Natural Product Type III Secretion System Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
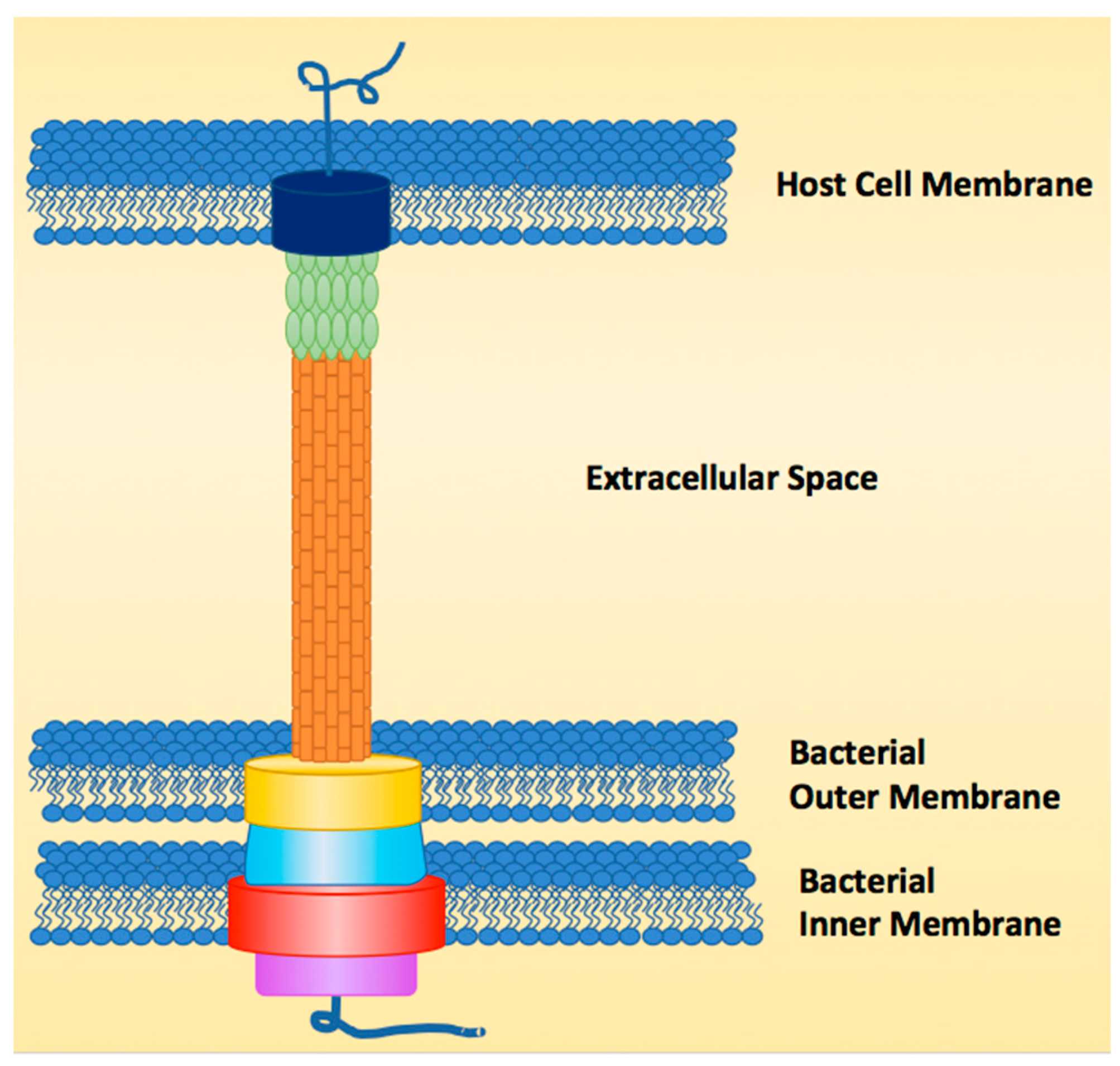
1. Introduction
2. Natural Products
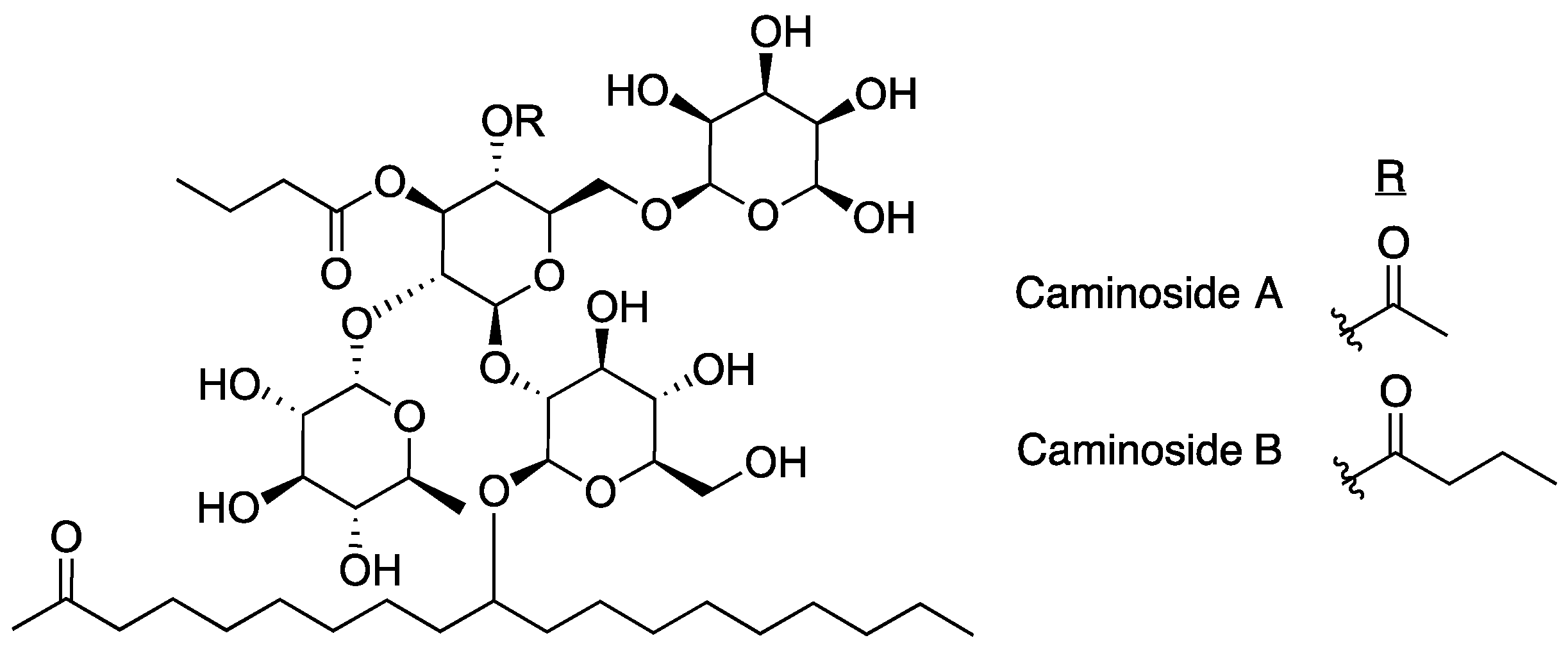
2.1. Caminosides
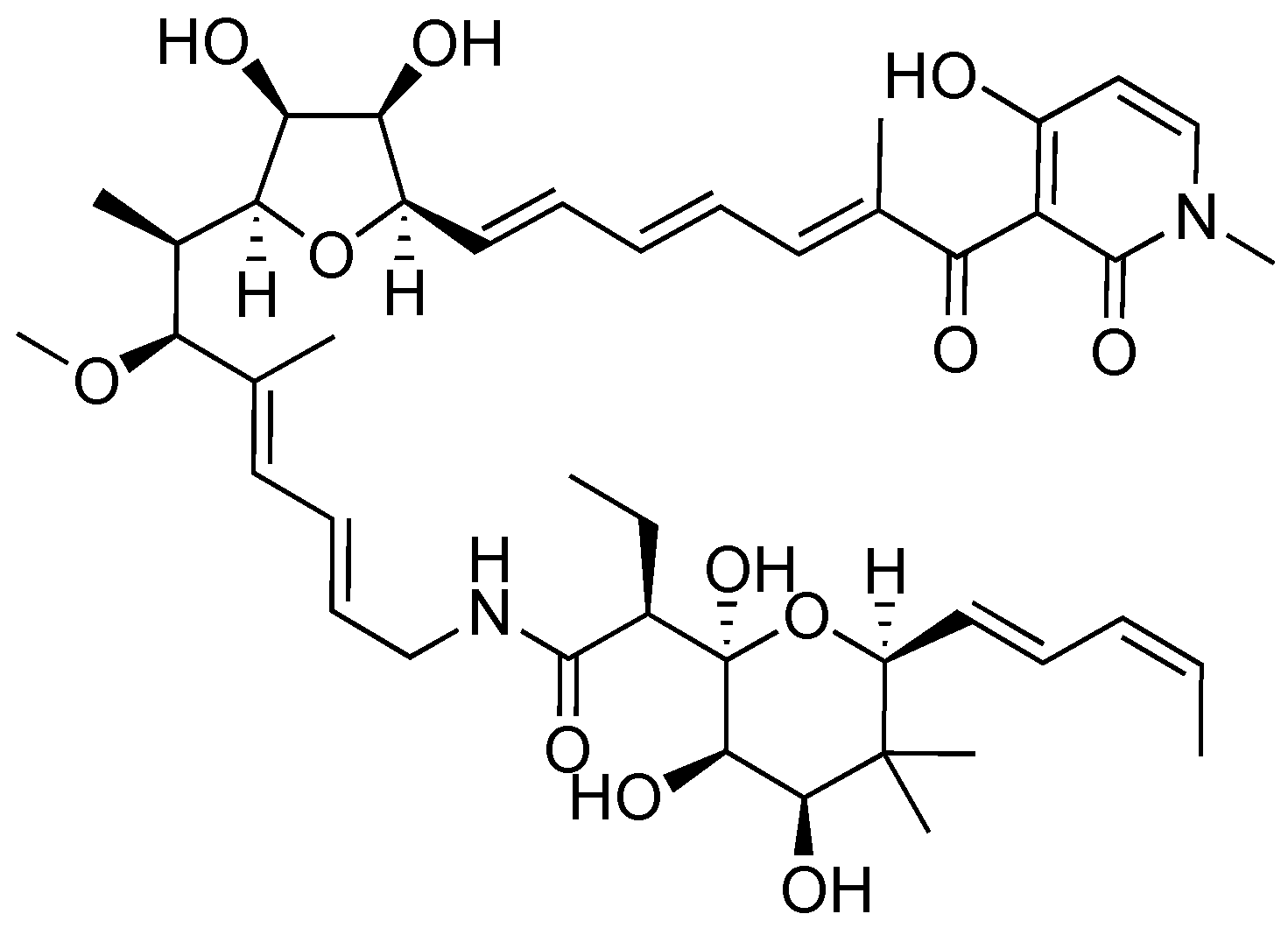
2.2. Aurodox
2.3. Piericidin A
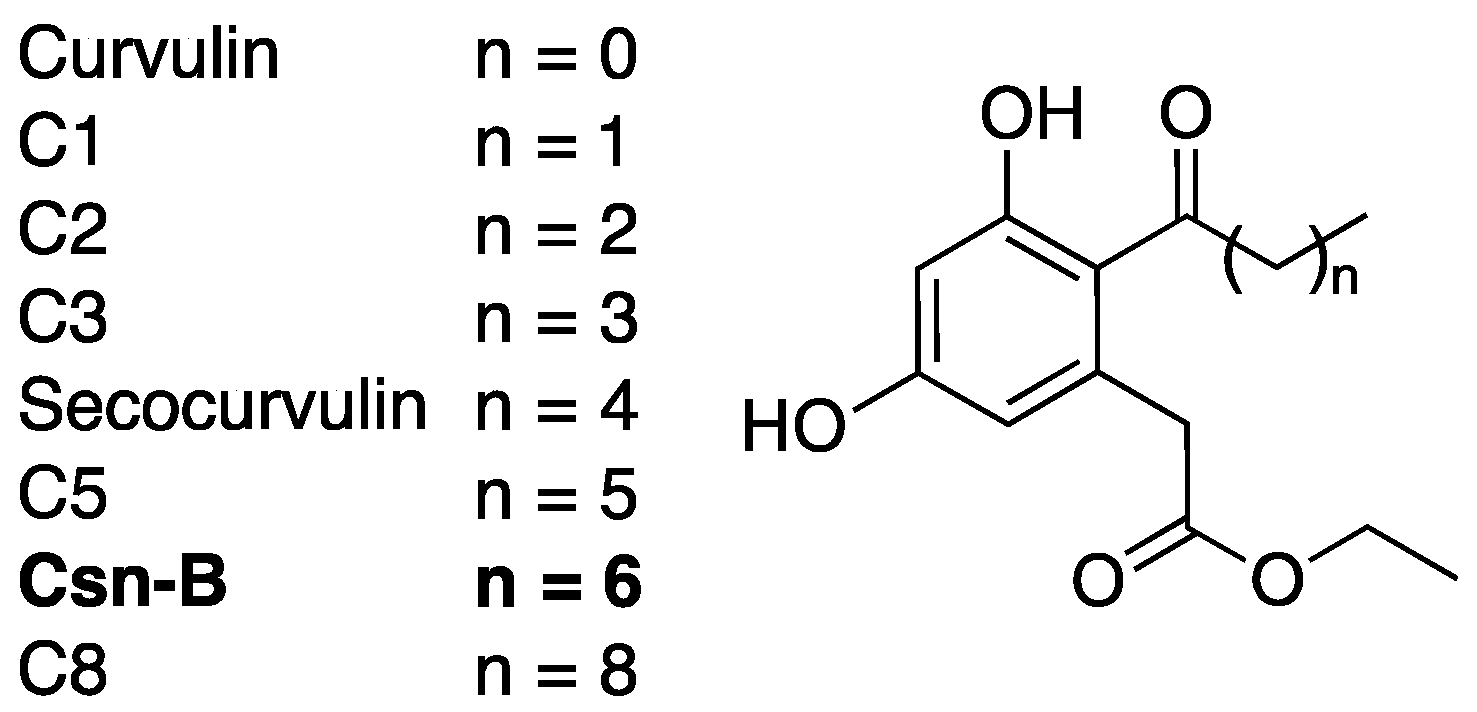
2.4. Cytosporone B
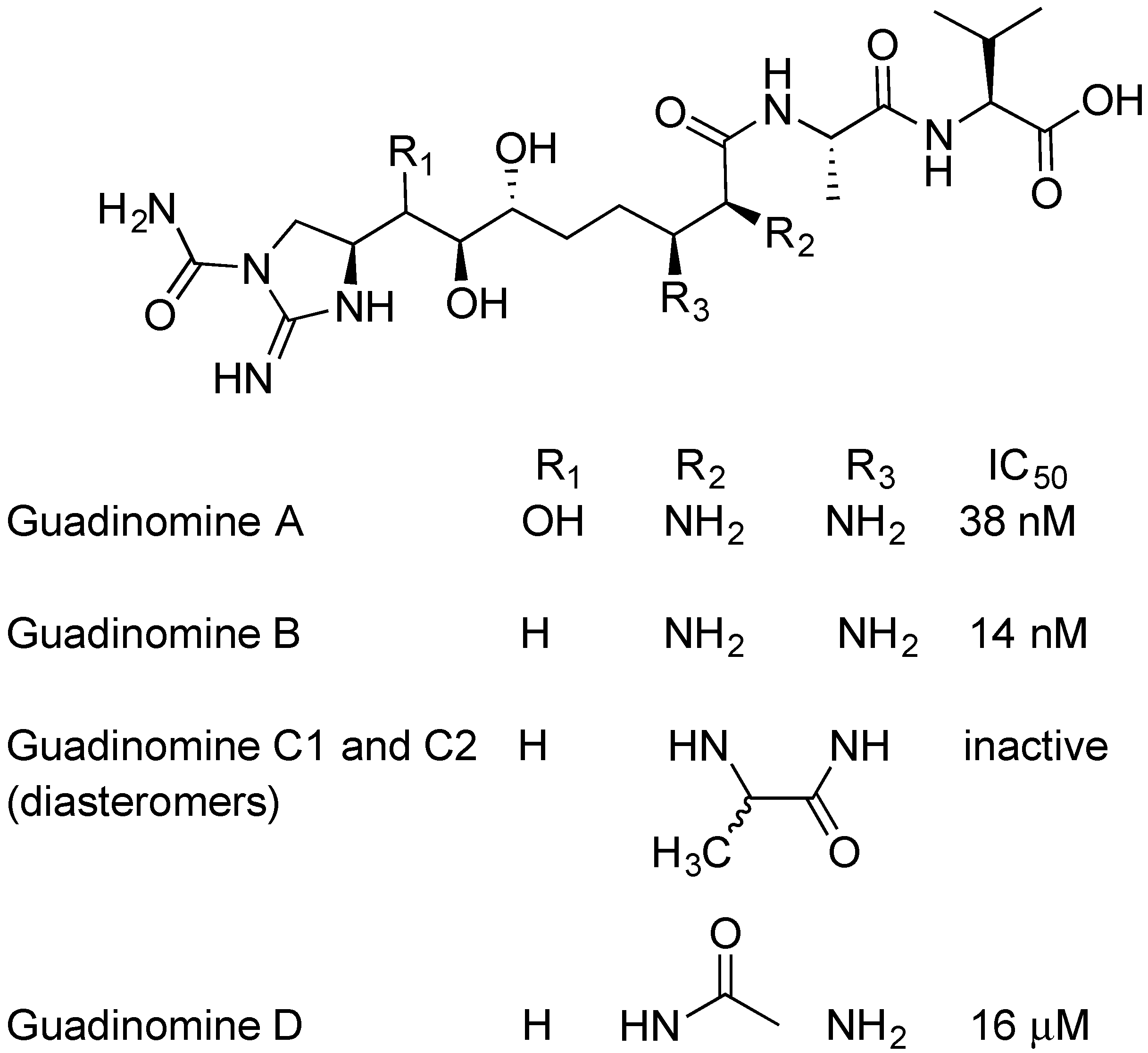
2.5. Guadinomines
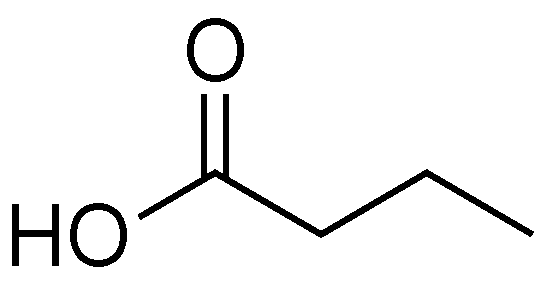
2.6. Butyric Acid
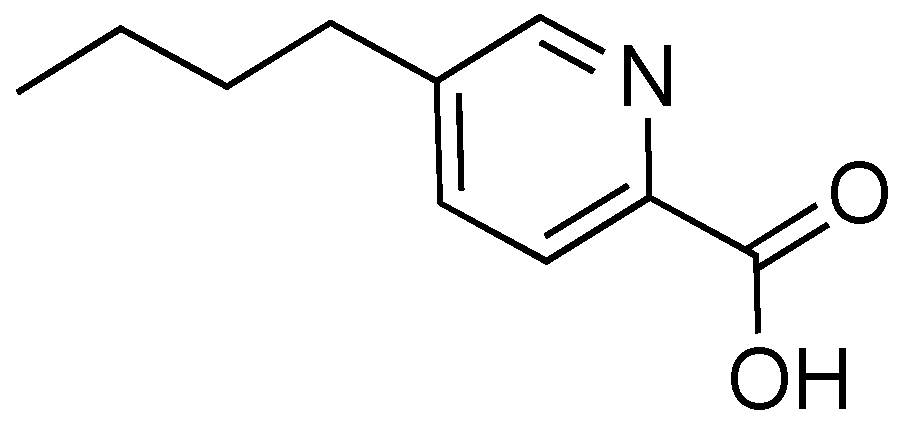
2.7. Fusaric Acid
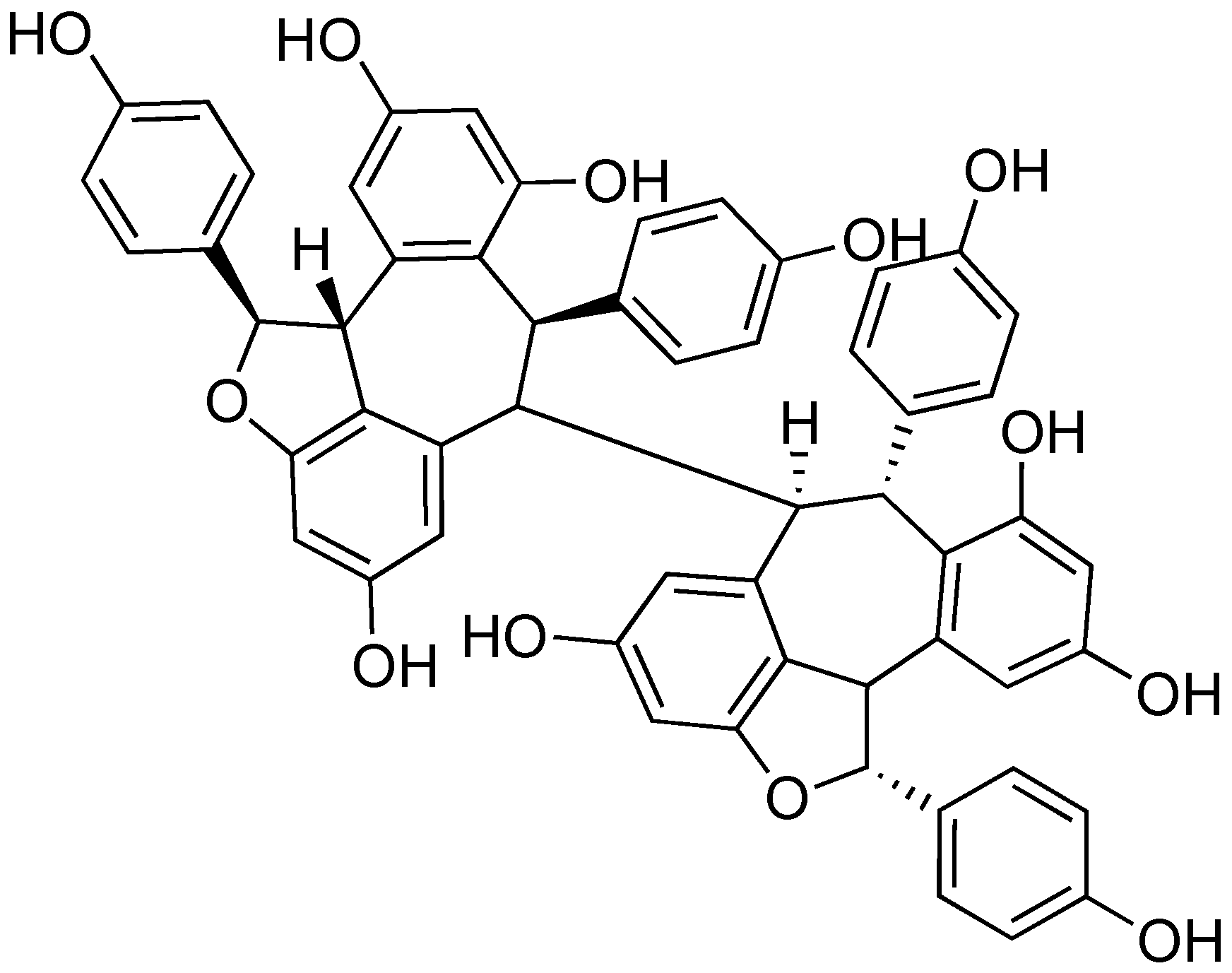
2.8. (-)-Hopeaphenol
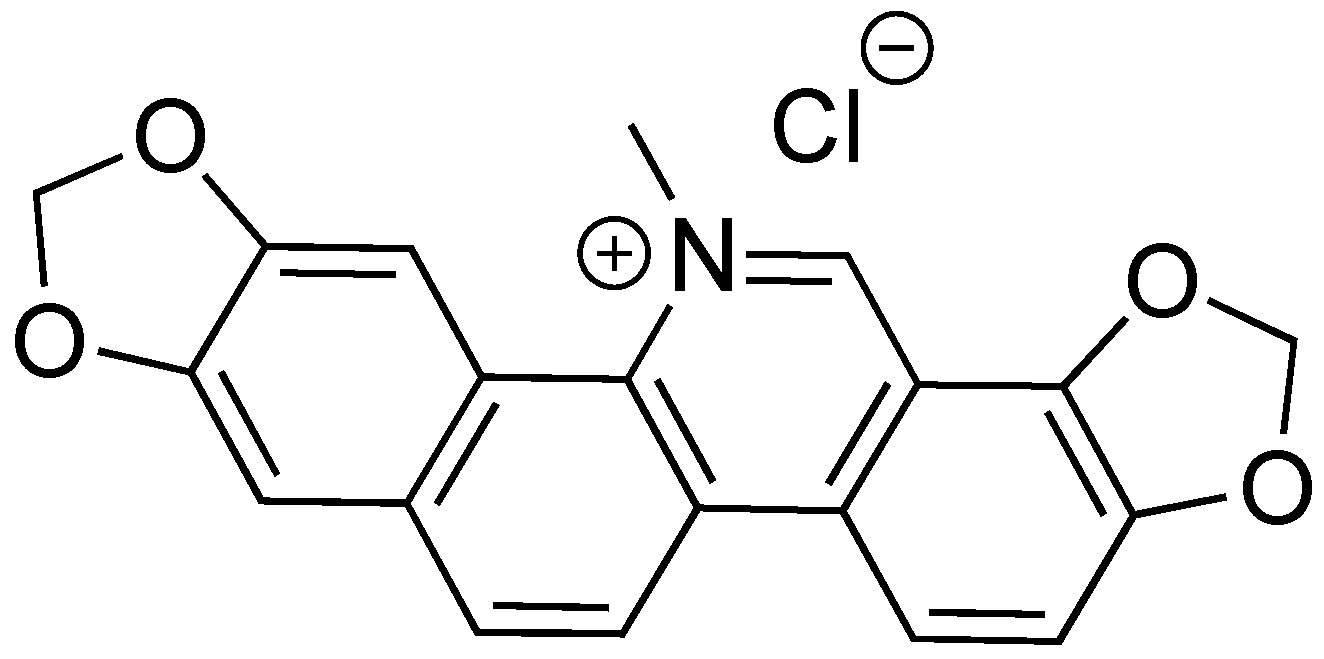
2.9. Sanguinarine Chloride
2.10. Thymol
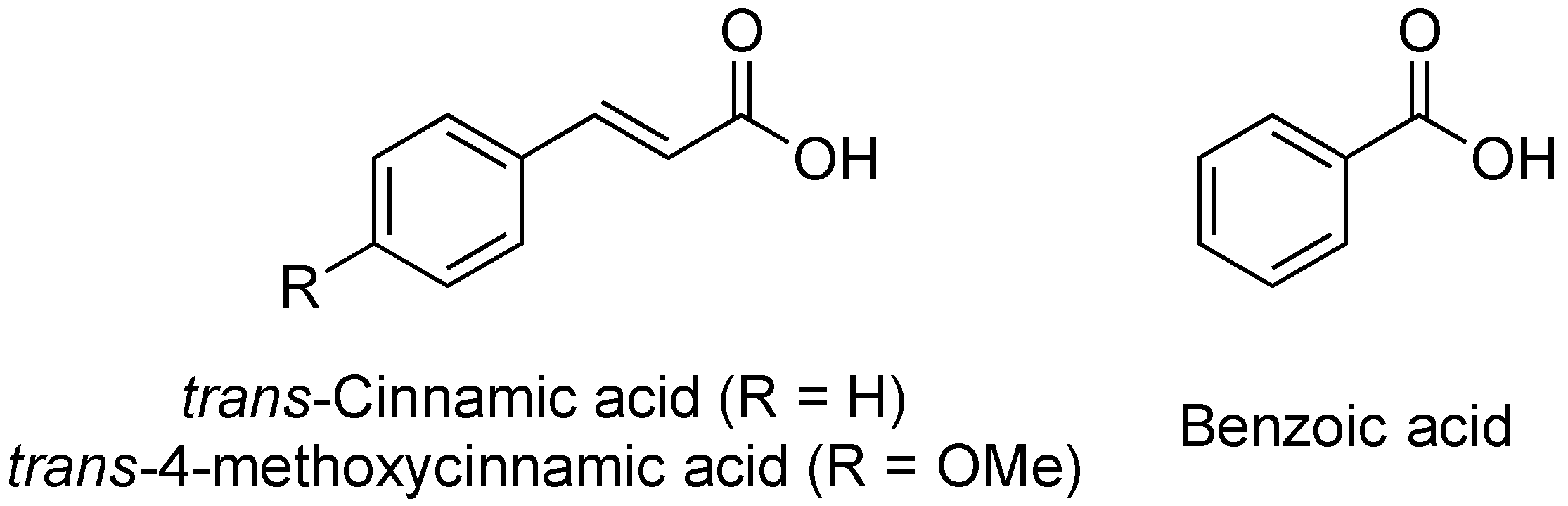
2.11. Cinnamic Acid and Derivatives
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salmond, G.P.C.; Reeves, P.J. Membrane traffic wardens and protein secretion in Gram-negative bacteria. Trends Biochem. Sci. 1993, 18, 7–12. [Google Scholar] [CrossRef]
- Cornelis, G.R.; Gijsegem, F.V. Assembly and function of type III secretory systems. Annu. Rev. Microbiol. 2000, 54, 735–774. [Google Scholar] [CrossRef] [PubMed]
- Grado, M.A.; Abe, A.G.; Gauthier, A.; Steele-Mortimer, O.; DeVinny, R.; Finlay, B.B. Identification of the intimin-binding domain of Tir of enteropathogenic Escherichia coli. Cell Microbiol. 1999, 1, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.G.; Giron, J.A.; Jerse, A.N.N.E.; Mcdaniel, T.K.; Donnenberg, M.S.; Kaper, J.B. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. USA 1995, 92, 7996–8000. [Google Scholar] [CrossRef]
- Franzin, F.M.; Sircili, M.P. Locus of enterocyte effacement: A pathogenicity island involved in the virulence of enteropathogenic and enterohemorrhagic Escherichia coli subjected to a complex network of gene regulation. BioMed Res. Int. 2015, 2015, 534738. [Google Scholar] [CrossRef]
- Croxen, M.A.; Finlay, B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 2010, 8, 26–38. [Google Scholar] [CrossRef]
- Dai, W.; Li, Z. Conserved type III secretion system exerts important roles in Chlamydia trachomatis. Int. J. Exp. Pathol. 2014, 7, 5404–5414. [Google Scholar]
- Bartra, S.S.; Lorica, C.; Qian, L.; Gong, X.; Bahnan, W. Chromosomally-encoded Yersinia pestis type III secretion effector proteins promote infection in cells and in mice. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Marketon, M.M.; Depaolo, R.W.; Debord, K.L. Plague bacteria target immune cells during infection. Science 2005, 309, 1739–1742. [Google Scholar] [CrossRef]
