Production of β-Lactamase Inhibitors by Streptomyces Species

 ,
,  and
and
Abstract
1. Introduction
2. Antibiotic Resistance
3. Producers of β-Lactamases
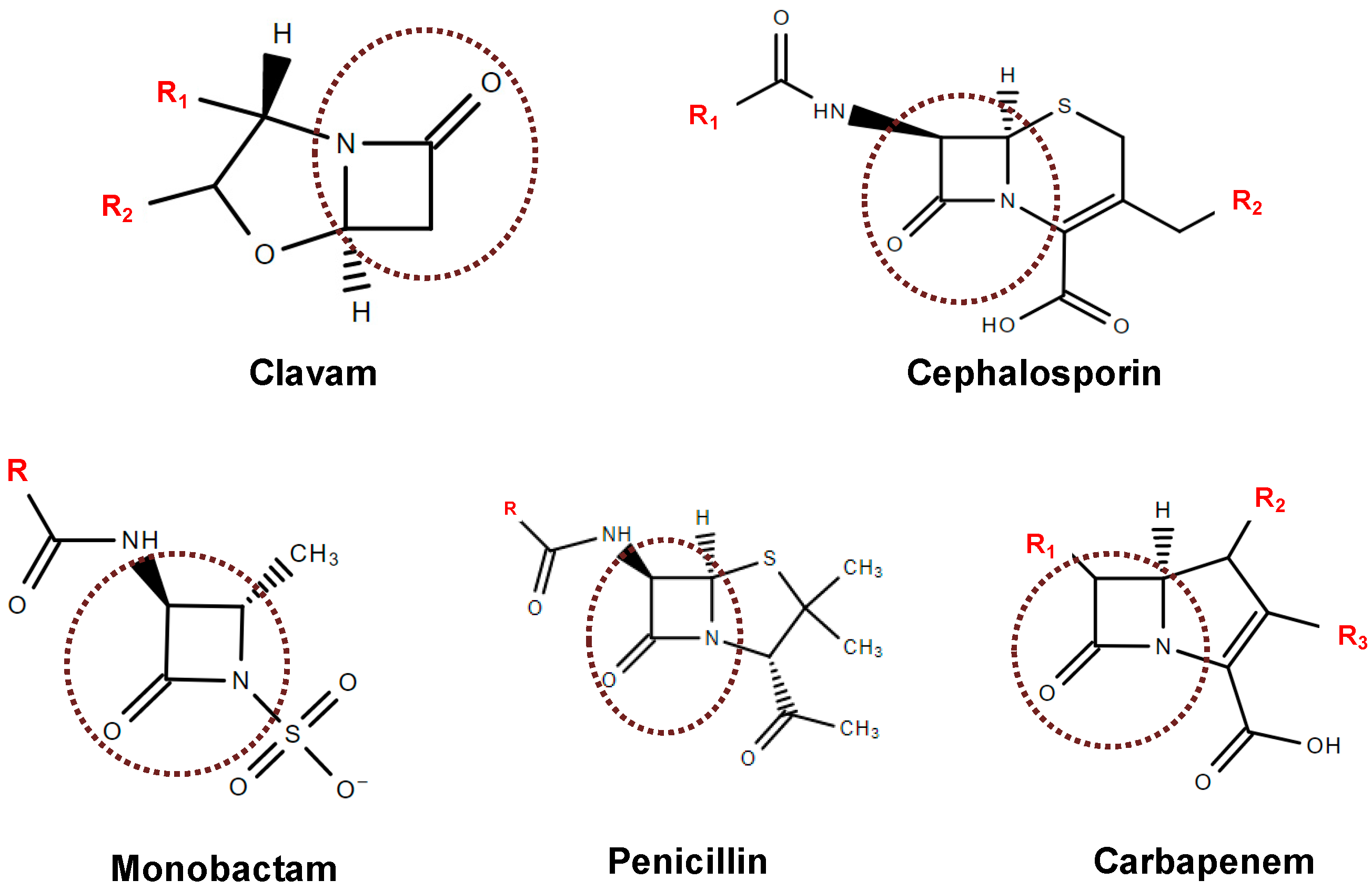
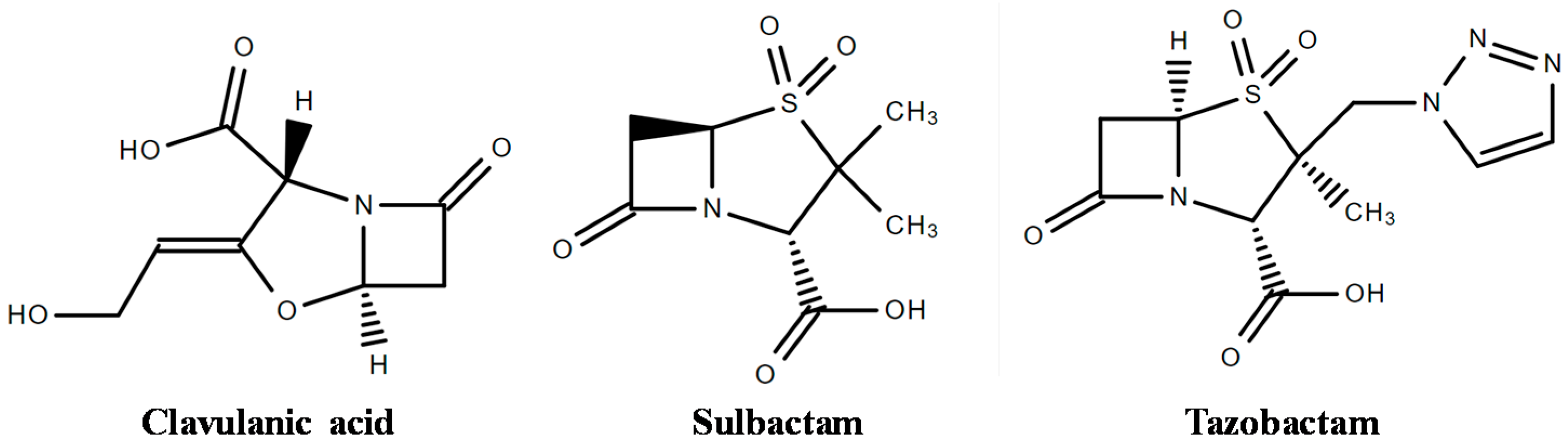
4. Inhibitors of β-Lactamases
5. Producers of β-Lactamases Inhibitors
5.1. Natural Microorganisms
5.2. Genetically-Modified Microorganisms
6. Production of β-Lactamase Inhibitors
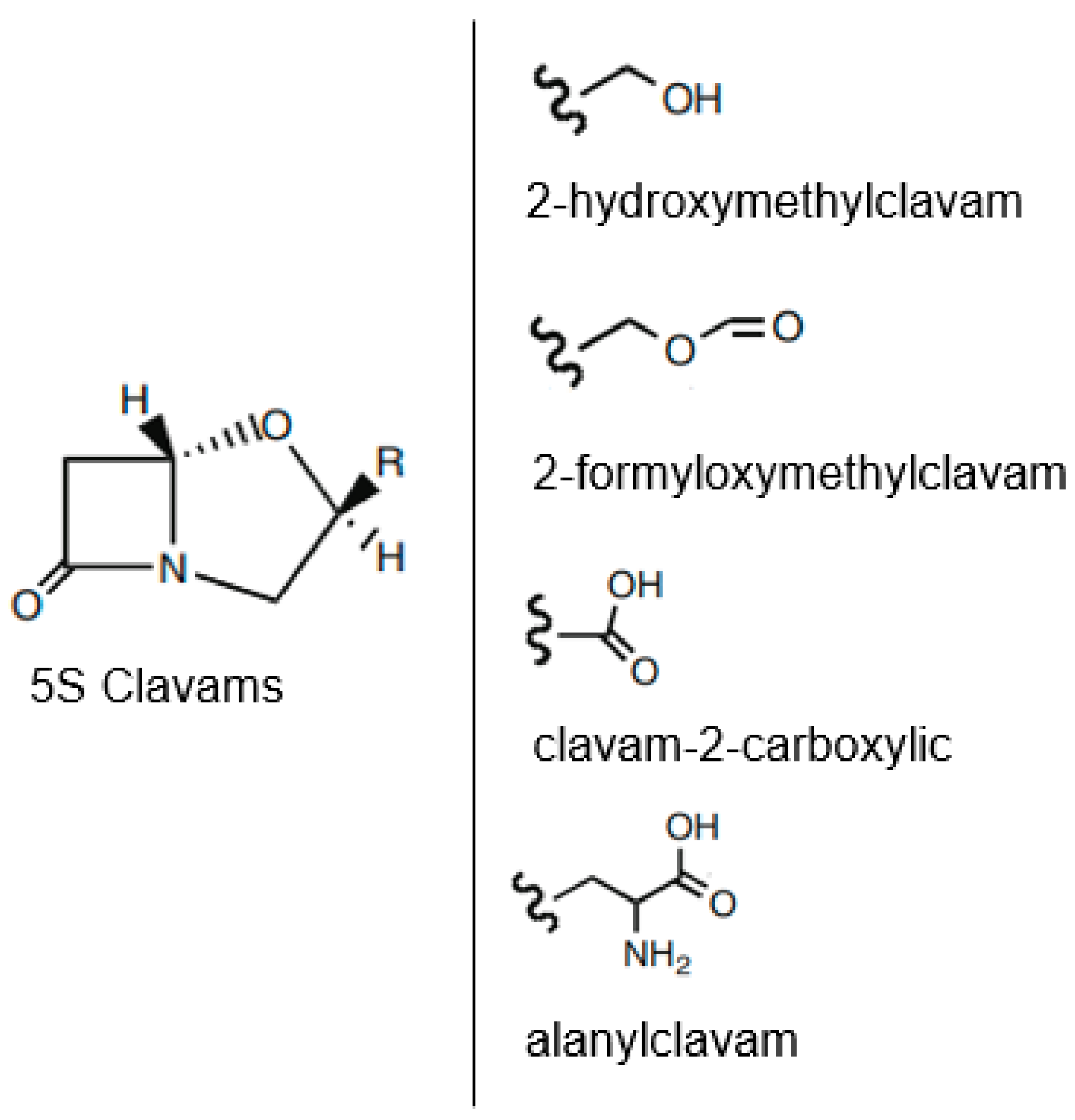
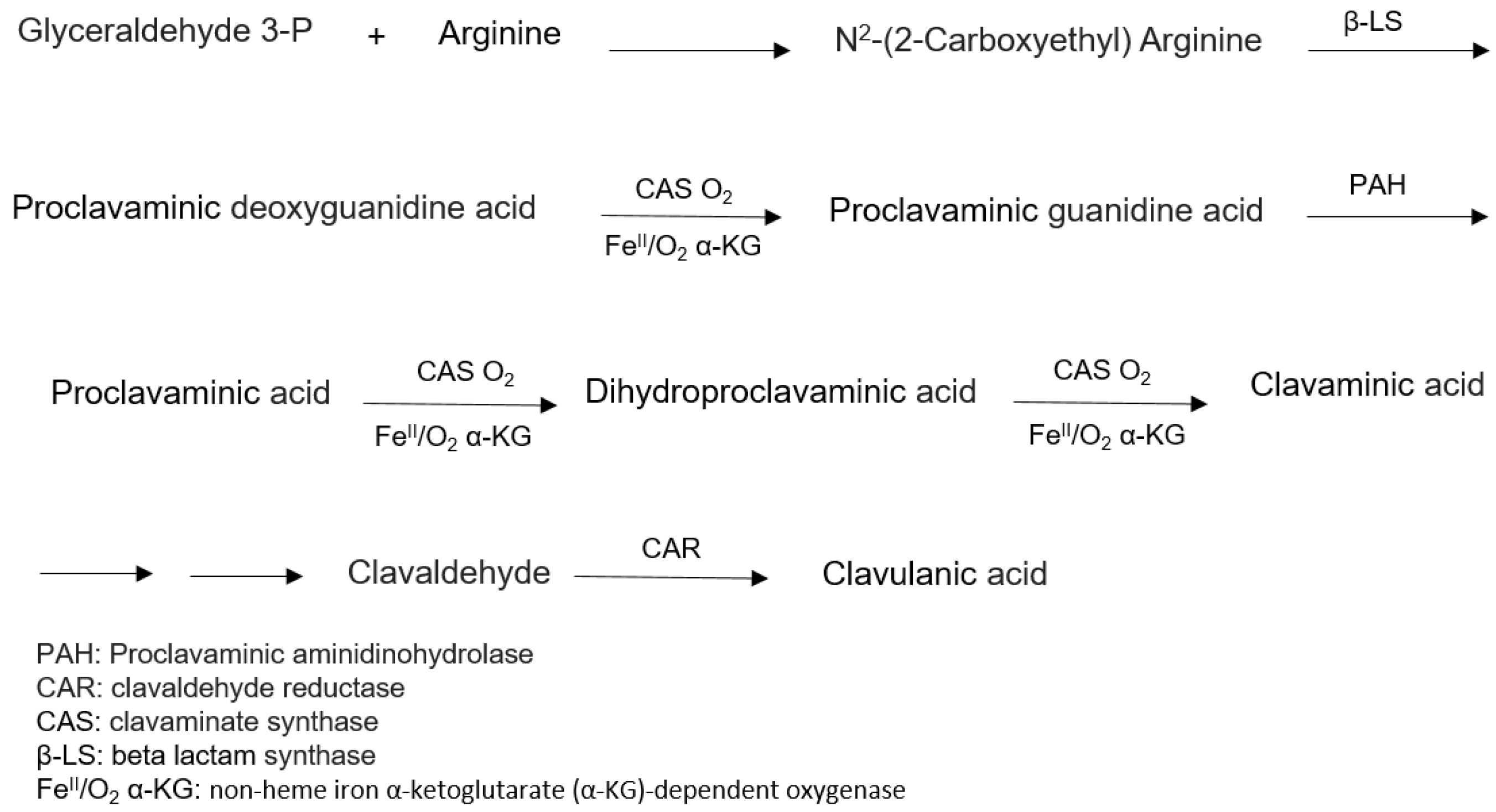
6.1. Biosynthesis of Clavulanic Acid
6.2. Production Process
6.2.1. Carbon Sources
6.2.2. Nitrogen Sources
6.2.3. Amino Acids as Supplements in Basal Medium
6.2.4. Salts in Basal Medium
6.2.5. Effect of pH
6.2.6. Extractive Fermentation of Clavulanic Acid
7. β-Lactamase Inhibitors in Clinical Practice
8. Commercial Use
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Procópio, R.E.; Silva, I.R.; Martins, M.K.; Azevedo, J.L.; Araújo, J.M. Antibiotics produced by Streptomyces. Braz. J. Infect. Dis. 2012, 16, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Radlinski, L.; Conlon, B.P. Antibiotic efficacy in the complex infection environment. Curr. Opin. Microbiol. 2018, 42, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.; Pal, S.; Sen, S. Anitibiotic production by Streptomyces hygroscopicus D1.5: Cultural effect. Rev. Microbiol. 1998, 29, 254–257. [Google Scholar] [CrossRef]
- Mohanraj, G.; Sekar, T. Isolation and screening of actinomycetes from marine sediments for their potential to produce antimicrobials. Int. J. Life Sci. Pharm. Res. 2013, 2, 115–126. [Google Scholar]
- Kirk, S.; Avogmpme-Rossa, C.A.; Bushell, M.E. Growth limiting substrate affects antibiotic production and associated metabolic flues in Streptomyces clavuligerus. Biotechnol. Lett. 2000, 22, 1803–1809. [Google Scholar] [CrossRef]
- Gouveia, E.R.; Baptista-Neto, A.; Badino, A.C., Jr.; Hokka, C.O. Optimisation of medium composition for clavulanic acid production by Streptomyces clavuligerus. Biotechnol. Lett. 2001, 23, 157–161. [Google Scholar] [CrossRef]
- Ortiz, S.C.A.; Hokka, C.O.; Badino, A.C., Jr. Utilization of soybean derivatives on clavulanic acid production by Streptomyces clavuligerus. Enzyme Microb. Technol. 2007, 40, 1071–1077. [Google Scholar] [CrossRef]
- Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Joslyn, D.A.; Burkholder, P.R. Chloromycetin, a new antibiotic from a soil actinomycete. Science 1947, 106, 417. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Kumar, R.; Gupta, S. Antibiotic production by microbes isolated from soil. Int. J. Pharm. Sci. Res. 2013, 4, 2967–2973. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harbor Perspect. Med. 2016, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.A. Extended-spectrum β-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 2001, 14, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.N.; Harper, P.B.; O’Callaghan, C.H. Principal beta–lactamases responsible for resistance to beta-lactam antibiotics in urinary tract infections. Antimicrob. Agents Chemother. 1980, 17, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Borodina, I.; Siebring, J.; Zhang, J.; Smith, C.P.; van Keulen, G.; Dijkhuizen, L.; Nielsen, J. Antibiotic Overproduction in Streptomyces coelicolor A3(2) Mediated by Phosphofructokinase Deletion. J. Biol. Chem. 2008, 283, 25186–25199. [Google Scholar] [CrossRef] [PubMed]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.P.; Chain, E. An enzyme from bacteria able to destroy penicillin. Rev. Infect. Dis. 1988, 10, 677–678. [Google Scholar] [CrossRef] [PubMed]
- Plough, H.H. Penicillin resistance of Staphylococcus aureus and its clinical implications. Am. J. Clin. Pathol. 1945, 15, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A., Jr.; Dietz, C.C. Penicillin resistant staphylococci. Proc. Soc. Exp. Biol. Med. 1945, 60, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, K.R. Studies on induced resistance to penicillin in pneumococcus type I. Acta Pathol. Microbiol. Scand. 1945, 22, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Gots, J.S. Production of extracellular penicillin-inactivating substances associated with penicillin resistance in Staphylococcus aureus. Proc. Soc. Exp. Biol. Med. 1945, 60, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Bohnhoff, M. Studies on the action of penicillin; development of penicillin resistance by gonococcus. Proc. Soc. Exp. Biol. Med. 1945, 60, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Waksman, S.A.; Reilly, H.C.; Schatz, A. Strain specificity and production of antibiotic substances: V. Strain resistance of bacteria to antibiotic substances, especially streptomycin. Proc. Natl. Acad. Sci. USA 1945, 31, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Usman, J.; Kaleem, F.; Omair, M.; Khalid, A.; Iqbal, M. Evaluation of different detection methods of biofilm formation in the clinical isolates. Braz. J. Infect. Dis. 2011, 15, 305–311. [Google Scholar] [CrossRef]
