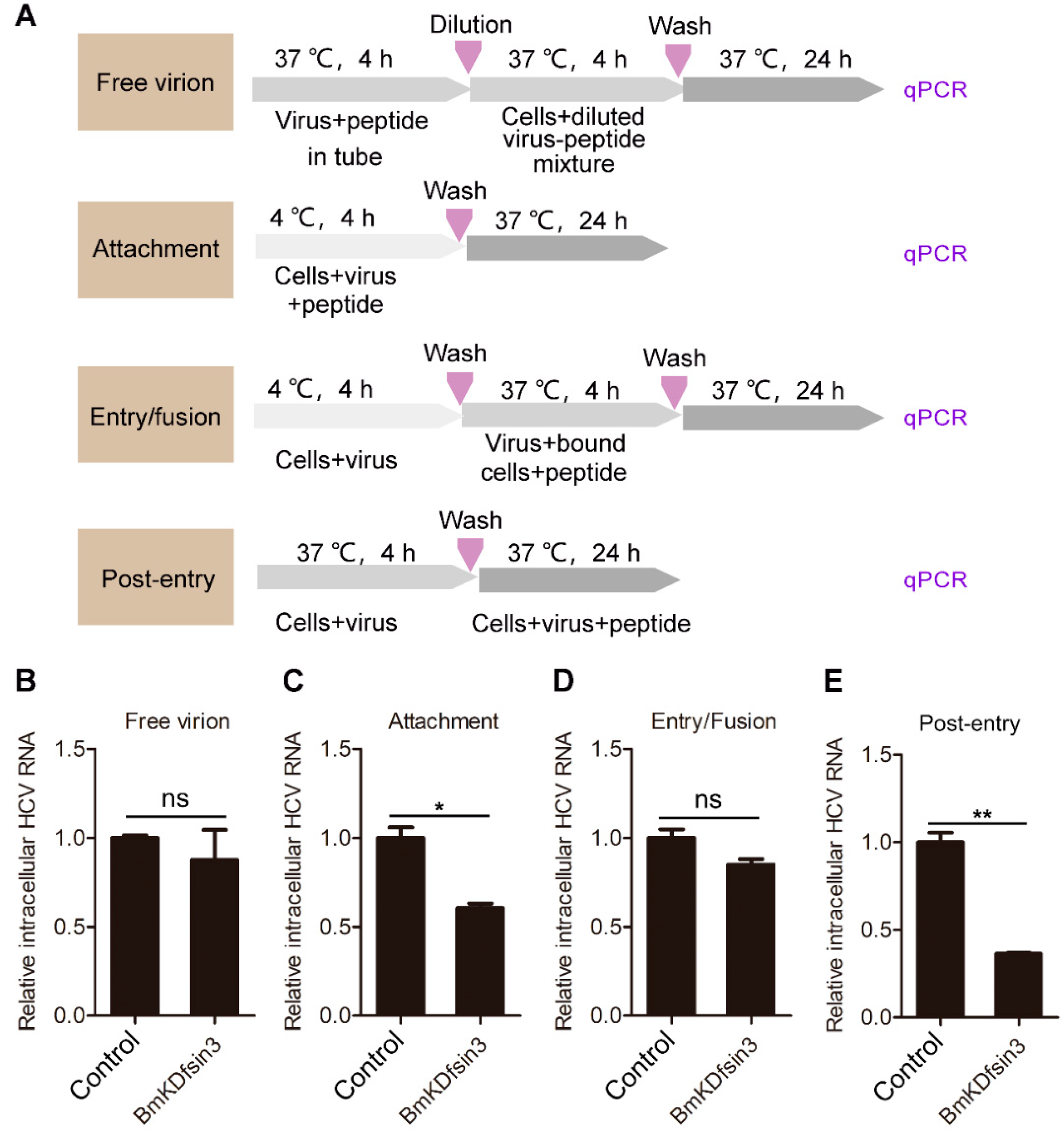
2.2. BmKDfsin3 Affects the Viral Attachment and Post-Entry Stages in HCV Infection Cycle
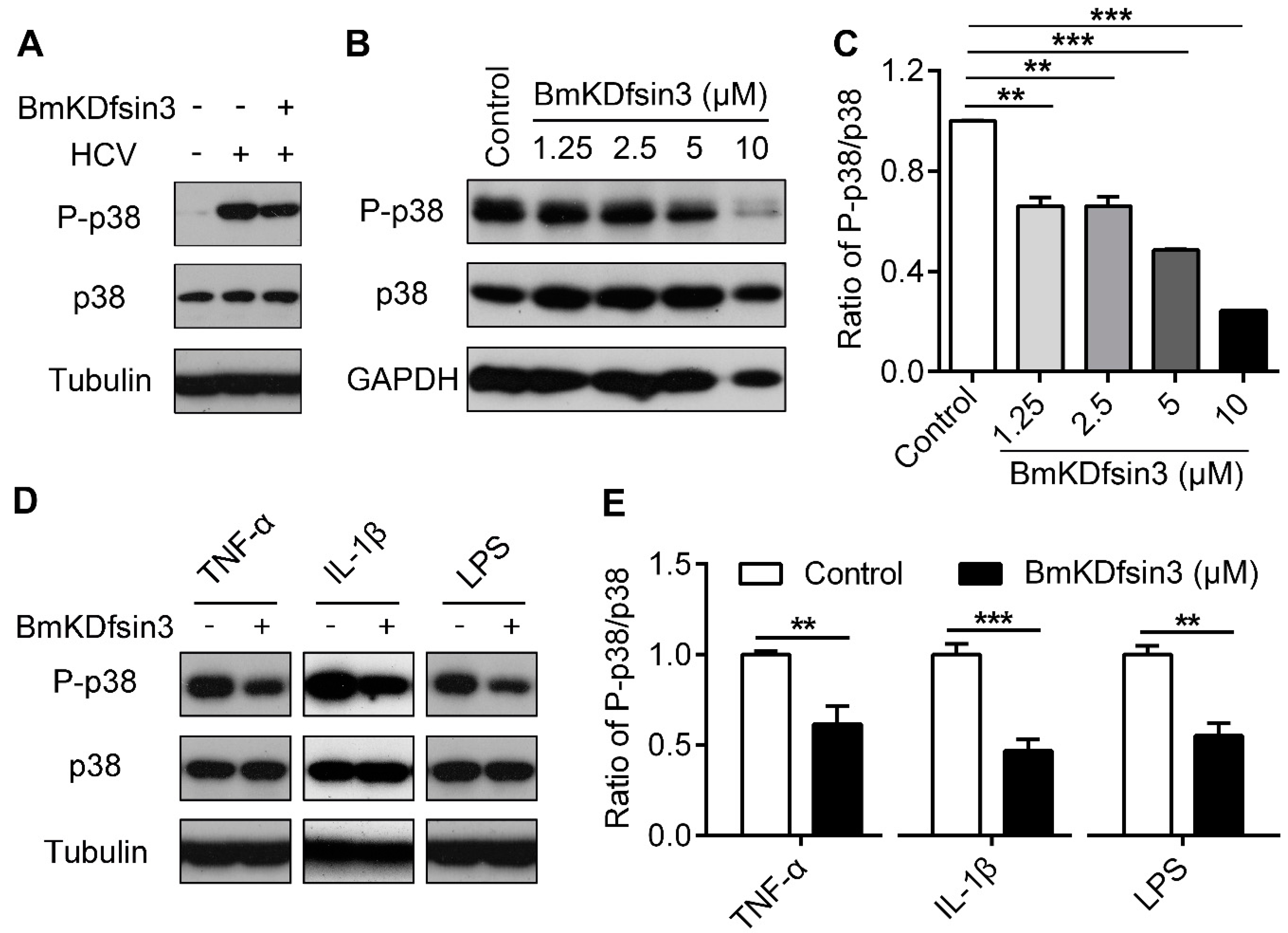
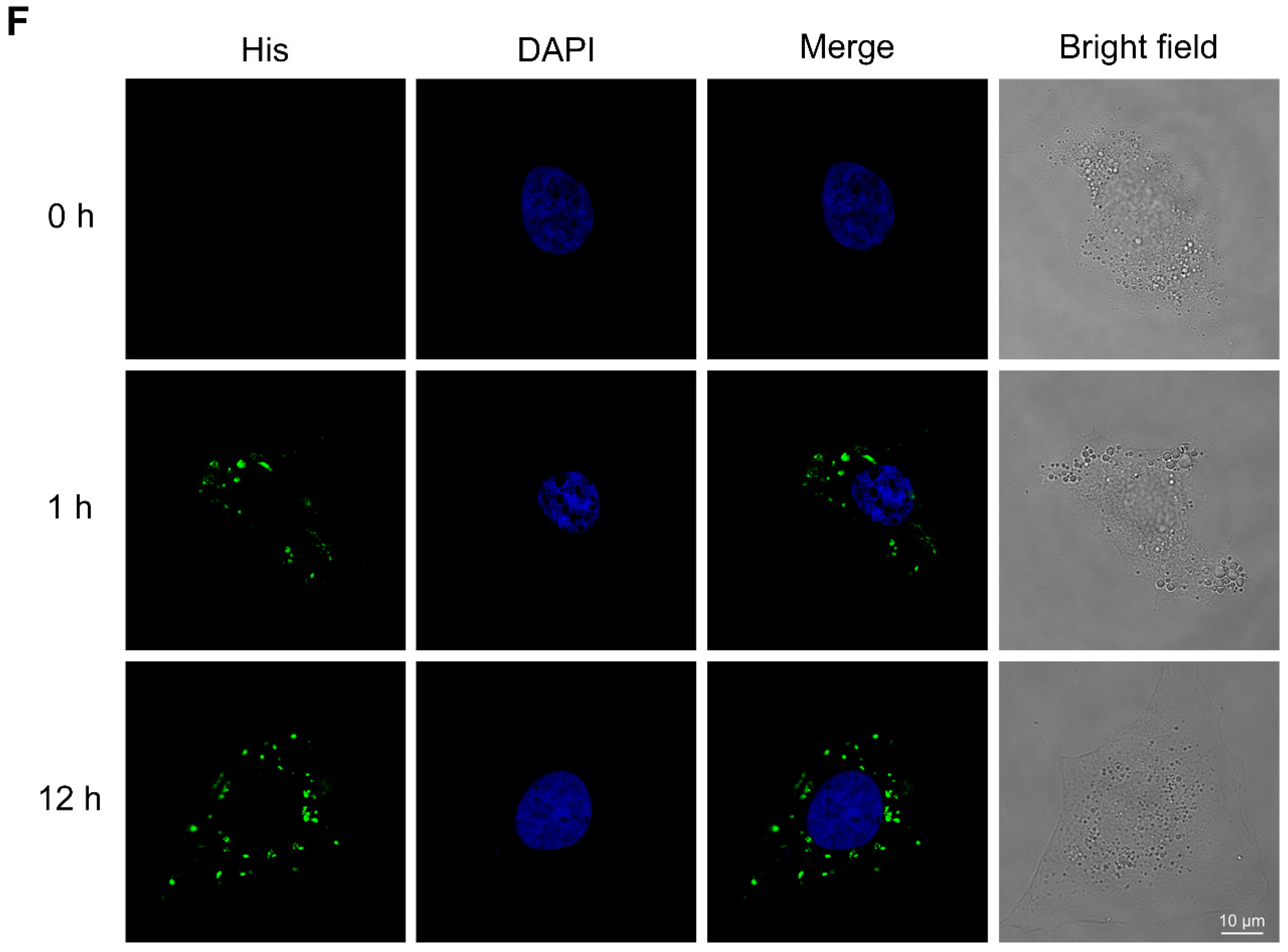
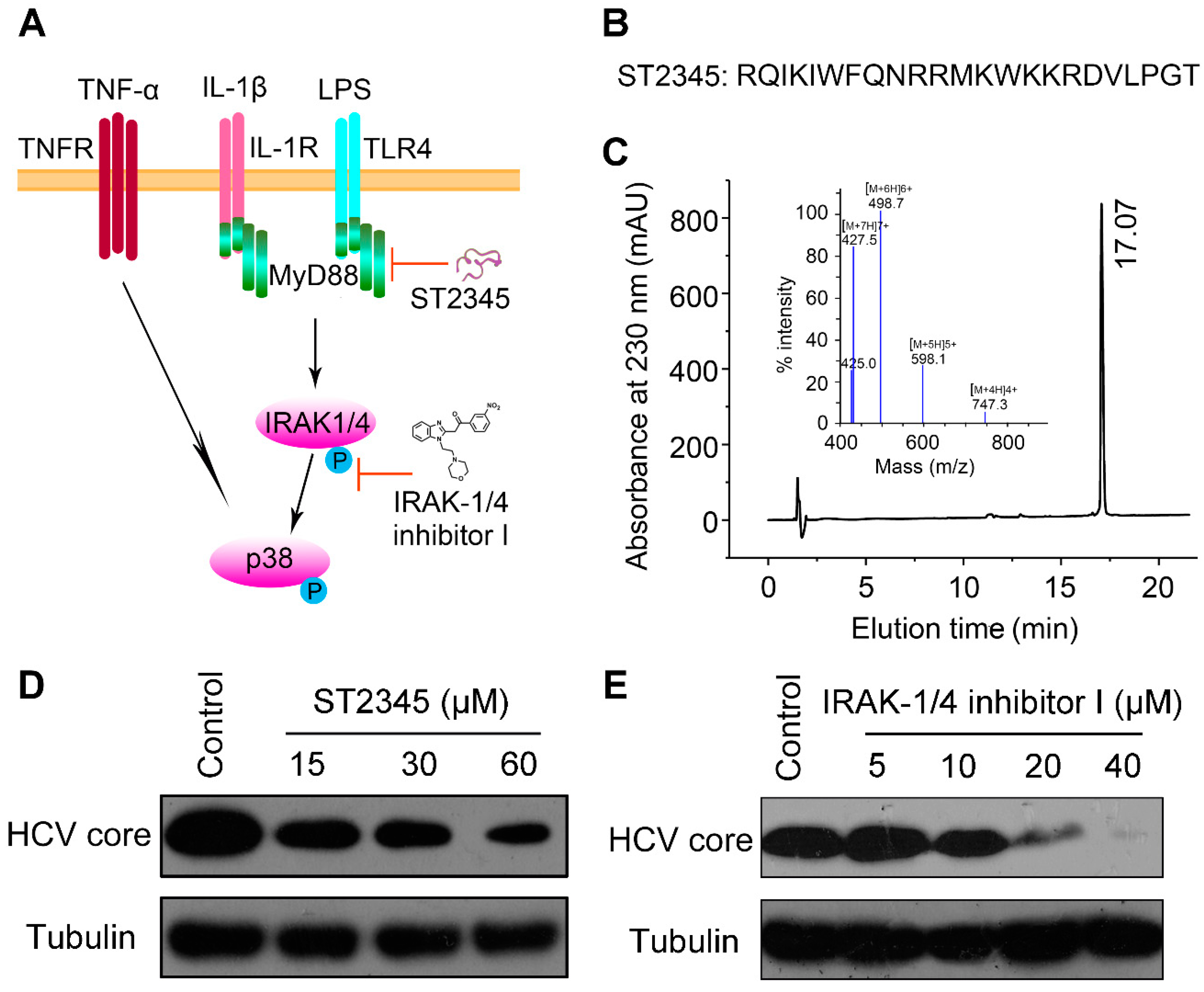
2.3. BmKDfsin3 Inhibits p38 Activation
2.4. Inhibition of p38 Activation Suppresses HCV Replication In Vitro
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cells and Viruses
4.3. MTT Assay
4.4. Confocal Microscopy
4.5. qPCR
4.6. Western Blotting
4.7. Peptide Oxidation and Purification
4.8. Statistics Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CC50 | 50% cytotoxic concentration. |
| CHIKV | chikungunya virus. |
| DAAs | direct-acting antivirals. |
| DAPI | 4′,6-diamidino-2-phenylindole. |
| DMSO | dimethyl sulfoxide. |
| EV71 | enterovirus 71. |
| GAPDH | glyceraldehyde-phosphate dehydrogenase. |
| HBV | hepatitis B virus. |
| HCV | hepatitis C virus. |
| HPLC | high-performance liquid chromatography. |
| HSV | herpes simplex virus. |
| IC50 | 50% inhibitory concentration. |
| IL-1β | interleukin-1β. |
| LPS | lipopolysaccharide. |
| MAPK | mitogen-activated protein (MAP) kinase. |
| MKK | MAP kinase kinase. |
| MOI | multiplicity of infection. |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide. |
| NC | nitrocellulose. |
| PBS | phosphate buffered saline. |
| PKC | protein kinase C. |
| RNA | ribonucleicacid. |
| RSV | respiratory syncytial virus. |
| qPCR | quantitative PCR. |
| SD | standard deviation. |
| TNF-α | tumor necrosis factor-α. |
References
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef]