- Crawford, J.A.; Kaper, J.B. The N-terminus of enteropathogenic Escherichia coli (EPEC) Tir mediates transport across bacterial and eukaryotic cell membranes. Mol. Microbiol. 2002, 46, 855–868. [Google Scholar] [CrossRef]
- Fasciano, A.C.; Shaban, L.; Mecsas, J. Promises and challenges of the type three secretion system injectisome as an antivirulence target. EcoSal Plus 2019. [Google Scholar] [CrossRef]
- Yang, J.; Hocking, D.M.; Cheng, C.; Dogovski, C.; Perugini, M.A.; Holien, J.K.; Parker, M.W.; Hartland, E.L.; Tauschek, M.; Robins-Browne, R.M. Disarming bacterial virulence through chemical inhibition of the DNA binding domain of an AraC-like transcriptional activator protein. J. Biol. Chem. 2013, 288, 31115–31126. [Google Scholar] [CrossRef]
- Marshall, N.C.; Finlay, B.B.; Marshall, N.C.; Finlay, B.B. Targeting the type III secretion system to treat bacterial infections. Expert Opin. Ther. Targets 2014, 18, 137–152. [Google Scholar] [CrossRef]
- Kimura, K.; Iwatsuki, M.; Nagai, T.; Matsumoto, A.; Takahashi, Y.; Shiomi, K.; Omura, S.; Abe, A. A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 2011, 64, 197–203. [Google Scholar] [CrossRef]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; Van Soest, R.; Andersen, R.J. Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org. Lett. 2002, 4, 4089–4092. [Google Scholar] [CrossRef]
- Kola, M.; Urba, K. Antibiotic selective pressure and development of bacterial resistance. Int. J. Antimicrob. Agents 2001, 17, 357–363. [Google Scholar] [CrossRef]
- Loquet, A.; Sgourakis, N.G.; Gupta, R.; Giller, K.; Riedel, D.; Goosmann, C.; Griesinger, C.; Kolbe, M.; Baker, D.; Becker, S.; et al. Atomic model of the type III secretion system needle. Nature 2012, 486, 276–279. [Google Scholar] [CrossRef]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef]
- Galán, J.E.; Waksman, G. Protein-injection machines in bacteria. Cell 2018, 172, 1306–1318. [Google Scholar] [CrossRef]
- Sekiya, K.; Ohishi, M.; Ogino, T.; Tamano, K.; Sasakawa, C.; Abe, A. Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl. Acad. Sci. USA 2001, 98, 11638–11643. [Google Scholar] [CrossRef]
- Bzdzion, L.; Krezel, H.; Wrzeszcz, K.; Grzegorek, I.; Nowinska, K.; Chodaczek, G.; Swietnicki, W. Design of small molecule inhibitors of type III secretion system ATPase EscN from enteropathogenic Escherichia coli. Acta Biochim. Pol. 2017, 64, 49–63. [Google Scholar] [CrossRef]