- Allen, H.K.; Donato, J.; Wang, H.H.; Cloud-Hansen, K.A.; Davies, J.; Handelsman, J. Call of the wild: Antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010, 8, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Petchiappan, A.; Chatterji, D. Antibiotic Resistance: Current Perspectives. ACS Omega 2017, 2, 7400–7409. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Microbiological effects of sublethal levels of antibiotics. Nat. Rev. Microbiol. 2014, 12, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, T.; Virta, M.; Laine, A.L. Antibiotic resistance in the wild: An eco-evolutionary perspective. Philos. Trans. R. Soc. B Biol. Sci. 2017, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Bignell, D.R.D.; Seipke, R.F.; Huguet-Tapia, J.C.; Chambers, A.H.; Parry, R.J.; Loria, R. Streptomyces scabies 87-22 contains a coronafacic acid-like biosynthetic cluster that contributes to plant–microbe interactions. Mol. Plant-Microbe Interact. 2010, 23, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P. Antibacterial discovery and development—The failure of success? Nat. Biotechnol. 2006, 24, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, H. Self-resistance in Streptomyces, with Special Reference to β-Lactam Antibiotics. Molecules 2016, 21, 605. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updat. 2018, 36, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Coulthurst, S.J.; Barnard, A.M.L.; Salmond, G.P.C. Regulation and biosynthesis of carbapenem antibiotics in bacteria. Nat. Rev. Microbiol. 2005, 3, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.C.; Pereira, J.F.B.; Haga, R.B.; Rangel-Yagui, C.O.; Teixeira, J.A.C.; Converti, A.; Pessoa, A., Jr. Stability of clavulanic acid under variable pH, ionic strength and temperature conditions. A new kinetic approach. Biochem. Eng. J. 2009, 45, 89–93. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, J. Beta-lactamase induction and cell wall metabolism in Gram-negative bacteria. Front. Microbiol. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.L. Changing patterns of infectious disease. Nature 2000, 406, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, 1–37. [Google Scholar]
- Wang, W.-J.; Wang, Q.; Zhang, Y.; Lu, R.; Zhang, Y.-L.; Yang, K.-W.; Lei, J.E. Characterization of β-lactamase activity using isothermal titration calorimetry. Biochim. Biophys. Acta 2017, 1861, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int. J. Antimicrob. 2015, 46, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Bonomo, R.A. New β-Lactamase inhibitors in the clinic. Infect. Dis. Clin. N. Am. 2016, 30, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A.; Medeiros, A. Functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Matsoso, P.; Pant, S.; Brower, C.; Røttingen, J.-A.; Klugman, K.; Davies, S. Access to effective antimicrobials: A worldwide challenge. Lancet 2016, 387, 168–175. [Google Scholar] [CrossRef]
- Bush, K. β-Lactamase inhibitors from laboratory to clinic. Clin. Microbiol. Rev. 1988, 1, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.S.C.; de Carvaiho Santos-Ebinuma, V.; Lopes, A.M.; de Oliveira Rangel-Yagui, C. Dextran sulfate/Triton X two-phase micellar systems as an alternative first purification step for clavulanic acid. Fluid Phase Equilib. 2015, 399, 80–86. [Google Scholar] [CrossRef]
- Akova, M. Sulbactam-containing b-lactamase inhibitor combinations. Clin. Microbiol. Infect. 2008, 14, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Jacqueline, C.; Howland, K.; Chesnel, L. In vitro activity of ceftolozane/tazobactam in combination with other classes of antibacterial agents. J. Glob. Antimicrob. Resist. 2017, 10, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Balci, C.; Uzun, O.; Arici, M.; Hayran, S.A.; Yuce, D.; Unal, S. Nephrotoxicity of piperacillin-tazobactam combined with vancomycin: Should it be a concern? Int. J. Antimicrob. Agents 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Fast, W.; Sutton, L.D. Metallo-β-lactamase: Inhibitors and reporter substrates. Biochim. Biophys. Acta 2013, 1834, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.; Boyd, A.; Ensor, G.J.; Evans, M.; Golden, M.; Linke, S.R.; Milne, D.; Murphy, R.; Telford, A.; Kalyan, Y.; et al. Development of a manufacturing route to avibactam, a β-lactamase inhibitor. Org. Process Res. Dev. 2016, 20, 1799–1805. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Ishikawa, J.; Hara, H.; Suzuki, H.; Ikenoya, M.; Ikeda, H.; Yamashita, A.; Hattori, M.; Horinouchi, S. Genome sequence of the streptomycin-producing microorganism Streptomyces griséus IFO 13350. J. Bacteriol. 2008, 190, 4050–4060. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, V.G.; Andreeva, I.P.; Rubtsova, M.Y.; Deygen, I.M.; Antipin, R.L.; Majouga, A.G.; Egorov, A.M.; Beshnova, D.A.; Kallio, J.; Hackenberg, C.; et al. Novel non-β-lactam inhibitor of b-lactamase TEM-171 based on acylated phenoxyaniline. Biochimie 2017, 132, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.M.; Distler, A.M.; Bonomo, R.A. Overcoming resistance to β-lactamase inhibitors: Comparing Sulbactam to novel inhibitors against clavulanate resistant SHV enzymes with substitutions at Ambler position 244. Biochemistry 2007, 46, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Saudagar, P.S.; Survase, S.A.; Singhal, R.S. Clavulanic acid: A review. Biotechnol. Adv. 2008, 26, 335–351. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.J.; Bycroft, B.W.; Salmond, G.P.C. Bacterial production of carbapenems and clavams: Evolution of β-lactam antibiotic pathways. Trends Microbiol. 1998, 6, 203–208. [Google Scholar] [CrossRef]
- Özcengiz, G.; Demain, A.L. Recent advances in the biosynthesis of penicillins, cephalosporins and clavams and its regulation. Biotechnol. Adv. 2013, 31, 287–311. [Google Scholar] [CrossRef] [PubMed]
- Hamed, R.B.; Gomez-Castellanos, J.R.; Henry, L.; Ducho, C.; McDonough, M.A.; Schofield, C.J. The enzymes of β-lactam biosynthesis. Nat. Prod. Rep. 2013, 30, 21–107. [Google Scholar] [CrossRef] [PubMed]