- Holly, M.K.; Diaz, K.; Smith, J.G. Defensins in viral infection and pathogenesis. Annu. Rev. Virol. 2017, 4, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Nemerow, G.R. Mechanism of adenovirus neutralization by human alpha-defensins. Cell Host Microbe 2008, 3, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Hou, J.; Chen, Q.; Yuan, W.; Cheng, B.; Sun, Y.; Jin, Y.; Ge, L.; Ben-Sasson, S.A.; Chen, J.; et al. Self-assembling myristoylated human alpha-defensin 5 as a next-generation nanobiotics potentiates therapeutic efficacy in bacterial infection. ACS Nano 2018, 12, 5284–5296. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liao, C.; Zhang, B.; Tolbert, W.D.; He, W.; Dai, Z.; Zhang, W.; Yuan, W.; Pazgier, M.; Liu, J.; et al. Human enteric alpha-defensin 5 promotes shigella infection by enhancing bacterial adhesion and invasion. Immunity 2018, 48, 1233–1244. [Google Scholar] [CrossRef]
- Pachon-Ibanez, M.E.; Smani, Y.; Pachon, J.; Sanchez-Cespedes, J. Perspectives for clinical use of engineered human host defense antimicrobial peptides. FEMS Microbiol. Rev. 2017, 41, 323–342. [Google Scholar] [CrossRef]
- Sable, R.; Durek, T.; Taneja, V.; Craik, D.J.; Pallerla, S.; Gauthier, T.; Jois, S. Constrained cyclic peptides as immunomodulatory inhibitors of the cd2:Cd58 protein-protein interaction. ACS Chem. Biol. 2016, 11, 2366–2374. [Google Scholar] [CrossRef]
- Estrada-Gomez, S.; Gomez-Rave, L.; Vargas-Munoz, L.J.; van der Meijden, A. Characterizing the biological and biochemical profile of six different scorpion venoms from the buthidae and scorpionidae family. Toxicon 2017, 130, 104–115. [Google Scholar] [CrossRef]
