Directed Evolution of a Mycobacteriophage



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
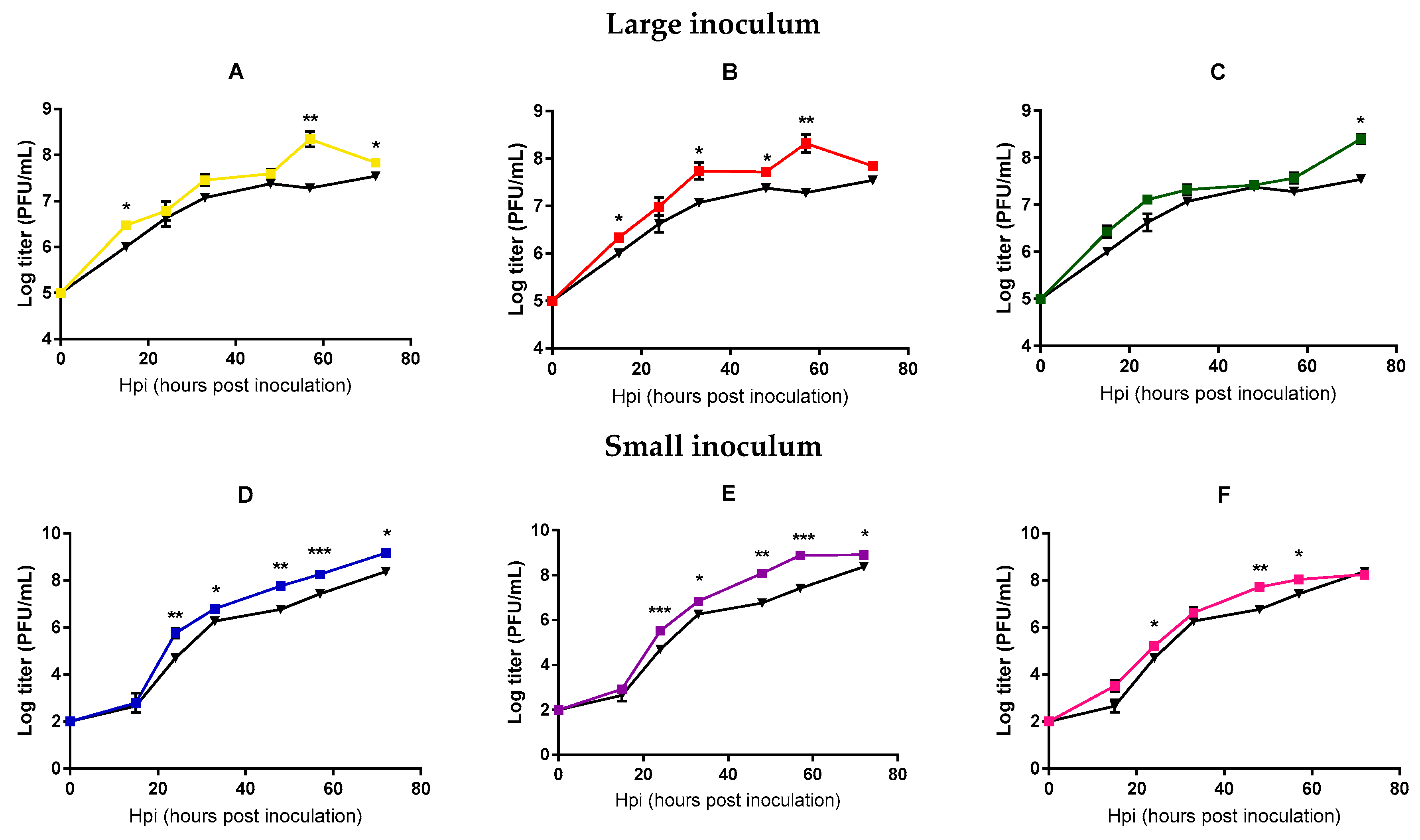
2.1. Directed Evolution
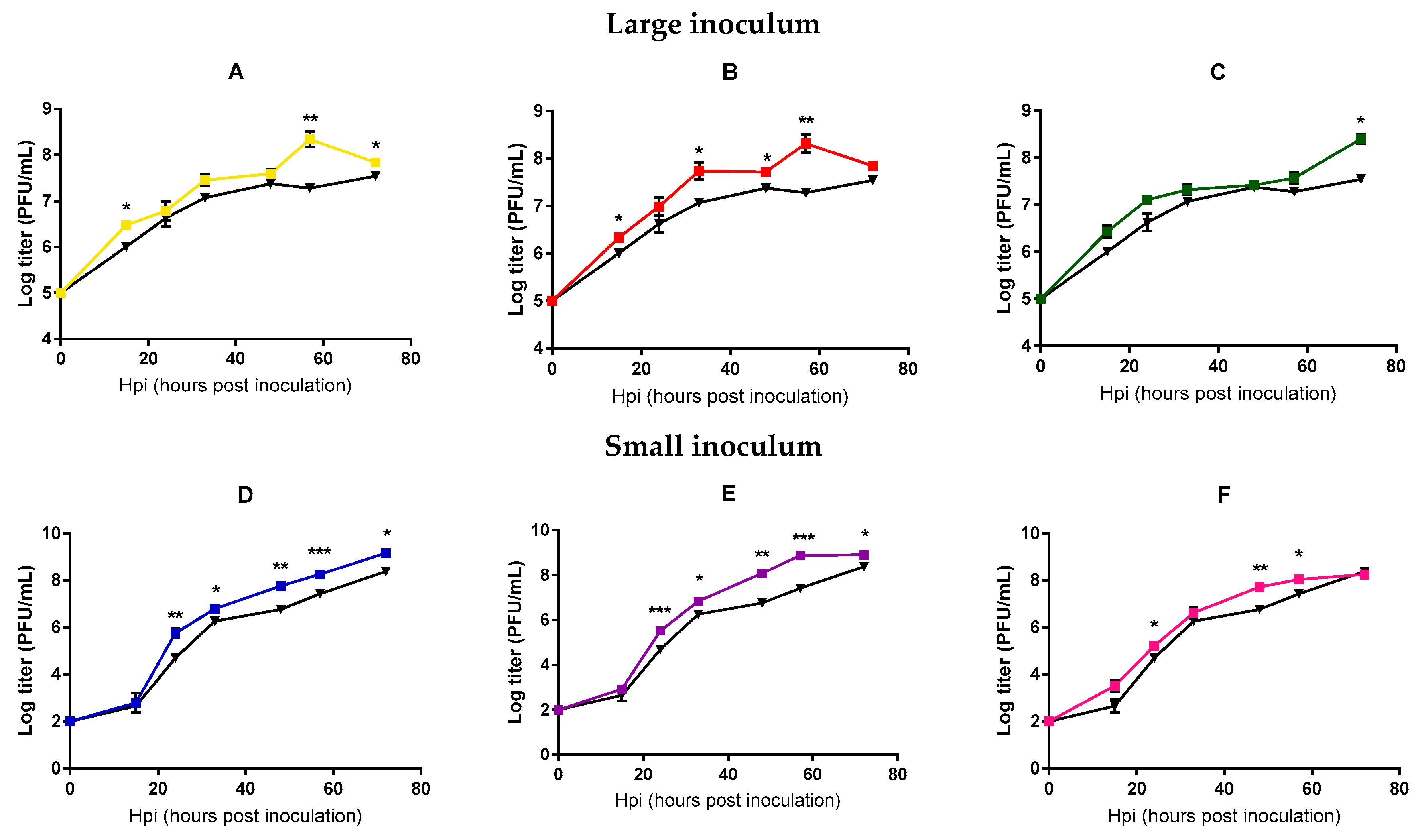