- Andrade, A.; Pardo, J.P.; Espinosa, N.; Pérez-Hernández, G.; González-Pedrajo, B. Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch. Biochem. Biophys. 2007, 468, 121–127. [Google Scholar] [CrossRef]
- Bergeron, J.R.C.; Fernández, L.; Wasney, G.A.; Vuckovic, M.; Reffuveille, F.; Hancock, R.E.W.; Strynadka, N.C.J. The structure of a type 3 secretion system (T3SS) ruler protein suggests a molecular mechanism for needle length sensing. J. Biol. Chem. 2016, 291, 1676–1691. [Google Scholar] [CrossRef]
- Munera, D.; Crepin, V.F.; Marches, O.; Frankel, G. N-terminal type III secretion signal of enteropathogenic Escherichia coli translocator proteins. J. Bacteriol. 2010, 192, 3534–3539. [Google Scholar] [CrossRef]
- Deng, W.; Yu, H.B.; Li, Y.; Brett, B. SepD/SepL-dependent secretion signals of the type III secretion system translocator proteins in enteropathogenic Escherichia coli. J. Bacteriol. 2015, 197, 1263–1275. [Google Scholar] [CrossRef]
- Shaw, R.K.; Cleary, J.; Murphy, M.S.; Frankel, G.; Knutton, S. Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: Role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect. Immun. 2005, 73, 1243–1251. [Google Scholar] [CrossRef]
- Park, D.; Lara-Tejero, M.; Waxham, M.N.; Li, W.; Hu, B. Visualization of the type III secretion-mediated Salmonella–host cell interface using cryo-electron tomography. eLife Sci. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Creasey, E.A.; Delahay, R.M.; Bishop, A.A.; Shaw, R.K.; Kenny, B.; Knutton, S.; Frankel, G. CesT Is a bivalent enteropathogenic Escherichia coli chaperone required for translocation of both Tir and Map. Mol. Microbiol. 2003, 47, 209–221. [Google Scholar] [CrossRef]
- Haack, K.R.; Robinson, C.L.; Miller, K.J.; Fowlkes, J.W.; Mellies, J.L. Interaction of Ler at the LEE5 (Tir) operon of enteropathogenic Escherichia coli. Infect. Immun. 2003, 71, 384–392. [Google Scholar] [CrossRef]
- Abe, A.; De Grado, M.; Pfuetzner, R.A.; Sánchez-Sanmartín, C.; Puente, Â.L.; Devinney, R.; Strynadka, N.C.; Finlay, B.B. Enteropathogenic Escherichia coli translocated intimin receptor, Tir, requires a specific chaperone for stable secretion. Mol. Microbiol. 1999, 33, 1162–1175. [Google Scholar] [CrossRef]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–666. [Google Scholar] [CrossRef]
- Galan, J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe 2009, 5, 571–579. [Google Scholar] [CrossRef]