- Bleackley, M.R.; Payne, J.A.; Hayes, B.M.; Durek, T.; Craik, D.J.; Shafee, T.M.; Poon, I.K.; Hulett, M.D.; van der Weerden, N.L.; Anderson, M.A. Nicotiana alata defensin chimeras reveal differences in the mechanism of fungal and tumor cell killing and an enhanced antifungal variant. Antimicrob. Agents Chemother. 2016, 60, 6302–6312. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Xie, Z.; Zhang, Q.; Li, Y.; Yang, F.; Chen, Z.; Li, W.; Cao, Z.; Wu, Y. Scorpion potassium channel-blocking defensin highlights a functional link with neurotoxin. J. Biol. Chem. 2016, 291, 7097–7106. [Google Scholar] [CrossRef]
- Feng, J.; Xie, Z.; Yang, W.; Zhao, Y.; Xiang, F.; Cao, Z.; Li, W.; Chen, Z.; Wu, Y. Human beta-defensin 1, a new animal toxin-like blocker of potassium channel. Toxicon 2016, 113, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zhang, Q.; Hong, W.; Xie, Y.; Liu, Y.; Li, W.; Wu, Y.; Cao, Z. A scorpion defensin bmkdfsin4 inhibits hepatitis b virus replication in vitro. Toxins 2016, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Hamdane, N.; Juhling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. Hcv-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology 2019, 156, 2313–2329. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ishii, K.; Aizaki, H.; Wakita, T. Hepatitis c viral life cycle. Adv. Drug Deliv. Rev. 2007, 59, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Spengler, U. Direct antiviral agents (DAAs)—A new age in the treatment of hepatitis c virus infection. Pharmacol. Ther. 2018, 183, 118–126. [Google Scholar] [CrossRef]
