Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities



Abstract
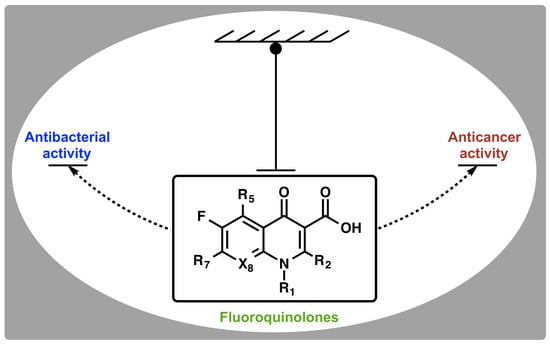
1. Introduction
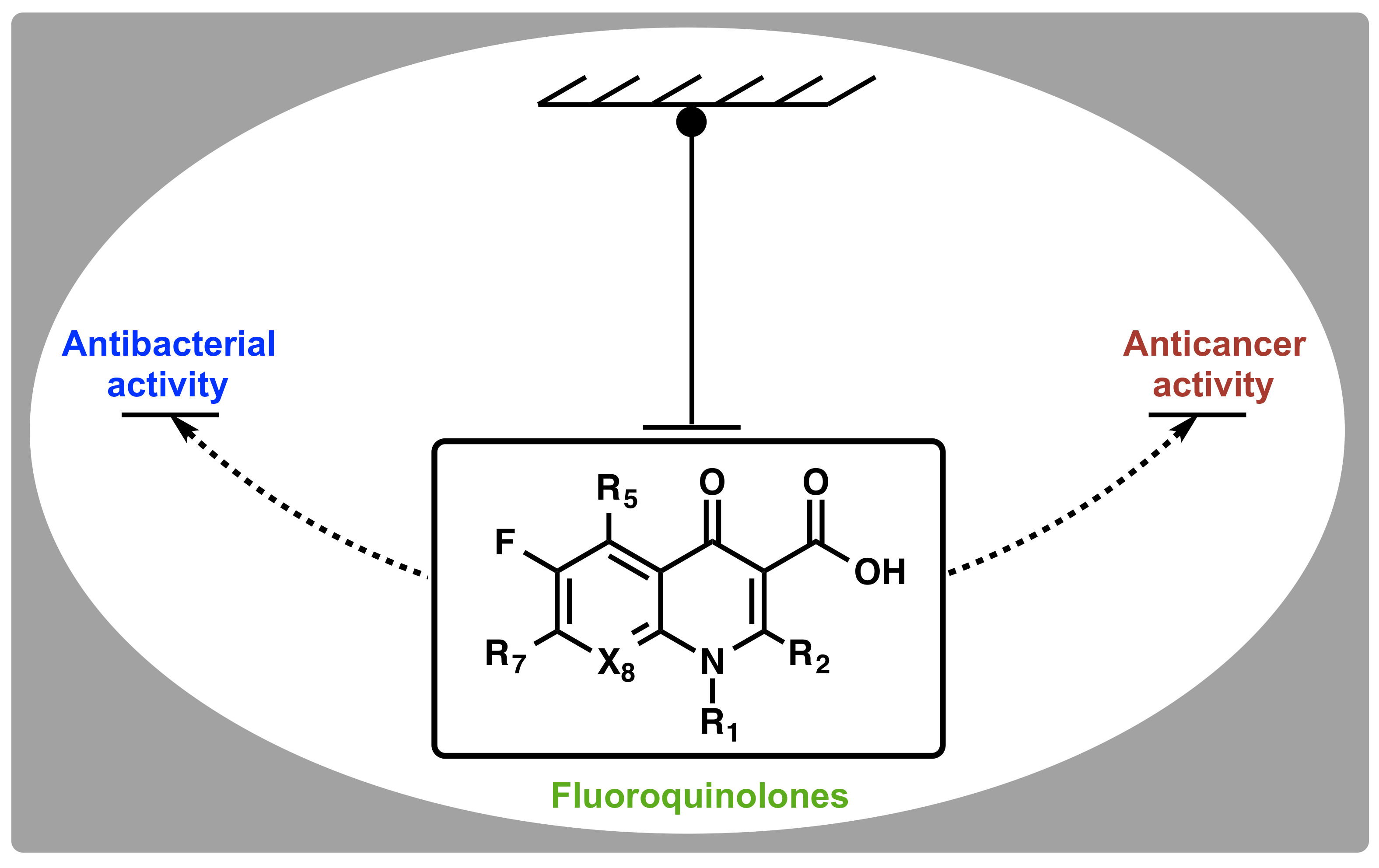
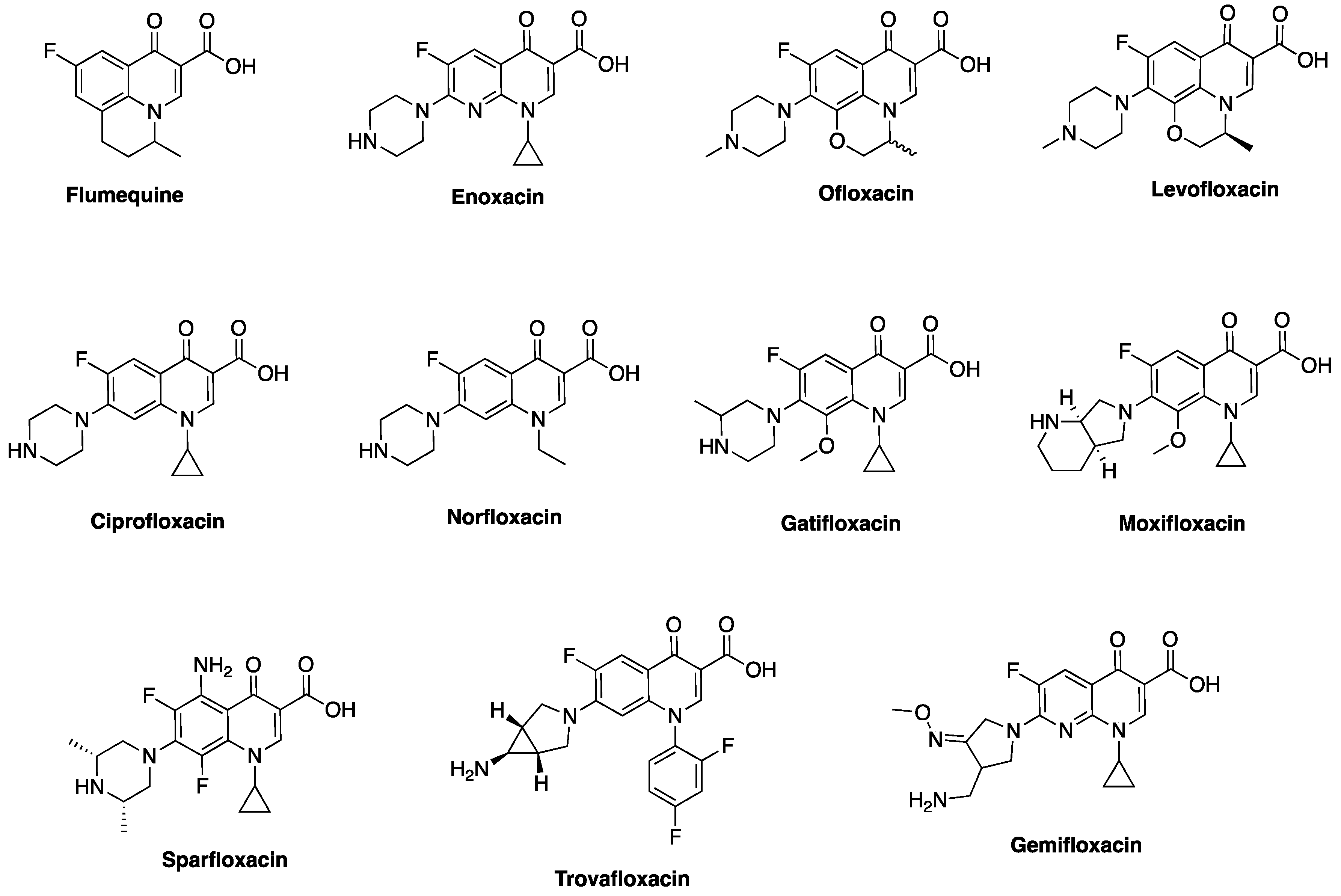
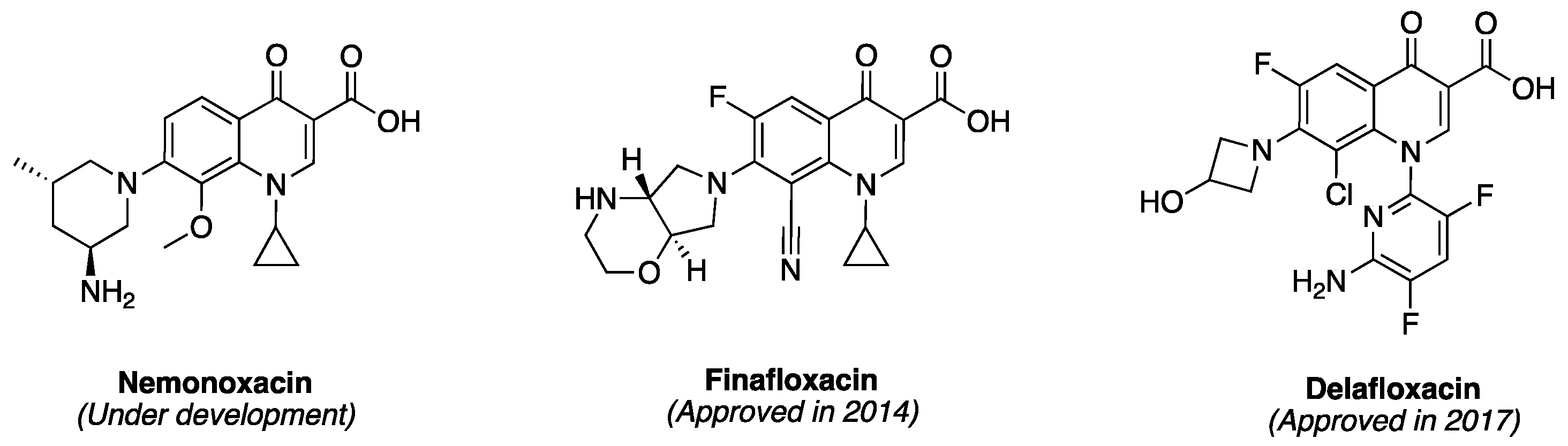
2. Discovery and Development of Fluoroquinolones
3. Classical Antimicrobial Activity of Fluoroquinolones
4. Non-Classical Antiproliferative Activity of Fluoroquinolones
4.1. Ciprofloxacin
4.2. Enoxacin
4.3. Moxifloxacin
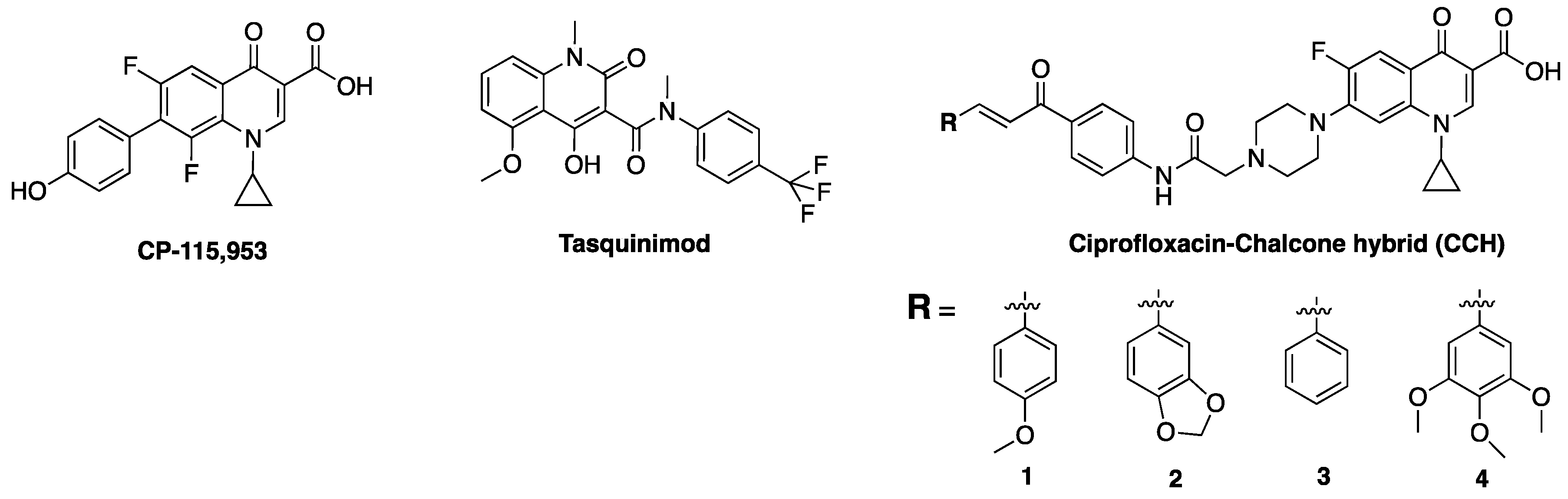
4.4. Other Quinolones
5. Topoisomerases as Targets for Fluoroquinolone Actions: Prokaryotes versus Eukaryotes
5.1. Prokaryotic Topoisomerase Type IIA
5.2. Eukaryotic Topoisomerase Type IIA
5.3. Interfacial Inhibition of Topoisomerases by Fluoroquinolones
6. Immunomodulatory Properties of Fluoroquinolones
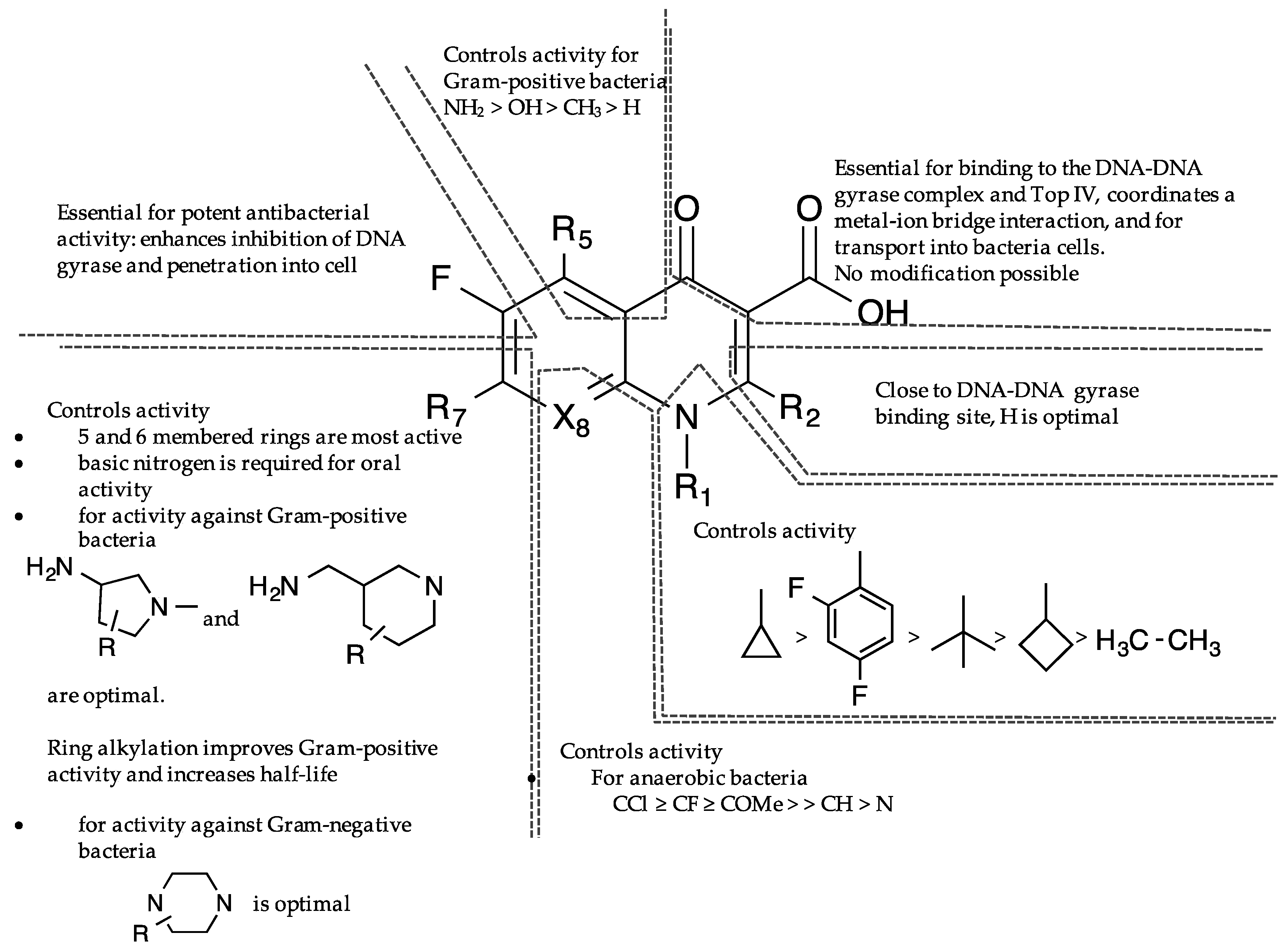
7. Selectivity and Amplification of Desirable Properties in Fluoroquinolones
8. Limitations to the Development and Use of Fluoroquinolones
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Becker, B.; Cooper, M.A. Aminoglycoside antibiotics in the 21st century. ACS Chem. Biol. 2013, 8, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A. A community-based approach to new antibiotic discovery. Nat. Rev. Drug Discov. 2015, 14, 587–588. [Google Scholar] [CrossRef] [PubMed]
- Mitscher, L.A. Bacterial topoisomerase inhibitors: Quinolone and pyridone antibacterial agents. Chem. Rev. 2005, 105, 559–592. [Google Scholar] [CrossRef] [PubMed]
- Ball, P. Quinolone generations: Natural history or natural selection? J. Antimicrob. Chemother. 2000, 46, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; Mcpherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug− enzyme interactions. ACS Chem. Biol. 2013, 8, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Cancer. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 5 September 2017).