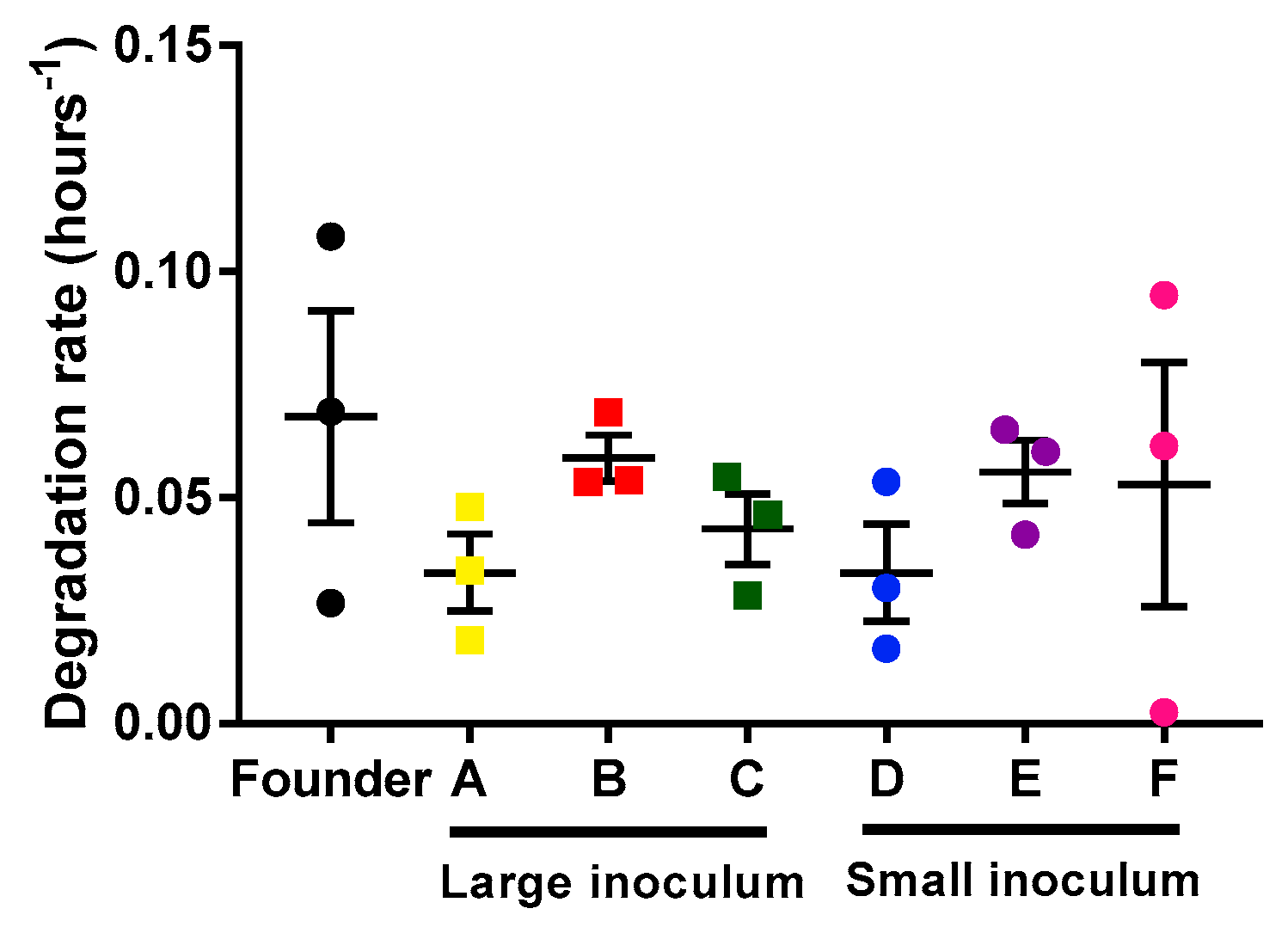
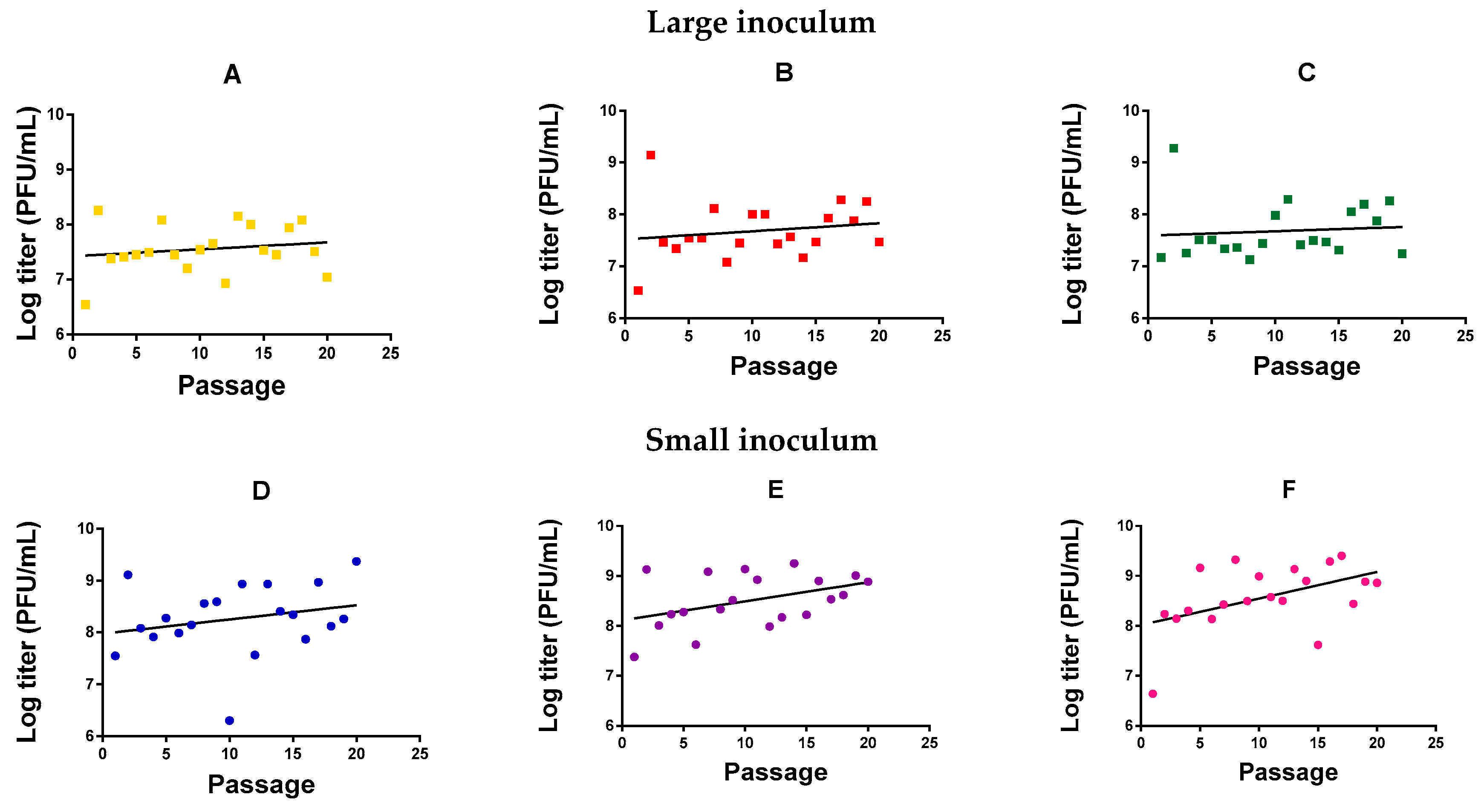
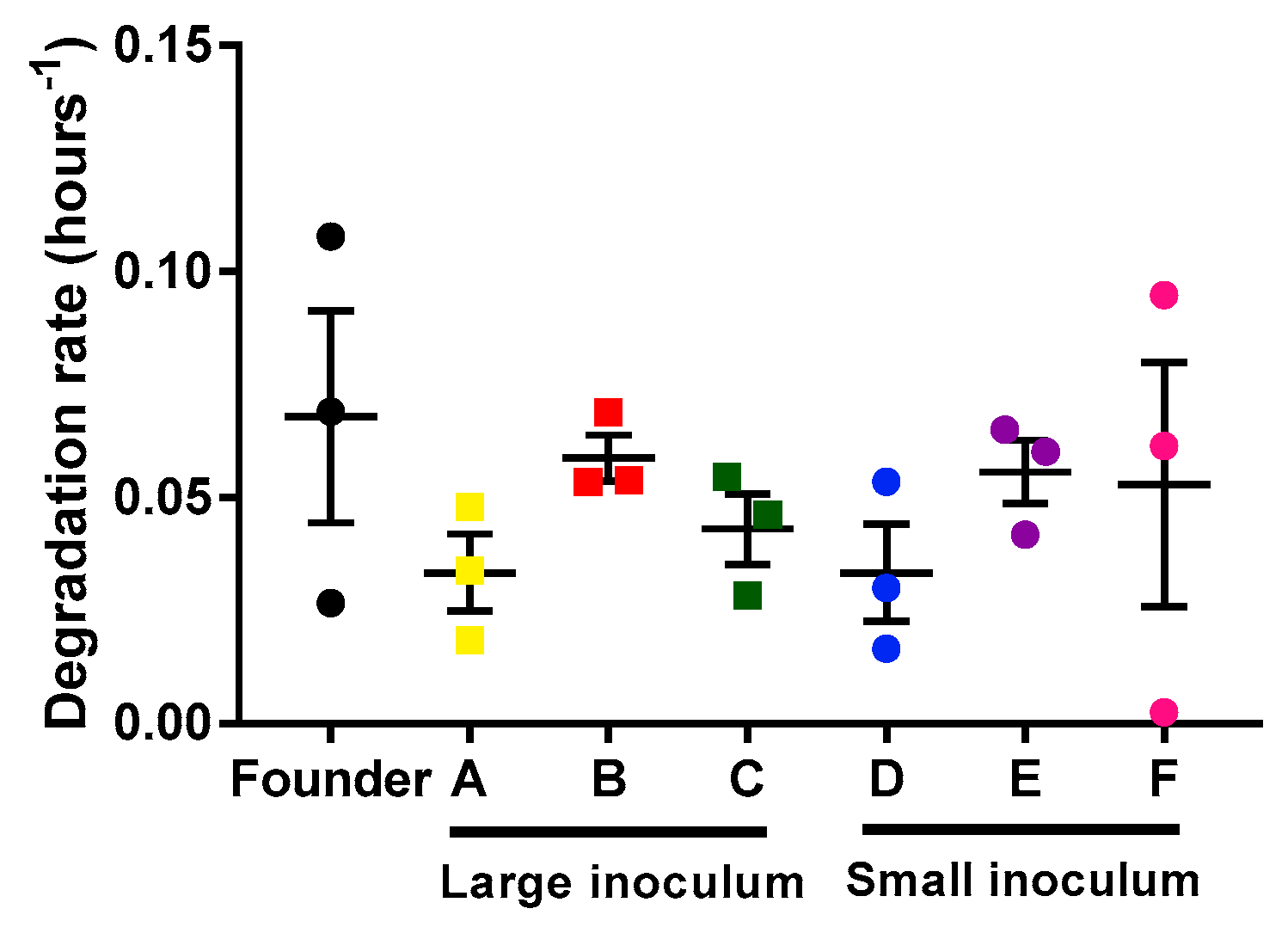
2.2. Analysis of Mycobacteriophage Growth Rate and Stability