- Gupta, S.; Rout, G.; Patel, A.H.; Mahanta, M.; Kalra, N.; Sahu, P.; Sethia, R.; Agarwal, A.; Ranjan, G.; Kedia, S.; et al. Efficacy of generic oral directly acting agents in patients with hepatitis c virus infection. J. Viral Hepat. 2018, 25, 771–778. [Google Scholar] [CrossRef]
- He, S.F.; Wang, W.; Ren, H.; Zhao, L.J.; Qi, Z.T. Interferon alpha and ribavirin collaboratively regulate p38 mitogen-activated protein kinase signaling in hepatoma cells. Cytokine 2013, 61, 801–807. [Google Scholar] [CrossRef]
- Varghese, F.S.; Thaa, B.; Amrun, S.N.; Simarmata, D.; Rausalu, K.; Nyman, T.A.; Merits, A.; McInerney, G.M.; Ng, L.F.P.; Ahola, T. The antiviral alkaloid berberine reduces chikungunya virus-induced mitogen-activated protein kinase signaling. J. Virol. 2016, 90, 9743–9757. [Google Scholar] [CrossRef]
- Xu, X.; Xu, Y.; Zhang, Q.; Yang, F.; Yin, Z.; Wang, L.; Li, Q. Porcine epidemic diarrhea virus infections induce apoptosis in vero cells via a reactive oxygen species (ros)/p53, but not p38 mapk and sapk/jnk signalling pathways. Vet. Microbiol. 2019, 232, 1–12. [Google Scholar] [CrossRef]
- Sloan, D.D.; Jerome, K.R. Herpes simplex virus remodels t-cell receptor signaling, resulting in p38-dependent selective synthesis of interleukin-10. J. Virol. 2007, 81, 12504–12514. [Google Scholar] [CrossRef]