- Nobary, S.G.; Jensen, S.E. A comparison of the clavam biosynthetic gene clusters in Streptomyces antibioticus Tü1718 and Streptomyces clavuligerus. Can. J. Microbiol. 2012, 58, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Jensen, S.L.; Lee, K.J. Clavulanic acid biosynthesis and genetic manipulation for its overproduction. Appl. Microbiol. Biotechnol. 2010, 88, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Malule, H.; Restrepo, A.; Cardona, W.; Junne, S.; Neubauer, P.; Rios-Estepa, R. Inversion of the stereochemical configuration (3S,5S)-clavaminic acid into (3R,5R)-clavulanic acid: A computationally-assisted approach based on experimental evidence. J. Theor. Biol. 2016, 395, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.; Zhong, C.; Zong, G.; Fua, J.; Pang, X.; Cao, G. Improvement of clavulanic acid production in Streptomyces clavuligerus F613-1 by using a claR-neo reporter strategy. Electron. J. Biotechnol. 2017, 28, 41–46. [Google Scholar] [CrossRef]
- Ünsaldı, E.; Kurt-Kızıldogan, A.; Voigt, B.; Becher, D.; Ozcengiz, G. Proteome-wide alterations in an industrial clavulanic acid producing strain of Streptomyces clavuligerus. Synth. Syst. Biotechnol. 2017, 2, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, K.; Anders, C.; Wong, A.; Mosher, R.H.; Beatty, P.H.; Brumlik, M.J.; Griffin, A.; Hughes, C.; Griffin, J.; Barton, B.; et al. 5S clavam biosynthetic genes are located in both the clavam and paralog gene clusters in Streptomyces clavuligerus. Chem. Biol. 2007, 14, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.L.L.; Badino, A.C. Overproduction of clavulanic acid by extractive fermentation. Electron. J. Biotechnol. 2015, 18, 154–160. [Google Scholar] [CrossRef]
- Haga, R.B.; Santos-Ebinuma, V.C.; Silva, M.S.C.; Pessoa, A., Jr.; Rangel-Yagui, C.O. Clavulanic acid partitioning in charged aqueous two-phase micellar systems. Sep. Purif. Technol. 2013, 273–278. [Google Scholar] [CrossRef]
- Viana-Marques, D.A.; Santos-Ebinuma, V.C.; Pessoa, A., Jr.; Porto, A.L.; Torres, B.R.; Converti, A. Effect of aeration and agitation on extractive fermentation of clavulanic acid by using aqueous two-phase system. Biotechnol. Prog. 2016, 32, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.C.S.; Souza, A.T.; Badino, A.C.; Pedrolli, D.B.; Cerri, M.O. Screening of medium constituents for clavulanic acid production by Streptomyces clavuligerus. Braz. J. Microbiol 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.B.S.; Taviot-Guého, C.; Leroux, F.; Rodrigues, M.I.; Maugeri Filho, F. Clavulanic acid separation on fixed bed columns of layered double hydroxides: Optimization of operating parameters using breakthrough curves. Process Biochem. 2016, 51, 509–516. [Google Scholar] [CrossRef]
- Kumar, P.S.; Duraipandiyan, V.; Ignacimuthu, S. Isolation, screening and partial purification of antimicrobial antibiotics from soil Streptomyces sp. SCA 7. Kaohsiung J. Med. Sci. 2014, 30, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Charusanti, P.; Fong, N.L.; Nagarajan, H.; Pereira, A.R.; Li, H.J.; Abate, E.A.; Su, Y.; Gerwick, W.H.; Palsson, B.O. Exploiting adaptive laboratory evolution of streptomyces clavuligerus for antibiotic discovery and overproduction. PLoS ONE 2012, 7, e033727. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Haque, S.; Singh, H.; Verma, J.; Vibha, K.; Singh, R.; Jawed, A.; Tripathi, C.K.M. Isolation, screening, and identification of novel isolates of Actinomycetes from India for antimicrobial applications. Front. Microbiol. 2016, 7, 1921. [Google Scholar] [CrossRef] [PubMed]
- Chater, K.F. Recent advances in understanding Streptomyces. F1000Research 2016, 5, 2795. [Google Scholar] [CrossRef] [PubMed]
- Al husnan, L.A.; Alkahtani, M.D.F. Molecular Identification of Streptomyces producing antibiotics and their antimicrobial activities. Ann. Agric. Sci. 2016, 61, 251–255. [Google Scholar] [CrossRef]
- Weber, T.; Charusanti, P.; Musiol-Kroll, E.M.; Jiang, X.; Tong, Y.; Kim, H.U.; Lee, S.Y. Metabolic engineering of antibiotic factories: New tools for antibiotic production in actinomycetes. Trends Biotechnol. 2015, 33, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, C.; Duraipandiyan, V.; Arund, Y.; Sangeetha, B.; Emi, N.; Al-Dhabi, N.A.; Ignacimuthu, S.; Inaguma, Y.; Okamoto, A.; Perumal, P.T. Isolation and characterization of 2-hydroxy-9,10-anthraquinone from Streptomyces olivochromogenes (ERINLG-261) with antimicrobial and antiproliferative properties. Rev. Bras. Farmacogn. 2016, 26, 285–295. [Google Scholar] [CrossRef]
- Martínez-Burgo, Y.; Álvarez-Álvarez, R.; Pérez-Redondo, R.; Liras, P. Heterologous expression of Streptomyces clavuligerus ATCC 27064 cephamycin C gene cluster. J. Biotechnol. 2014, 186, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Bellão, C.; Antonio, T.; Araujo, M.L.G.C.; Badino, A.C. Production of clavulanic acid and cephamycin C by Streptomyces clavuligerus under different fed-batch conditions. Braz. J. Chem. Eng. 2013, 30, 257–266. [Google Scholar] [CrossRef]
- Cavallieri, A.P.; Baptista, A.S.; Leite, C.A.; Araujo, M.L.G.C. A case study in flux balance analysis: Lysine, a cephamycin C precursor, can also increase clavulanic acid production. Biochem. Eng. J. 2016, 112, 42–53. [Google Scholar] [CrossRef]