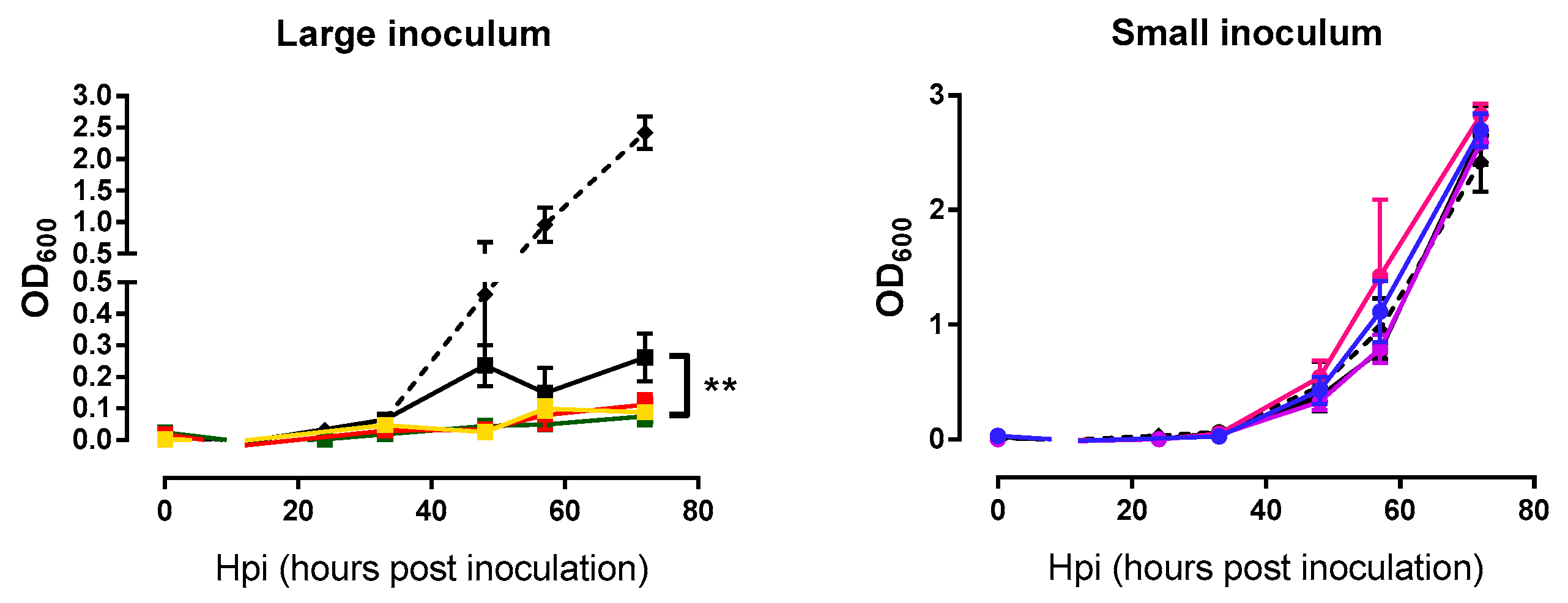
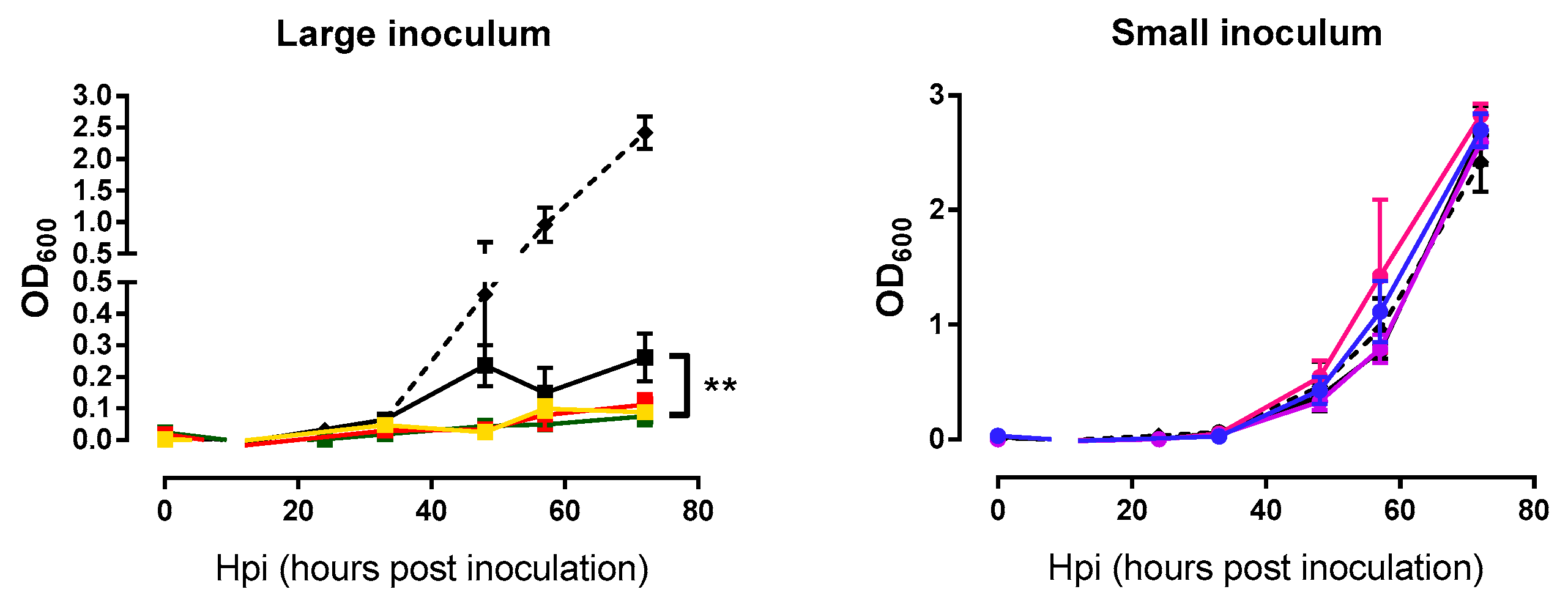
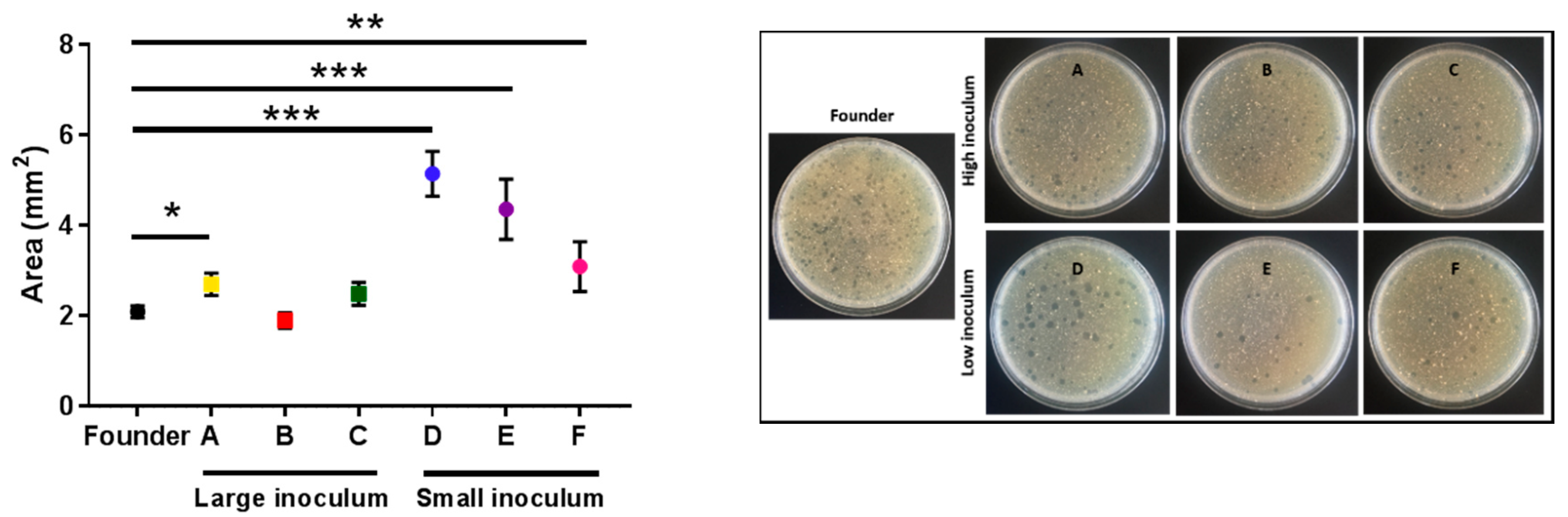
2.3. Analysis of Bacterial Lysis Efficiency
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Mycobacteriophage
4.3. Evolution
4.4. Growth Curves
4.5. Mycobacteriophage Degradation Rates
4.6. Plaque Sizes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Górski, A.; Miedzybrodzki, R.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Rogóz, P.; Jończyk-Matysiak, E.; Dabrowska, K.; Majewska, J.; Borysowski, J. Phage Therapy: Combating Infections with Potential for Evolving from Merely a Treatment for Complications to Targeting Diseases. Front. Microbiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2017. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 24 April 2019).