- Golubeva, Y.A.; Sadik, A.Y.; Ellermeier, J.R.; Slauch, J.M. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 2012, 190, 79–90. [Google Scholar] [CrossRef]
- Figueira, R.; Holden, D.W. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 2012, 2, 1147–1161. [Google Scholar] [CrossRef]
- Winstanley, C.; Hart, C.A. Type III secretion systems and pathogenicity islands. J. Med. Microbiol. 2001, 50, 116–126. [Google Scholar] [CrossRef]
- Wang, H.; Avican, K.; Fahlgren, A.; Erttmann, S.F.; Nuss, A.M.; Dersch, P.; Fallman, M.; Edgren, T.; Wolf-Watz, H. Increased plasmid copy number is essential for Yersinia T3SS function and virulence. Science 2016, 353, 492–495. [Google Scholar] [CrossRef]
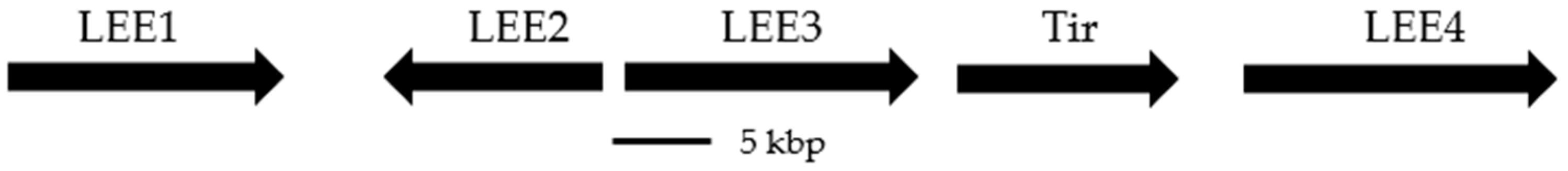
- Mcdaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef]
- Elliott, S.J.; Sperandio, V.; Giron, J.A.; Wainwright, L.; Hutcheson, S.W.; McDaniel, T.K. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 2000, 68, 6115–6126. [Google Scholar] [CrossRef]
- Deng, W.; Li, Y.; Vallance, B.A.; Finlay, B.B. Locus of enterocyte effacement from Citrobacter rodentium: Sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Am. Soc. Microbiol. 2001, 69, 6323–6335. [Google Scholar] [CrossRef]
- Vidal, J.E.; Navarro-García, F. EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell. Microbiol. 2008, 10, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xiuwen, H.; Yu, B. First total synthesis of caminoside A, an antimicrobial glycolipid from sponge. Synlett 2005, 3, 437–440. [Google Scholar] [CrossRef]
- Zhang, Z.; Zong, C.; Song, G.; Lv, G.; Chun, Y.; Wang, P.; Ding, N.; Li, Y. Total synthesis of caminoside B, a novel antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Carbohydr. Res. 2010, 345, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Lehr, H.H.; Teitel, S.; Maehr, H.; Grunberg, E. A new antibiotic X-5108 of Streptomyces origin. I. Production, isolation and properties. J. Antibiot. 1973, 26, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chinali, G. Synthetic Analogs of aurodox and kirromycin active on elongation factor Tu from Escherichia coli. J. Antibiot. 1981, 34, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Warawa, J.; Finlay, B.B.; Kenny, B. Type III secretion-dependent hemolytic activity of enteropathogenic Escherichia coli. Infect. Immun. 1999, 67, 5538–5540. [Google Scholar]
- Swimm, A.I.; Kalman, D. Cytosolic extract induces Tir translocation and pedestals in EPEC-infected red blood cells. PLoS Pathog. 2008, 4, e4. [Google Scholar] [CrossRef]