- Li, R.; Townsend, C.A. Rational strain improvement for enhanced clavulanic acid production by genetic engineering of the glycolytic pathway in Streptomyces clavuligerus. Metab. Eng. 2006, 8, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Olano, C.; Lombó, F.; Méndez, C.; Salas, J.A. Improving production of bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab. Eng. 2008, 10, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Adrio, J.L.; Demain, A.L. Recombinant organisms for production of industrial products. Bioeng. Bugs 2010, 1, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Hirata, D.B.; Oliveira, J.H.H.L.; Leão, K.V.; Rodrigues, M.I.; Ferreira, A.G.; Giulietti, M.; Barboza, M.; Hokka, C.O. Precipitation of clavulanic acid from fermentation broth with potassium 2-ethyl hexanoate salt. Sep. Purif. Technol. 2009, 66, 598–605. [Google Scholar] [CrossRef]
- Higgens, C.E.; Kastner, R.E. Streptomyces clavuligerus sp. nova b-lactam antibiotic producer. Int. J. Syst. Evol. Microbiol. 1971, 21, 326–331. [Google Scholar] [CrossRef]
- Brown, A.G.; Butterworth, D.; Cole, M.; Hanscomb, G.; Hood, J.D.; Reading, C.; Rolinson, G.N. Naturally occurring beta-lactamase inhibitors with antibacterial activity. J. Antibiot. 1976, 29, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.R. Optimization and scale up of industrial fermentation processes. Appl. Microbiol. Biotechnol. 2005, 68, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.H.L.; Granato, A.C.; Hirata, D.B.; Hokka, C.O.; Barboza, M. Clavulanic acid and cephamicin C: A perspective of the biosynthesis, isolation and action mechanism. Quim. Nova 2009, 32, 2142–2150. [Google Scholar] [CrossRef]
- Townsend, C.A. New reactions in clavulanic acid biosynthesis. Curr Opin Chem Biol. 2002, 6, 583–589. [Google Scholar] [CrossRef]
- Ser, H.L.; Law, J.W.F.; Chaiyakunapruk, N.; Jacob, S.A.; Palanisamy, U.D.; Chan, K.G.; Goh, B.H.; Lee, L.H. Fermentation conditions that affect clavulanic acid production in Streptomyces clavuligerus: A systematic review. Front. Microbiol. 2016, 7, 522. [Google Scholar] [CrossRef] [PubMed]
- Domingues, L.C.G.; Teodoro, J.C.; Hokka, C.O.; Badino, A.C.; Araujo, M.L.G.C. Optimisation of the glycerol-to-ornithine molar ratio in the feed medium for the continuous production of clavulanic acid by Streptomyces clavuligerus. Biochem. Eng. J. 2010, 53, 7–11. [Google Scholar] [CrossRef]
- Saudagar, P.S.; Singhal, R.S. Optimization of nutritional requirements and feeding strategies for clavulanic acid production by Streptomyces clavuligerus. Bioresour. Technol. 2007, 98, 2010–2017. [Google Scholar] [CrossRef] [PubMed]
- Viana, D.A.; Carneiro-Cunha, M.N.; Araújo, J.M.; Barros-Neto, B.; Lima-Filho, J.L.; Converti, A.; Pessoa-Júnior, A.; Porto, A.L.F. Screening of variables influencing the clavulanic acid production by Streptomyces DAUFPE 3060 strain. Appl. Biochem. Biotechnol. 2010, 160, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.A.V.; Carneiro da Cunha, M.N.; Araújo, J.M.; Lima-Filho, J.L.; Converti, A.; Pessoa, A., Jr.; Porto, A.L.F. Optimization of clavulanic acid production by Streptomyces DAUFPE 3060 by response surface methodology. Braz. J. Microbiol. 2011, 42, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.L.L.; Badino, A.C. Production of clavulanic acid by Streptomyces clavuligerus in batch cultures without and with glycerol pulses under different temperature conditions. Biochem. Eng. J. 2012, 69, 1–7. [Google Scholar] [CrossRef]
- Marques, D.A.V.; Santos-Ebinuma, V.C.; Oliveira, P.M.S.; Lima, G.M.S.; Araújo, J.M.; Lima-Filho, J.L.; Converti, A.; Pessoa-Júnior, A.; Porto, A.L.F. Screening of wild type Streptomyces isolates able to overproduce clavulanic acid. Braz. J. Microbiol. 2014, 45, 919–928. [Google Scholar] [CrossRef]
- Saudagar, P.S.; Singhal, R.S. A statistical approach using L25 orthogonal array method to study fermentative production of clavulanic acid by Streptomyces clavuligerus MTCC1142. Appl. Biochem. Biotechnol. 2007, 136, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Salem-Berkhit, M.M.; Alanazi, F.K.; Alsarra, I.A. Improvement and enhancement of clavulanic acid production in Streptomyces clavuligerus using vegetable oils. Afr. J. Biotechnol. 2010, 9, 6806–6812. [Google Scholar]
- Chen, K.C.; Lin, Y.H.; Tsai, C.M.; Hsieh, C.H.; Houng, J.Y. Optimization of glycerol feeding for clavulanic acid production by Streptomyces clavuligerus with glycerol feeding. Biotechnol. Lett. 2002, 24, 455–458. [Google Scholar] [CrossRef]
- Thakur, R.; Roy, M.K.; Dutta, N.N.; Bezbaruah, R.L. Coordinate production of cephamycin c and clavulanic acid by Streptomyces clavuligerus. Indian J. Exp. Biol. 1999, 37, 1031–1033. [Google Scholar] [PubMed]
- Teodoro, J.C.; Baptista-Neto, A.; Araujo, M.L.G.D.C.; Hokka, C.O.; Badino, A.C. Influence of glycerol and ornithine feeding on clavulanic acid production by Streptomyces clavuligerus. Braz. J. Chem. Eng. 2010, 27, 499–506. [Google Scholar] [CrossRef]
- Vasconcelos, E.S.; Lima, V.A.; Goto, L.S.; Cruz-Hernández, I.L.; Hokka, C.O. Clavulanic acid production by the MMS 150 mutant obtained from wild type Streptomyces clavuligerus ATCC 27064. Braz. J. Microbiol. 2014, 44, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Baggaley, K.H.; Brown, A.G.; Schofield, C.J. Chemistry and biosynthesis of clavulanic acid and other clavams. Nat. Prod. Rep. 1997, 14, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Ives, P.R.; Bushell, M.E. Manipulation of the physiology of clavulanic acid production in Streptomyces clavuligerus. Microbiology 1997, 143, 3573–3579. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Yang, B.; Ren, J.; Dong, M.L.; Liang, D.; Xu, A.L. Optimization of medium composition for the production of clavulanic acid by Streptomyces clavuligerus. Process Biochem. 2005, 40, 1161–1166. [Google Scholar] [CrossRef]