- Peng, H.; Shi, M.; Zhang, L.; Li, Y.; Sun, J.; Zhang, L.; Wang, X.; Xu, X.; Zhang, X.; Mao, Y.; et al. Activation of jnk1/2 and p38 mapk signaling pathways promotes enterovirus 71 infection in immature dendritic cells. BMC Microbiol. 2014, 14, 147. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhu, X.; Ma, T.; Wang, J.; Wang, F.; Zhang, S. The p38 mapk nf-kappab pathway, not the erk pathway, is involved in exogenous hiv-1 tat-induced apoptotic cell death in retinal pigment epithelial cells. Int. J. Biochem. Cell Biol. 2013, 45, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Olvera, I.; Chavez-Salinas, S.; Medina, F.; Ludert, J.E.; del Angel, R.M. Jnk phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 2010, 396, 30–36. [Google Scholar] [CrossRef]
- Huang, H.; Chan, H.; Wang, Y.Y.; Ouyang, D.Y.; Zheng, Y.T.; Tam, S.C. Trichosanthin suppresses the elevation of p38 mapk, and bcl-2 induced by hsv-1 infection in vero cells. Life Sci. 2006, 79, 1287–1292. [Google Scholar] [CrossRef]
- Su, A.R.; Qiu, M.; Li, Y.L.; Xu, W.T.; Song, S.W.; Wang, X.H.; Song, H.Y.; Zheng, N.; Wu, Z.W. Bx-795 inhibits hsv-1 and hsv-2 replication by blocking the jnk/p38 pathways without interfering with pdk1 activity in host cells. Acta Pharmacol. Sin. 2017, 38, 402–414. [Google Scholar] [CrossRef]
- Shen, L.; Yang, Q.; He, Y.; Zou, X.; Cao, Z. Bmk nt1-induced neurotoxicity is mediated by pkc/camk-dependent erk1/2 and p38 activation in primary cultured cerebellar granule cells. Toxicology 2019, 421, 22–29. [Google Scholar] [CrossRef]
- Wang, X.G.; Zhu, D.D.; Li, N.; Huang, Y.L.; Wang, Y.Z.; Zhang, T.; Wang, C.M.; Wang, B.; Peng, Y.; Ge, B.Y.; et al. Scorpion venom heat-resistant peptide is neuroprotective against cerebral ischemia-reperfusion injury in association with the nmda-mapk pathway. Neurosci. Bull. 2019. [Google Scholar] [CrossRef]