- Samaddar, S.; Grewal, R.K.; Sinha, S.; Ghosh, S.; Roy, S.; Das Gupta, S.K. Dynamics of Mycobacteriophage-Mycobacterial Host Interaction: Evidence for Secondary Mechanisms for Host Lethality. Appl. Environ. Microbiol. 2016, 82, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Owczarek, B.; Międzybrodzki, R.; Łusiak-Szelchowska, M.; Łodej, N.; Górski, A. Phage-Phagocyte Interactions and Their Implications for Phage Application as Therapeutics. Viruses 2017, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Calap, P.; Georgel, P.; Bahram, S. Back to the Future: Bacteriophages as Promising Therapeutic Tools. HLA 2016, 87, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Housby, J.N.; Mann, N.H. Phage Therapy. Drug Discov. Today 2009, 14, 536–540. [Google Scholar] [CrossRef]
- Weber-Dabrowska, B.; Jończyk-Matysiak, E.; Zaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front Microbiol. 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.S. Phage Therapy-Constraints and Possibilities. Ups. J. Med. Sci. 2014, 119, 192–198. [Google Scholar] [CrossRef]
- Ul Haq, I.; Chaudhry, W.N.; Akhtar, M.N.; Andleeb, S.; Qadri, I. Bacteriophages and Their Implications on Future Biotechnology: A Review. Virol. J. 2012, 9, 9. [Google Scholar] [CrossRef]
- Kingwell, K. Bacteriophage Therapies Re-Enter Clinical Trials. Nat. Rev. Drug Discov. 2015, 14, 515–516. [Google Scholar] [CrossRef]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage Therapy: An Alternative to Antibiotics in the Age of Multi-Drug Resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.A.; Chandry, P.S.; Kaur, M.; Kocharunchitt, C.; Bowman, J.P.; Fox, E.M. Novel Biocontrol Methods for Listeria Monocytogenes Biofilms in Food Production Facilities. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- The Actinobacteriophage Database. Available online: http://phagesdb.org/ (accessed on 24 April 2019).