- Teodoro, J.C.; Baptista-Neto, A.; Cruz-Hernandez, I.L.; Hokka, C.O.; Badino, A.C. Influence of feeding conditions on clavulanic acid production in fed-batch cultivation with medium containing glycerol. Appl. Microbiol. Biotechnol. 2006, 72, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Liras, P.; Martin, J.F. Dissociation of cephamycin and clavulanic acid biosynthesis in Streptomyces clavuligerus. Appl. Microbiol. Biotechnol. 1984, 20, 318–325. [Google Scholar] [CrossRef]
- Lee, P.C.; Ho, C.C. Production of clavulanic acid and cephamycin C by Streptomyces clavuligerus in palm-oil medium. World J. Microbiol. Biotechnol. 1996, 12, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Large, K.P.; Ison, A.P.; Williams, D.J. The effect of agitation rate on lipid utilisation and clavulanic acid production in Streptomyces clavuligerus. J. Biotechnol. 1998, 63, 111–119. [Google Scholar] [CrossRef]
- Maranesi, G.L.; Baptista-Neto, A.; Hokka, C.O.; Badino, A.C. Utilization of vegetable oil in the production of clavulanic acid by Streptomyces clavuligerus ATCC 27064. World J. Microbiol. Biotechnol. 2005, 21, 509–514. [Google Scholar] [CrossRef]
- Efthimiou, G.; Thumser, A.E.; Avignone−Rossa, C.A. A novel finding that Streptomyces clavuligerus can produce the antibiotic clavulanic acid using olive oil as a sole carbon source. J. Appl. Microbiol. 2008, 105, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.O.; Shin, C.H.; Park, H.W.; Kim, C.W. An approach to strain improvement and enhanced production of clavulanic acid in Streptomyces clavuligerus. Biosci. Biotech. Biochem. 2009, 73, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Stowell, J.D. The application of oils and fats in antibiotic processes. In Carbon Substrates in Biotechnology; Stowell, J.D., Beardsmore, A.J., Keevil, C.W., Woodward, J.R., Eds.; IRL Press: Oxford, UK, 1987; pp. 139–159. [Google Scholar]
- Mayer, A.F.; Deckwer, W.D. Simultaneous production and decomposition of clavulanic acid during Streptomyces clavuligerus cultivations. Appl. Microbiol. Biotechnol. 1996, 45, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.S.; Chang, C.S. Production of protease from a new alkalophilic Bacillus sp. I-312 grow on soybean meal: Optimization and some properties. Process Biochem. 2005, 40, 1263–1270. [Google Scholar] [CrossRef]
- Romero, J.; Liras, P.; Martín, J.F. Utilization of ornithine and arginine as specific precursors of clavulanic acid. Appl. Environ. Microbiol. 1986, 52, 892–897. [Google Scholar] [PubMed]
- Romero, J.; Liras, P.; Martín, J.F. Isolation and biochemical characterization of Streptomyces clavuligerus mutants in the biosynthesis of clavulanic acid and cephamycin C. Appl. Microbiol. Biotechnol. 1988, 27, 510–516. [Google Scholar] [CrossRef]
- Townsend, C.A.; Ho, M.F. Biosynthesis of clavulanic acid: Origin of C3 unit. J. Am. Chem. Soc. 1985, 107, 1065–1066. [Google Scholar] [CrossRef]
- De la Fuente, J.L.; Martin, J.F.; Liras, P. New type of hexameric ornithine carbamoyltransferase with arginase activity in the cephamycin producers Streptomyces clavuligerus and Nocardia lactamdurans. Biochem. J. 1996, 15, 173–179. [Google Scholar]
- Valentine, B.P.; Bailey, C.R.; Doherty, A.; Morris, J.; Elson, S.W.; Baggaley, K.H.; Nicholson, N.H. Evidence that arginine is a later metabolic intermediate than ornithine in the biosynthesis of clavulanic acid by Streptomyces clavuligerus. Chem. Soc. Chem. Commun. 1993, 15, 1210–1211. [Google Scholar] [CrossRef]
- Chen, K.C.; Lin, Y.H.; Wu, J.Y.; Hwang, S.C.J. Enhancement of clavulanic acid production in Streptomyces clavuligerus with ornithine feeding. Enzyme Microb. Technol. 2003, 32, 152–156. [Google Scholar] [CrossRef]
- Barboza, M.; Almeida, R.M.; Hokka, C.O. Kinetic studies on clavulanic acid recovery by ion exchange chromatography. Bioseparation 2001, 10, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Albertsson, P.A. Partition of Cell Particles and Macromolecules, 3rd ed.; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Lima, C.A.; Freitas, A.C.V., Jr.; Lima Filho, J.L.; Converti, A.; Viana Marques, D.A.; Carneiro-da-Cunha, M.G.; Porto, A.L.F. Two-phase partitioning and partial characterization of a collagenase from Penicillium aurantiogriseum URM4622: Application to collagen hydrolysis. Biochem. Eng. J. 2013, 75, 64–71. [Google Scholar] [CrossRef]
- Saravanan, S.; Rao, J.R.; Nair, B.U.; Ramasami, T. Aqueous two-phase poly(ethylene glycol)–poly(acrylic acid) system for protein partitioning: Influence of molecular weight, pH and temperature. Process Biochem. 2008, 43, 905–911. [Google Scholar] [CrossRef]
- Viana Marques, D.A.; Pessoa-Júnior, A.; Lima-Filho, J.L.; Converti, A.; Perego, P.; Porto, A.L.F. Extractive fermentation of clavulanic acid by Streptomyces DAUFPE 3060 using aqueous two-phase system. Biotechnol. Prog. 2011, 27, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.S.; Cuel, M.F.; Barreto, V.O.; Kwong, W.H.; Hokka, C.O.; Barboza, M. Separation of clavulanic acid from fermented broth of aminoacids by an aqueous two-phase system and ion-exchange adsorption. New Biotechnol. 2012, 29, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Dong, J.; Yang, S.T. Butanol production from wood pulping hydrolysate in na integrated fermentation-gas stripping process. Bioresour. Technol. 2013, 143, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Park, H.R.; Park, D.J.; Lee, H.B.; Kim, Y.B.; Kim, C.J. Improved production of teicoplanin using adsorbent resin in fermentations. Lett. Appl. Microbiol. 2003, 37, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Bersanetti, P.A.; Almeida, R.M.; Barboza, M.; Araújo, M.L.G.; Hokka, C.O. Kinetic studies on clavulanic acid degradation. Biochem. Eng. J. 2005, 23, 31–36. [Google Scholar] [CrossRef]