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Higgins, P.; Mayer, S.; Fluit, A.; Dalhoff, A. Activity of quinolones against gram-positive cocci: Mechanisms of drug action and bacterial resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 647–659. [Google Scholar] [PubMed]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar] [PubMed]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Bourikas, L.A.; Kolios, G.; Valatas, V.; Notas, G.; Drygiannakis, I.; Pelagiadis, I.; Manousou, P.; Klironomos, S.; Mouzas, I.A.; Kouroumalis, E. Ciprofloxacin decreases survival in HT-29 cells via the induction of TGF-beta1 secretion and enhances the anti-proliferative effect of 5-fluorouracil. Br. J. Pharmacol. 2009, 157, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Elsea, S.H.; Osheroff, N.; Nitiss, J.L. Cytotoxicity of quinolones toward eukaryotic cells. J. Biol. Chem. 1992, 267, 13150–13153. [Google Scholar] [PubMed]
- Elsea, S.H.; Mcguirk, P.R.; Gootz, T.D.; Moynihan, M.; Osheroffl, N. Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: Correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential. Antimicrob. Agents Chemother. 1993, 37, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Dayam, R.; Al-Mawsawi, L.Q.; Zawahir, Z.; Witvrouw, M.; Debyser, Z.; Neamati, N. Quinolone 3-carboxylic acid pharmacophore: Design of second generation HIV-1 integrase inhibitors. J. Med. Chem. 2008, 51, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.; Muccioli, G.G.; Millet, R.; Goossens, J.F.; Farce, A.; Chavatte, P.; Poupaert, J.H.; Lambert, D.M.; Depreux, P.; Hénichart, J.P. Novel 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new CB2 cannabinoid receptors agonists: Synthesis, pharmacological properties and molecular modeling. J. Med. Chem. 2006, 49, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Benetti, V.; Castelli, M.P.; Cavallini, T.; Lazzarotti, S.; Pibiri, F.; Saccomanni, G.; Tuccinardi, T.; Vannacci, A.; Martinelli, A.; Ferrarini, P.L. Design, synthesis, and biological evaluation of new 1,8-naphthyridin-4(1H)-on-3-carboxamide and quinolin-4(1H)-on-3-carboxamide derivatives as CB2 selective agonists. J. Med. Chem. 2006, 49, 5947–5957. [Google Scholar] [CrossRef] [PubMed]
- Kahnberg, P.; Howard, M.H.; Liljefors, T.; Nielsen, M.; Nielsen, E.Ø.; Sterner, O.; Pettersson, I. The use of a pharmacophore model for identification of novel ligands for the benzodiazepine binding site of the GABA A receptor. J. Mol. Graph. Model. 2004, 23, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lager, E.; Andersson, P.; Nilsson, J.; Pettersson, I.; Nielsen, E.O.; Nielsen, M.; Sterner, O.; Liljefors, T. 4-Quinolone derivatives: High-affinity ligands at the benzodiazepine site of brain GABAA receptors. Synthesis, pharmacology, and pharmacophore modeling. J. Med. Chem. 2006, 49, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Lee, J.; Jung, H.Y.; Kim, M.J.; Lim, S.H.; Yeo, H.T.; Choi, E.C.; Yoon, E.J.; Kim, K.W.; Cha, J.H.; et al. Identification, biological activity, and mechanism of the anti-ischemic quinolone analog. Bioorganic Med. Chem. 2007, 15, 6517–6526. [Google Scholar] [CrossRef] [PubMed]
- Lucero, B.D.; Gomes, C.R.; Frugulhetti, I.C.; Faro, L.V.; Alvarenga, L.; De Souza, M.C.; De Souza, T.M.; Ferreira, V.F. Synthesis and anti-HSV-1 activity of quinolonic acyclovir analogues. Bioorg. Med. Chem. Lett. 2006, 16, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J. Pharmacology of polymyxins: New insights into an “old” class of antibiotics. Future Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Bud, R. Penicillin: Triumph and Tragedy; Slinn, J., Ed.; Oxford University Press: Oxford, UK, 2009; ISBN 978-19-925406-4. [Google Scholar]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, P.R. 1,8-Naphthyridine derivatives. A new class of chemotherapeutic agents. J. Med. Pharm. Chem. 1962, 5, 1063–1068. [Google Scholar] [CrossRef]