- Cao, Z.; Yu, Y.; Wu, Y.; Hao, P.; Di, Z.; He, Y.; Chen, Z.; Yang, W.; Shen, Z.; He, X.; et al. The genome of mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 2013, 4, 2602. [Google Scholar] [CrossRef]
- Qi, M.; Elion, E.A. Map kinase pathways. J. Cell Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef]
- Loiarro, M.; Sette, C.; Gallo, G.; Ciacci, A.; Fanto, N.; Mastroianni, D.; Carminati, P.; Ruggiero, V. Peptide-mediated interference of tir domain dimerization in myd88 inhibits interleukin-1-dependent activation of nf-{kappa}b. J. Biol. Chem. 2005, 280, 15809–15814. [Google Scholar] [CrossRef]
- Powers, J.P.; Li, S.; Jaen, J.C.; Liu, J.; Walker, N.P.; Wang, Z.; Wesche, H. Discovery and initial sar of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg. Med. Chem. Lett. 2006, 16, 2842–2845. [Google Scholar] [CrossRef] [PubMed]
- Klotman, M.E.; Chang, T.L. Defensins in innate antiviral immunity. Nat. Rev. Immunol. 2006, 6, 447–456. [Google Scholar] [CrossRef]
- Conibear, A.C.; Craik, D.J. The chemistry and biology of theta defensins. Angew. Chem. 2014, 53, 10612–10623. [Google Scholar] [CrossRef]
- Wilmes, M.; Stockem, M.; Bierbaum, G.; Schlag, M.; Gotz, F.; Tran, D.Q.; Schaal, J.B.; Ouellette, A.J.; Selsted, M.E.; Sahl, H.G. Killing of staphylococci by theta-defensins involves membrane impairment and activation of autolytic enzymes. Antibiotics 2014, 3, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Mattar, E.H.; Almehdar, H.A.; Uversky, V.N.; Redwan, E.M. Virucidal activity of human alpha- and beta-defensins against hepatitis c virus genotype 4. Mol. Biosyst. 2016, 12, 2785–2797. [Google Scholar] [CrossRef]