- Panas, P.; Lopes, C.; Cerri, M.O.; Ventura, S.P.; Santos-Ebinuma, V.C.; Pereira, J.F. Purification of clavulanic acid produced by Streptomyces clavuligerus via submerged fermentation using polyethylene glycol/cholinium chloride aqueous two-phase systems. Fluid Phase Equilib. 2017, 450, 42–50. [Google Scholar] [CrossRef]
- Smith, T.; Wolff, K.A.; Nguyen, L. Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 2013, 374, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T. Inhibiting Mycobacterium tuberculosis within and without. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wlodarchak, N.; Teachout, N.; Procknow, R.; Beczkiewicz, J.; Schaenzer, A.; Satyshur, K.; Pavelka, M.; Zuercher, B.; Drewry, D.; Sauer, J.D.; et al. Repurposed kinase inhibitors and β-lactams as a novel therapy for antibiotic resistant. BioRxiv 2017. [Google Scholar] [CrossRef]
- Diacon, A.H.; van der Merwe, L.; Barnard, M.; von Groote-Bidlingmaier, F.; Lange, C.; Garcia-Basteiro, A.L.; Sevene, E.; Ballell, L.; Barros-Aguirre, D. Beta-Lactams against tuberculosis—New trick for an old dog? N. Engl. J. Med. 2016, 375, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Ramon-Garcia, S.; Gonzalez Del Rio, R.; Villarejo, A.S.; Sweet, G.D.; Cunningham, F.; Barros, D.; Ballell, L.; Mendoza-Losana, A.; Ferrer-Bazaga, S.; Thompson, C.J. Repurposing clinically approved cephalosporins for tuberculosis therapy. Sci. Rep. 2016, 6, 34293. [Google Scholar] [CrossRef] [PubMed]
- Veziris, N.; Truffot, C.; Mainardi, J.L.; Jarlier, V. Activity of carbapenems combined with clavulanate against murine tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2597–2600. [Google Scholar] [CrossRef] [PubMed]
- Nitsch-Osuch, A.; Kuchar, E.; Zycinska, K.; Gyrczuk, E.; Miskiewicz, K.; Korzeniewski, K. Implementation of hospital’s antibiotic policy decreases antimicrobial use in the general pediatric ward. Adv. Exp. Med. Biol. 2015, 857, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.; Go, E.; Mariano, N.; Berger, B.J.; Avraham, I.; Rubin, D.; Rahal, J.J. Effect of sulbactam on infections caused by imipenem-resistant Acinetobacter calcoaceticus biotype anitratus. J. Infect. Dis. 1993, 167, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Poustchi, S.; Cunningham, D.; Toscani, M.; Nguyen, J.; Lim, J.; Ding, Y.; Nahass, R.G. Clinical and economic impact of empirical extended-infusion piperacillin–Tazobactam in a community medical center. Ann. Pharmacother. 2015, 49, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.J.; Flamm, R.K.; Sader, H.S.; Jones, R.N. Antimicrobial activity of ceftolozane–Tazobactam tested against Enterobacteriaceae and Pseudomonas aeruginosa with various resistance patterns isolated in U.S. hospitals (2011–2012). Antimicrob. Agents Chemother. 2013, 57, 6305–6310. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.A.; Nicolau, D.P. Susceptibility profile of ceftolozane/tazobactam and other parenteral antimicrobials against Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa from US hospitals. Clin. Ther. 2015, 37, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Farrell, D.J.; Castanheira, M.; Flamm, R.K.; Jones, R.N. Antimicrobial activity of ceftolozane/tazobactam tested against Pseudomonas aeruginosa and Enterobacteriaceae with various resistance patterns isolated in European hospitals (2011–12). J. Antimicrob. Chemother. 2014, 69, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Estabrook, M.; Bussell, B.; Clugston, S.L.; Bush, K. In vitro activity of ceftolozane-tazobactam as determined by broth dilution and agar diffusion assays against recent U.S. Escherichia coli isolates from 2010 to 2011 carrying CTX-M-type extended-spectrum β-lactamases. J. Clin. Microbiol. 2014, 52, 4049–4052. [Google Scholar] [PubMed]
- Solomkin, J.; Hershberger, E.; Miller, B.; Popejoy, M.; Friedland, I.; Steenbergen, J.; Yoon, M.; Collins, S.; Yuan, G.; Barie, P.S.; et al. Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: Results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin. Infect. Dis. 2015, 60, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Gautam, V.; Singhal, L.; Ray, P. Antimicrobial activity of cefepime-tazobactam combination tested against clinical isolates of Enterobacteriaceae. J. Antibiot. 2014, 67, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Mangani, S.; Jahic, H.; Benvenuti, M.; Durand-Reville, T.F.; De Luca, F.; Ehmann, D.E.; Rossolini, G.M.; Alm, R.A.; et al. Molecular basis of selective inhibition and slow reversibility of avibactam against class D carbapenemases: A structure-guided study of OXA-24 and OXA-48. ACS Chem. Biol. 2015, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Estabrook, M.; Jacoby, G.A.; Nichols, W.W.; Testa, R.T.; Bush, K. In vitro susceptibility of characterized β-lactamase-producing strains tested with avibactam combinations. Antimicrob. Agents Chemother. 2015, 59, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, P.; Girard, A.M.; Miossec, C.; Pace, J.; Coleman, K. In vitro antibacterial activity of the ceftazidime–avibactam combination against Enterobacteriaceae, including strains with well characterized β-lactamases. Antimicrob. Agents Chemother. 