- Simpson, N.; Shaw, R.; Crepin, V.F.; Mundy, R.; Fitzgerald, A.J.; Straatman-Iwanowska, A.; Knutton, S.; Frankel, G. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: Implication for cell signaling and diarrhea. Mol. Microbiol. 2006, 60, 349–363. [Google Scholar] [CrossRef]
- McHugh, R.E.; O’Boyle, N.; Connoly, J.P.R.; Hoskisson, P.A.; Roe, A.J. Characterization of the mode of action of aurodox, a type III secretion system inhibitor from Streptomyces goldiniensis. Infect. Immun. 2019, 87, 1–12. [Google Scholar] [CrossRef]
- Tamura, S.; Takahashi, N.; Miyamoto, S.; Mori, R.; Suzuki, S.; Nagatsu, J. Isolation and physiological activities of piericidin A, a natural insecticide produced by Streptomyces. Agric. Biol. Chem. 1963, 27, 576–582. [Google Scholar] [CrossRef]
- Hall, C.; Wu, M.; Crane, F.L. Piericidin A: A new inhibitor of mitochondrial electron transport. Biochem. Biophys. Res. Commun. 1966, 25, 373–377. [Google Scholar] [CrossRef]
- Kroiss, J.; Kaltenpoth, M.; Schneider, B.; Schwinger, M.; Hertweck, C.; Maddula, R.K.; Strohm, E.; Svatoš, A. Symbiotic Streptomycetes provide antibiotic combination prophylaxis for wasp offspring. Nat. Chem. Biol. 2010, 6, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.C.; Wong, R.; Dupzyk, A.J.; Bray, W.M.; Linington, R.G. Inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob. Agents Chemother. 2014, 58, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Auerbuch, V.; Golenbock, D.T.; Isberg, R.R. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 2009, 5, e1000686. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.M.; Duncan, M.C.; Johnson, K.S.; Diepold, A.; Lam, H.; Dupzyk, A.J.; Martin, L.R.; Wong, R.; Armitage, J.P.; Linington, R.G. Piericidin A1 blocks Yersinia Ysc type III secretion system needle assembly. Am. Soc. Microbiol. 2017, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Du, X.; Chen, H.; Liu, J.; Zhao, B.; Huang, D.; Li, G.; Xu, Q.; Zhang, M.; Weimer, B.C.; et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 2008, 4, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Cao, X.; Rico-bautista, E.; Yu, J.; Chen, L.; Chen, J.; Bobkov, A.; Wolf, D.A.; Zhang, X.; Dawson, M.I. Cytosporone B on cancer cell viability. Med. Chem. Commun. 2013, 4, 332–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, H.; Liu, Q.; Li, L.; Wang, W.; Yao, L.; Yang, M.; Liu, B.; Chen, W.; Zhan, Y.; Zhang, M.; et al. The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating wnt signaling. Gastrointrotestinal Neoplasia 2012, 61, 714–724. [Google Scholar]