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “discovery story. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef] [PubMed]
- Domagala, J.M. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother. 1994, 33, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Bertino, J.; Fish, D. The safety profile of the fluoroquinolones. Clin. Ther. 2000, 22, 798–817. [Google Scholar] [CrossRef]
- Naber, K.G.; Hollauer, K.; Kirchbauer, D.; Witte, W. In vitro activity of gatifloxacin compared with gemifloxacin, moxifloxacin, trovafloxacin, ciprofloxacin and ofloxacin against uropathogens cultured from patients with complicated urinary tract infections. Int. J. Antimicrob. Agents 2000, 16, 239–243. [Google Scholar] [CrossRef]
- Stein, G.E.; Goldstein, E.J.C. Fluoroquinolones and anaerobes. Clin. Infect. Dis. 2006, 42, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.D. Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 1987, 51, 341–350. [Google Scholar] [PubMed]
- Mingeot-Leclercq, M.P.; Glupczynski, Y.; Tulkens, P.M. Aminoglycosides: Activity and resistance. Antimicrob. Agents Chemother. 1999, 43, 727–737. [Google Scholar] [PubMed]
- Owens, R.C.; Ambrose, P.G. Clinical use of the fluoroquinolones. Med. Clin. N. Am. 2000, 84, 1447–1469. [Google Scholar] [CrossRef]
- Oliphant, C.M.; Green, G.M. Quinolones: A comprehensive review. Am. Fam. Physician 2002, 65, 455–464. [Google Scholar] [PubMed]