2015, 59, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Mills, J.C.; Costello, S.E.; Jones, R.N.; Sader, H.S. Ceftazidime-avibactam activity tested against Enterobacteriaceae isolates from U.S. hospitals (2011 to 2013) and characterization of β-lactamase-producing strains. Antimicrob. Agents Chemother. 2015, 59, 3509–3517. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, J.; Melchers, M.J.; van Mil, A.C.; Nichols, W.W.; Mouton, J.W. In vitro activity of ceftazidime-avibactam combination in in vitro checkerboard assays. Antimicrob. Agents Chemother. 2015, 59, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, F.; Zhao, C.; Wang, Z.; Nichols, W.W.; Testa, R.; Li, H.; Chen, H.; He, W.; Wang, Q.; et al. In vitro activities of ceftazidime-avibactam and aztreonam-avibactam against 372 Gram-negative bacilli collected in 2011 and 2012 from 11 teaching hospitals in China. Antimicrob. Agents Chemother. 2014, 58, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Flamm, R.K.; Farrell, D.J.; Jones, R.N. Antimicrobial activity of ceftazidime-avibactam against Gram-negative organisms collected from U.S. medical centers in 2012. Antimicrob. Agents Chemother. 2014, 58, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Sader, H.S.; Farrell, D.J.; Jones, R.N. Ceftazidime-avibactam and comparator agents tested against urinary tract isolates from a global surveillance program (2011). Diagn. Microbiol. Infect. Dis. 2014, 80, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Farrell, D.J.; Sader, H.S.; Jones, R.N. Ceftazidime/avibactam activity tested against Gram-negative bacteria isolated from bloodstream, pneumonia, intra-abdominal and urinary tract infections in US medical centres (2012). J. Antimicrob. Chemother. 2014, 69, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Flamm, R.K.; Mendes, R.E.; Farrell, D.J.; Jones, R.N. Ceftazidime/avibactam tested against Gram-negative bacteria from intensive care unit (ICU) and non-ICU patients, including those with ventilator-associated pneumonia. Int. J. Antimicrob. Agents 2015, 46, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Castanheira, M.; Mendes, R.E.; Flamm, R.K.; Farrell, D.J.; Jones, R.N. Ceftazidime-avibactam activity against multidrug-resistant Pseudomonas aeruginosa isolated in U.S. medical centers in 2012 and 2013. Antimicrob. Agents Chemother. 2015, 59, 3656–3659. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Canton, R.; Giani, T.; Morosini, M.I.; Nichols, W.W.; Seifert, H.; Stefanik, D.; Rossolini, G.M.; Nordmann, P. In vitro activity of ceftazidime, ceftaroline and aztreonam alone and in combination with avibactam against European Gram-negative and Gram-positive clinical isolates. Int. J. Antimicrob. Agents 2015, 45, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Biedenbach, D.J.; Kazmierczak, K.; Bouchillon, S.K.; Sahm, D.F.; Bradford, P.A. In vitro activity of aztreonam–avibactam against a global collection of Gram-negative pathogens from 2012 and 2013. Antimicrob. Agents Chemother. 2015, 59, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Zhang, J.; Maharjan, S.; Doumith, M.; Woodford, N. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2011, 55, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Sader, H.S.; Farrell, D.J.; Flamm, R.K.; Jones, R.N. Activity of ceftaroline and comparator agents tested against Staphylococcus aureus from patients with bloodstream infections in US medical centres (2009–2013). J. Antimicrob. Chemother. 2015, 70, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Farrell, D.J.; Sader, H.S.; Jones, R.N. Antimicrobial activity of ceftaroline combined with avibactam tested against bacterial organisms isolated from acute bacterial skin and skin structure infections in United States medical centers (2010–2012). Diagn. Microbiol. Infect. Dis. 2014, 78, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Blizzard, T.A.; Chen, H.; Kim, S.; Wu, J.; Bodner, R.; Gude, C.; Imbriglio, J.; Young, K.; Park, Y.W.; Ogawa, A.; et al. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin®. Bioorg. Med. Chem. Lett. 2014, 24, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Morinaka, A.; Tsutsumi, Y.; Yamada, M.; Suzuki, K.; Watanabe, T.; Abe, T.; Furuuchi, T.; Inamura, S.; Sakamaki, Y.; Mitsuhashi, N.; et al. OP0595, a new diazabicyclooctane: Mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam ‘enhancer’. J. Antimicrob. Chemother. 2015, 70, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Quale, J.; Landman, D. Activity of meropenem combined with RPX7009, a novel β-lactamase inhibitor, against Gram-negative clinical isolates in New York City. Antimicrob. Agents Chemother. 2015, 59, 4856–4860. [Google Scholar] [CrossRef] [PubMed]
- Crandon, J.L.; Nicolau, D.P. Comparative potency of cefepime (FEP) and cefepime/AAI101 (FEP/AAI101) against highly resistant Enterobacteriaceae. In Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), Washington, DC, USA, 5–9 September 2014. [Google Scholar]