- Salvatore, M.; Garcia-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. Alpha-defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.L.; Vargas, J., Jr.; DelPortillo, A.; Klotman, M.E. Dual role of alpha-defensin-1 in anti-hiv-1 innate immunity. J. Clin. Investig. 2005, 115, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Zhang, R.; Hong, W.; Cheng, Y.; Wang, H.; Lang, Y.; Ji, Z.; Wu, Y.; Li, W.; Xie, Y.; et al. Histidine-rich modification of a scorpion-derived peptide improves bioavailability and inhibitory activity against hsv-1. Theranostics 2018, 8, 199–211. [Google Scholar] [CrossRef]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995, 270, 7420–7426. [Google Scholar] [CrossRef]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A map kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Heo, J.; Yi, C.M.; Ban, J.; Lee, N.J.; Lee, N.R.; Kim, S.W.; Kim, N.J.; Inn, K.S. A novel p38 mitogen activated protein kinase (mapk) specific inhibitor suppresses respiratory syncytial virus and influenza a virus replication by inhibiting virus-induced p38 mapk activation. Biochem. Biophys. Res. Commun. 2016, 477, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Xu, C.; Wu, C.; Zhu, W.; Yang, R.; Chen, X. Compensatory mutations in ns3 and ns5a proteins enhance the virus production capability of hepatitis c reporter virus. Virus Res. 2009, 145, 63–73. [Google Scholar] [CrossRef]