- Hooper, D.C.; Wolfson, J.S. The Fluoroquinolones: Pharmacology, clinical uses, and toxicities in humans. Antimicrob. Agents Chemother. 1985, 28, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V.; Young, J.A.H.; Wingard, J.R. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.E. Pharmacokinetics and pharmacodynamics of newer fluoroquinolones. Clin. Infect. Dis. 1996, 23, S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, J.M. A review of the comparative in-vitro activities of 12 antimicrobial agents, with a focus on five new respiratory quinolones’. J. Antimicrob. Chemother. 1999, 43, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhao, X.; Domagala, J.; Drlica, K. Effect of fluoroquinolone concentration on selection of resistant mutants of Mycobacterium bovis BCG and Staphylococcus aureus. Antimicrob. Agents Chemother. 1999, 43, 1756–1758. [Google Scholar] [PubMed]
- Zhanel, G.G.; Ennis, K.; Vercaigne, L.; Walkty, A.; Gin, A.S.; Embil, J.; Smith, H.; Hoban, D.J. A critical review of the fluoroquinolones. Drugs 2002, 62, 13–59. [Google Scholar] [CrossRef] [PubMed]
- Gorityala, B.K.; Guchhait, G.; Fernando, D.M.; Deo, S.; McKenna, S.A.; Zhanel, G.G.; Kumar, A.; Schweizer, F. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem. Int. Ed. Engl. 2016, 55, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Diggle, S.P.; Matthijs, S.; Wright, V.J.; Fletcher, M.P.; Ram Chhabra, S.; Lamont, I.L.; Kong, X.; Hider, R.C.; Cornelis, P.; Cá mara, M.; et al. The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chem. Biol. 2007, 14, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Fontaine, S.; Adam, H.; Schurek, K.; Mayer, M.; Noreddin, A.M.; Gin, A.S.; Rubinstein, E.; Hoban, D.J. A review of new fluoroquinolones: Focus on their use in respiratory tract infections. Treat. Respir. Med. 2006, 5, 437–465. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.H. The role of moxifloxacin in tuberculosis therapy. Eur. Respir. Rev. 2016, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Diacon, A.H.; Everitt, D.; van Niekerk, C.; Donald, P.R.; Burger, D.A.; Schall, R.; Spigelman, M.; Conradie, A.; Eisenach, K.; et al. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: A phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385, 1738–1747. [Google Scholar] [PubMed]