- Crandon, J.L.; Nicolau, D.P. In vivo activities of simulated human doses of cefepime and cefepime–AAI101 against multidrug-resistant Gram-negative Enterobacteriaceae. Antimicrob. Agents Chemother. 2015, 59, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Chekan, J.R.; Cogan, D.P.; Nair, S.K. Molecular basis for resistance against phosphonate antibiotics and herbicides. MedChemComm 2016, 7, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Sogi, M.K.; Lien, K.A.; Johnson, J.R.; Krogan, N.J.; Stanley, S.A. The tyrosine kinase inhibitor Gefitinib restricts Mycobacterium tuberculosis growth through increased lysosomal biogenesis and modulation of cytokine signaling. ACS Infect. Dis. 2017, 3, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Kapur, R. Kinase inhibitors in clinical practice: An expanding world. J. Allergy Clin. Immunol. 2017, 141, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.C.S.; Franken, L.; Voller, S.; Rentmeister, H.; Grosch, B. Breaking through the Wall: A Call for Concerted Action on Antibiotics Research and Development; The Boston Consulting Group: Berlin, Germany, 2017. [Google Scholar]
- The Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; The Review on Antimicrobial Resistance: London, UK, 2014; pp. 1–20. [Google Scholar]
- Bush, K. Antibacterial drug discovery in the 21st century. Clin. Microbiol. Infect. 2004, 10, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Projan, S.J. Why is big pharma getting out of antibacterial drug discovery? Drug Discov. Today 2003, 6, 427–430. [Google Scholar] [CrossRef]
- Boggs, A.F.; Miller, G.H. Antibacterial drug discovery: Is small pharma the solution? Clin. Microbiol. Infect. 2004, 10, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, J.S.; Towse, A.; Mestre-Ferrandiz, J. Incentives for New Drugs to Tackle Anti-Microbial Resistance; Office of Health Economics: London, UK, 2017. [Google Scholar]
- The Pew Charitable Trusts. Antibiotics Currently in Clinical Development; The Pew Charitable Trusts: Philadelphia, PA, USA, 2017. [Google Scholar]
- Simpkin, V.L.; Renwick, M.J.; Kelly, R.; Mossialos, E. Incentivising innovation in antibiotic drug discovery and development: Progress, challenges and next steps. J. Antibiot. 2017, 70, 1087–1096. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Group According Bush, Jacoby, and Medeiros * | Ambler Class * | Hydrolyzed β-Lactam Antibiotics | |
|---|---|---|---|
| 1 | C | Cephalosporins, benzylpenicillin, cephamycins | |
| 1e | C | Ceftazidime and often other oxyimino-β-lactams | |
| 2 | 2a | A | Benzylpenicillin |
| 2b | A | Benzylpenicillin and cephalosporins | |
| 2be | A | Cefotaxime, ceftazidime, ceftriaxone, cefepime, aztreonam | |
| 2br | A | Resistance to clavulanic acid, sulbactam, and tazobactam | |
| 2ber | A | Oxyimino-β-lactams combined with resistance to clavulanic acid, sulbactam, and tazobactam | |
| 2c | A | Carbenicillin | |
| 2ce | A | Carbenicillin, cefepime, and cefpirome | |
| 2d | D | Cloxacillin or oxacillin | |
| 2de | D | Cloxacillin or oxacillin, and oxyimino-β-lactams | |
| 2df | D | Cloxacillin or oxacillin and carbapenems | |
| 2e | A | Cephalosporins inhibited by clavulanic acid | |
| 2f | A | Carbapenems, oxyimino-β-lactams, cephamycins | |
| 3 | B | Carbapenems | |
| 4 | Unknown | Carbapenems | |
| Genetic Method | Secondary Metabolite |
|---|---|
| Protoplast fusion | Penicillin G, cephalosporin C, cephamycin C, clavulanic acid, indolizomycin, rifamycins |
| Metabolic engineering | Antibiotics (penicillin G, cephalosporin C, cephamycin C, clavulanic acid, semisynthetic cephalosporins) |
| Transposition | Daptomycin, tylosin |
| Combinatorial biosynthesis | Erythromycins, tetracenomycins, tylosin, spiramycins |
| Genome mining | Echinosporamicin-type antibiotics, antifungal compounds (ECO-02301), and so on |
| β–Lactamase Inhibitor | β–Lactam Antibiotic | Development Status | References |
|---|---|---|---|
| Clavulanic acid | Amoxicillin | Approved by FDA, EMA | [139] |
| Sulbactam | Ampicillin | Approved by FDA, EMA | [140] |
| ETX2514 | Phase 1 trials completed in 2017 | [34] | |
| Tazobactam | Piperacillin | Approved by FDA, EMA | [141] |
| Ceftolozane | Approved in 2014 by FDA and in 2015 by EMA | [34,142,143,144,145,146] | |
| Cefepime | Used in Asia | [147] | |
| Avibactam | Ceftazidime | Approved in 2015 by FDA and in 2016 by EMA | [34,148,149,150,151,152,153,154,155,156,157,158,159] |
| Aztreonam | Phase 2 in progress | [149,153,160,161] | |
| Ceftaroline | Phase 2 in progress | [149,159,162,163] | |
| Relebactam (MK7655) | Imipenem (+ cilastatin) | Phase 3, cUTI (in progress) Phase 2 cIAI (completed) | [164,165] |
| Vaborbactam (RPX7009) | Meropenem | Approved in 2017 by FDA | [34,166] |
| AAI101 | Cefepime | Phase 2 initiated in 2017 | [34,167,168] |
| RG6080 (OP0595, FPI-1459) | Unknown | Phase 1 complete | [165] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viana Marques, D.D.A.; Machado, S.E.F.; Ebinuma, V.C.S.; Duarte, C.D.A.L.; Converti, A.; Porto, A.L.F. Production of β-Lactamase Inhibitors by Streptomyces Species. Antibiotics 2018, 7, 61. https://doi.org/10.3390/antibiotics7030061
Viana Marques DDA, Machado SEF, Ebinuma VCS, Duarte CDAL, Converti A, Porto ALF. Production of β-Lactamase Inhibitors by Streptomyces Species. Antibiotics. 2018; 7(3):61. https://doi.org/10.3390/antibiotics7030061
Chicago/Turabian StyleViana Marques, Daniela De Araújo, Suellen Emilliany Feitosa Machado, Valéria Carvalho Santos Ebinuma, Carolina De Albuquerque Lima Duarte, Attilio Converti, and Ana Lúcia Figueiredo Porto. 2018. "Production of β-Lactamase Inhibitors by Streptomyces Species" Antibiotics 7, no. 3: 61. https://doi.org/10.3390/antibiotics7030061
APA StyleViana Marques, D. D. A., Machado, S. E. F., Ebinuma, V. C. S., Duarte, C. D. A. L., Converti, A., & Porto, A. L. F. (2018). Production of β-Lactamase Inhibitors by Streptomyces Species. Antibiotics, 7(3), 61. https://doi.org/10.3390/antibiotics7030061