- Li, F.; Lang, Y.; Ji, Z.; Xia, Z.; Han, Y.; Cheng, Y.; Liu, G.; Sun, F.; Zhao, Y.; Gao, M.; et al. A scorpion venom peptide ev37 restricts viral late entry by alkalizing acidic organelles. J. Biol. Chem. 2019, 294, 182–194. [Google Scholar] [CrossRef]
- Shen, B.; Song, J.; Zhao, Y.; Zhang, Y.; Liu, G.; Li, X.; Guo, X.; Li, W.; Cao, Z.; Wu, Y. Triintsin, a human pathogenic fungus-derived defensin with broad-spectrum antimicrobial activity. Peptides 2018, 107, 61–67. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Sun, F.; Li, S.; Gao, M.; Wang, L.; Sarhan, M.; Abdel-Rahman, M.A.; Li, W.; Kwok, H.F.; Wu, Y.; et al. Inhibitory Activity of a Scorpion Defensin BmKDfsin3 against Hepatitis C Virus. Antibiotics 2020, 9, 33. https://doi.org/10.3390/antibiotics9010033
Cheng Y, Sun F, Li S, Gao M, Wang L, Sarhan M, Abdel-Rahman MA, Li W, Kwok HF, Wu Y, et al. Inhibitory Activity of a Scorpion Defensin BmKDfsin3 against Hepatitis C Virus. Antibiotics. 2020; 9(1):33. https://doi.org/10.3390/antibiotics9010033
Chicago/Turabian StyleCheng, Yuting, Fang Sun, Songryong Li, Minjun Gao, Luyao Wang, Moustafa Sarhan, Mohamed A. Abdel-Rahman, Wenxin Li, Hang Fai Kwok, Yingliang Wu, and et al. 2020. "Inhibitory Activity of a Scorpion Defensin BmKDfsin3 against Hepatitis C Virus" Antibiotics 9, no. 1: 33. https://doi.org/10.3390/antibiotics9010033
APA StyleCheng, Y., Sun, F., Li, S., Gao, M., Wang, L., Sarhan, M., Abdel-Rahman, M. A., Li, W., Kwok, H. F., Wu, Y., & Cao, Z. (2020). Inhibitory Activity of a Scorpion Defensin BmKDfsin3 against Hepatitis C Virus. Antibiotics, 9(1), 33. https://doi.org/10.3390/antibiotics9010033