- Lee, S.; Li, X.; Khan, S.; Safe, S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin. Ther. Targets 2011, 15, 195–206. [Google Scholar] [CrossRef]
- Li, J.; Lv, C.; Sun, W.; Li, Z.; Han, X.; Li, Y.; Shen, Y. Cytosporone B, an inhibitor of the type III secretion system of Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2013, 57, 2191–2198. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, L.; Zhang, X.; Ji, X.; Ding, X.; Shen, X.; Jiang, H.; Liu, H. Total synthesis of cytosporone B. Chin. J. Chem. 2010, 28, 1041–1043. [Google Scholar] [CrossRef]
- Iwatsuki, M.; Uchida, R.; Yoshijima, H.; Ui, H.; Shiomi, K.; Matsumoto, A.; Takahashi, Y.; Abe, A.; Tomoda, H.; Satoshi, O. Guadinomines, type III secretion system inhibitors, produced by Streptomyces sp. K01-0509 I. Taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2008, 61, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Uchida, R.; Yoshijima, H.; Ui, H.; Shiomi, K.; Kim, Y.; Hirose, T.; Sunazuka, T.; Abe, A.; Tomoda, H. Guadinomines, type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. II. Physico-chemical properties and structure elucidation. J. Antibiot. 2008, 61, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Sunazuka, T.; Tsuchiya, S.; Tanaka, T.; Kojima, Y.; Mori, R.; Iwatsuki, M.; Ōmura, S. Total synthesis and determination of the absolute configuration of guadinomines B and C2. Chem. Eur. J. 2008, 14, 8220–8238. [Google Scholar] [CrossRef]
- Holmes, T.C.; May, A.E.; Zaleta-Rivera, K.; Ruby, J.G.; Skewes-Cox, P.; Fischbach, M.A.; Derisi, J.L.; Iwatsuki, M.; Satoshi, O.; Khosla, C. Molecular insights into the biosynthesis of guadinomine: A type III secretion system inhibitor. J. Am. Chem. Soc. 2012, 134, 17797–17806. [Google Scholar] [CrossRef]
- Bohnhoff, B.Y.M.; Miller, C.P.; Martin, W.R. Resistance of the mouse’s intestinal tract to experimental Salmonella infection I. Factors which interfere with the initiation of infection. J. Exp. Med. 1964, 120, 805–816. [Google Scholar] [CrossRef]
- Sun, Y.; O’Riordan, M.X. Regulation of bacterial pathogenesis by intestinal short-chain fatty acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar]
- Nakanishi, N.; Tashiro, K.; Kuhara, S.; Hayashi, T.; Sugimoto, N.; Tobe, T. Regulation of virulence by butyrate sensing in enterohaemorrhagic Escherichia coli. Microbiology 2009, 155, 521–530. [Google Scholar] [CrossRef]