- Lima-Junior, J.D.; Viana-Niero, C.; Conde Oliveira, D.V.; Machado, G.E.; Rabello, M.C.D.S.; Martins-Junior, J.; Martins, L.F.; Digiampietri, L.A.; Da Silva, A.M.; Setubal, J.C.; et al. Characterization of Mycobacteria and Mycobacteriophages Isolated from Compost at the São Paulo Zoo Park Foundation in Brazil and Creation of the New Mycobacteriophage Cluster U. BMC Microbiol. 2016, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Molecular Genetics of Mycobacteriophages. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Jacobs-Sera, D.; Marinelli, L.J.; Bowman, C.; Broussard, G.W.; Guerrero Bustamante, C.; Boyle, M.M.; Petrova, Z.O.; Dedrick, R.M.; Pope, W.H.; Modlin, R.L.; et al. On the Nature of Mycobacteriophage Diversity and Host Preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Klann, A.G.; Belanger, A.E.; Abanes-de Mello, A.; Lee, J.Y.; Hatfull, G.F. Characterization of the DnaG Locus in Mycobacterium smegmatis Reveals Linkage of DNA Replication and Cell Division. J. Bacteriol. 1998, 180, 65–72. [Google Scholar]
- Domingo-Calap, P.; Cuevas, J.M.; Sanjuán, R. The Fitness Effects of Random Mutations in Single-Stranded DNA and RNA Bacteriophages. PLoS Genet. 2009, 5, 1–7. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Sanjuán, R. Experimental evolution of RNA versus DNA viruses. Evolution (NY) 2011, 65, 2987–2994. [Google Scholar] [CrossRef]
- Pal, C.; Maciá, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with Viruses Drives the Evolution of Bacterial Mutation Rates. Nature 2007, 450, 1079–1981. [Google Scholar] [CrossRef]
- Betts, A.; Gray, C.; Zelek, M.; MacLean, R.C.; King, K.C. High Parasite Diversity Accelerates Host Adaptation and Diversification. Science 2018, 360, 907–911. [Google Scholar] [CrossRef]
- Wichman, H.A.; Badgett, M.R.; Scott, L.A.; Boulianne, C.M.; Bull, J.J. Different Trajectories of Parallel Evolution During Viral Adaptation. Science 1999, 285, 422–424. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Miralles, R.; Gerrish, P.J.; Moya, A.; Elena, S.F. Clonal Interference and the Evolution of RNA Viruses. Science 1999, 285, 1745–1747. [Google Scholar] [CrossRef] [PubMed]
- Arjan, G.J.; de Visser, M.; Zeyl, C.W.; Gerrish, P.J.; Blanchard, J.L.; Lenski, R.E. Diminishing Returns from Mutation Supply Rate in Asexual Populations. Science 1999, 283, 404–406. [Google Scholar] [CrossRef]
- Kick, B.; Hensler, S.; Praetorius, F.; Dietz, H.; Weuster-Botz, D. Specific Growth Rate and Multiplicity of Infection Affect High-Cell-Density Fermentation with Bacteriophage M13 for ssDNA Production. Biotechnol. Bioeng. 2017, 114, 777–784. [Google Scholar] [CrossRef]
- Sun, C.L.; Barrangou, R.; Thomas, B.C.; Horvath, P.; Fremaux, C.; Banfield, J.F. Phage Mutations in Response to CRISPR Diversification in a Bacterial Population. Environ. Microbiol. 2012, 15, 463–470. [Google Scholar] [CrossRef]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonté, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage Response to CRISPR-Encoded Resistance in Streptococcus Thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Buckling, A.; Hall, A.R. Experimental Evolution and Bacterial Resistance: (Co)Evolutionary Costs and Trade-Offs as Opportunities in Phage Therapy Research. Bacteriophage 2015, 5, e1050153. [Google Scholar] [CrossRef]
- Broxmeyer, L.; Sosnowska, D.; Miltner, E.; Chacón, O.; Wagner, D.; McGarvey, J.; Barletta, R.G.; Bermudez, L.E. Killing of Mycobacterium Avium and Mycobacterium Tuberculosis by a Mycobacteriophage Delivered by a Nonvirulent Mycobacterium: A Model for Phage Therapy of Intracellular Bacterial Pathogens. J. Infect. Dis. 2002, 186, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, J.; Papavinasasundaram, K.; Galán, B.; Sassetti, C.M.; García, J.L. Unravelling the Pleiotropic Role of the MceG ATPase in Mycobacterium Smegmatis. Environ. Microbiol. 2017, 19, 2564–2576. [Google Scholar] [CrossRef]
- Piuri, M.; Rondón, L.; Urdániz, E.; Hatfull, G.F. Generation of Affinity-Tagged Fluoromycobacteriophages by Mixed Assembly of Phage Capsids. Appl. Environ. Microbiol. 2013, 79, 5608–5615. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cebriá-Mendoza, M.; Sanjuán, R.; Domingo-Calap, P. Directed Evolution of a Mycobacteriophage. Antibiotics 2019, 8, 46. https://doi.org/10.3390/antibiotics8020046
Cebriá-Mendoza M, Sanjuán R, Domingo-Calap P. Directed Evolution of a Mycobacteriophage. Antibiotics. 2019; 8(2):46. https://doi.org/10.3390/antibiotics8020046
Chicago/Turabian StyleCebriá-Mendoza, María, Rafael Sanjuán, and Pilar Domingo-Calap. 2019. "Directed Evolution of a Mycobacteriophage" Antibiotics 8, no. 2: 46. https://doi.org/10.3390/antibiotics8020046
APA StyleCebriá-Mendoza, M., Sanjuán, R., & Domingo-Calap, P. (2019). Directed Evolution of a Mycobacteriophage. Antibiotics, 8(2), 46. https://doi.org/10.3390/antibiotics8020046