- Scheld, W.M. Maintaining fluoroquinolone class efficacy: Review of influencing factors. Emerg. Infect. Dis. 2003, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pestova, E.; Millichap, J.J.; Noskin, G.A.; Peterson, L.R. Intracellular targets of moxifloxacin: A comparison with other fluoroquinolones. J. Antimicrob. Chemother. 2000, 45, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Bast, D.J.; Low, D.E.; Duncan, C.L.; Kilburn, L.; Mandell, L.A.; Davidson, R.J.; De Azavedo, J.C. Fluoroquinolone resistance in clinical isolates of Streptococcus pneumoniae: Contributions of type II topoisomerase mutations and efflux to levels of resistance. Antimicrob. Agents Chemother. 2000, 44, 3049–3054. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Walters, M.; Hisanaga, T.; Zhanel, G.G.; Hoban, D.J. Mutant prevention concentrations for single-step fluoroquinolone-resistant mutants of wild-type, efflux-positive, or ParC or GyrA mutation-containing Streptococcus pneumoniae isolates. Antimicrob. Agents Chemother. 2004, 48, 3954–3958. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Kishii, R.; Takei, M.; Hosaka, M. Contributions of the 8-methoxy group of gatifloxacin to resistance selectivity, target preference, and antibacterial activity against Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Kishii, R.; Takei, M.; Fukuda, H.; Hayashi, K.; Hosaka, M. Contribution of the 8-methoxy group to the activity of gatifloxacin against type II topoisomerases of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2003, 47, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Frémaux, A.; Sissia, G.; Geslin, P. In-vitro bacteriostatic activity of levofloxacin and three other fluoroquinolones against penicillin-susceptible and penicillin-resistant Streptococcus pneumoniae. J. Antimicrob. Chemother. 