- Pomare, E.W.; Branch, H.W.J.; Naylor, C.P.E.; Macfarlane, T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar]
- Takao, M.; Yen, H.; Tobe, T. LeuO Enhances butyrate-induced virulence expression through a positive regulatory loop in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 2014, 93, 1302–1313. [Google Scholar]
- Baek, C.; Wang, S.; Roland, K.L.; Curtiss, R., III. Leucine-responsive regulatory protein (Lrp) acts as a virulence repressor in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2009, 191, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Cordone, A.; Lucchini, S.; De Felice, M.; Ricca, E. Direct and indirect control of Lrp on LEE pathogenicity genes of Citrobacter rodentium. Fed. Eur. Microbiol. Soc. 2011, 325, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Crutcher, F.K.; Puckhaber, L.S.; Stipanovic, R.D.; Bell, A.A.; Nichols, R.L.; Lawrence, K.S.; Liu, J. Microbial resistance mechanisms to the antibiotic and phytotoxin fusaric acid. J. Chem. Ecol. 2017, 43, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Gaumman, E. Fusaric acid as a wilt toxin. Phytopathology 1957, 47, 342–357. [Google Scholar]
- Dong, X.; Ling, N.; Wang, M.; Shen, Q.; Guo, S. Fusaric acid is a crucial factor in the disturbance of leaf water imbalance in Fusarium-infected banana plants. Plant. Physiol. Biochem. 2012, 60, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Reveglia, P.; Cinelli, T.; Cimmino, A.; Masi, M.; Evidente, A. The main phytotoxic metabolite produced by a strain of Fusarium oxysporum inducing grapevine plant declining in Italy. Nat. Prod. Res. 2017, 32, 2398–2407. [Google Scholar] [CrossRef]
- Li, J.; Sun, W.; Guo, Z.; Lu, C.; Shen, Y. Fusaric acid modulates type three secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Res. Commun. 2014, 449, 455–459. [Google Scholar] [CrossRef]
- Zetterström, C.E.; Hasselgren, J.; Salin, O.; Davis, R.A.; Quinn, R.J. The resveratrol tetramer (-)-hopeaphenol inhibits type III secretion in the Gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLoS ONE 2013, 8, e81969. [Google Scholar] [CrossRef]
- Subramanian, R.; Raj, V.; Manigandan, K.; Elangovan, N. Antioxidant activity of hopeaphenol isolated from Shorea roxburghii stem bark extract. J. Taibah Univ. Sci. 2015, 9, 237–244. [Google Scholar] [CrossRef]
- International Union for Conservation of Nature (IUCN) Red List. Available online: https://www.iucnredlist.org/search?query=anisoptera&searchType=species (accessed on 5 September 2019).
- Babich, H.; Zuckerbraun, H.L.; Barber, I.B.; Babich, S.B.; Borenfreund, E. Cytotoxicity of sanguinarine chloride to cultured human cells from oral tissue. Pharmacol. Toxicol. 1996, 78, 397–403. [Google Scholar] [CrossRef]
- Mondal, A.; Gandhi, A.; Fimognari, C.; Atanasov, A.G.; Bishayee, A. Alkaloids for cancer prevention and therapy: Current progress and future perspectives. Eur. J. Pharmacol. 2019, 858. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Wang, T.; Deng, X.; Chu, X. Natural compound sanguinarine chloride targets the type III secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Rep. 2018, 14, 149–154. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Qiu, J.; Luo, Z.; Deng, X. The herbal compound thymol protects mice from lethal infection by Salmonella typhimurium. Front. Microbiol. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Ghannadi, A.; Ebrahim, S.; Kabouche, A. Thymus Fontanesii Boiss. & Reut: A potential source of thymol-rich essential oil in North Africa. Zeitschrift fur Naturforschung C 2004, 59, 187–189. [Google Scholar]
- Yang, S.; Peng, Q.; Francisco, M.S.; Wang, Y.; Zeng, Q.; Yang, C. Type III secretion system genes of Dickeya dadantii 3937 are induced by plant phenolic acids. PLoS ONE 2008, 3, e2973. [Google Scholar] [CrossRef]
- Khokhani, D.; Zhang, C.; Li, Y.; Wang, Q.; Zeng, Q.; Yamazaki, A.; Hutchins, W.; Zhou, S.; Chen, X.; Yang, C. Discovery of plant phenolic compounds that act as type III secretion system inhibitors or inducers of the fire blight pathogen, Erwinia amylovora. Appl. Environ. Microbiol. 2013, 79, 5424–5436. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pendergrass, H.A.; May, A.E. Natural Product Type III Secretion System Inhibitors. Antibiotics 2019, 8, 162. https://doi.org/10.3390/antibiotics8040162
Pendergrass HA, May AE. Natural Product Type III Secretion System Inhibitors. Antibiotics. 2019; 8(4):162. https://doi.org/10.3390/antibiotics8040162
Chicago/Turabian StylePendergrass, Heather A., and Aaron E. May. 2019. "Natural Product Type III Secretion System Inhibitors" Antibiotics 8, no. 4: 162. https://doi.org/10.3390/antibiotics8040162
APA StylePendergrass, H. A., & May, A. E. (2019). Natural Product Type III Secretion System Inhibitors. Antibiotics, 8(4), 162. https://doi.org/10.3390/antibiotics8040162