1999, 43, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Citron, D.M.; Merriam, C.V.; Tyrrell, K.; Warren, Y. Activity of gatifloxacin compared to those of five other quinolones versus aerobic and anaerobic isolates from skin and soft tissue samples of human and animal bite wound infections. Antimicrob. Agents Chemother. 1999, 43, 1475–1479. [Google Scholar] [PubMed]
- Poole, R.M. Nemonoxacin: First global approval. Drugs 2014, 74, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K. Finafloxacin: First global approval. Drugs 2015, 75, 687–693. [Google Scholar] [CrossRef] [PubMed]
- US FDA Highlights of Prescribing Information for Baxdela. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208610s000,208611s000lbl.pdf (accessed on 14 September 2017).
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.R. Quinolone molecular structure-activity relationships: What we have learned about improving antimicrobial activity. Clin. Infect. Dis. 2001, 33, S180–186. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L. Molecular mechanisms of DNA gyrase inhibition by quinolone antibacterials. Adv. Pharmacol. 1994, 29, 285–304. [Google Scholar]
- Bhanot, S.K.; Singh, M.; Chatterjee, N.R. The chemical and biological aspects of fluoroquinolones: Reality and dreams. Curr. Pharm. Des. 2001, 7, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N.; Osheroff, N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: Mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.S.; Gould, K.A.; Fisher, L.M. Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 2009, 53, 3822–3831. [Google Scholar] [CrossRef] [PubMed]
- Oppegard, L.M.; Streck, K.R.; Rosen, J.D.; Schwanz, H.A.; Drlica, K.; Kerns, R.J.; Hiasa, H. Comparison of in vitro activities of fluoroquinolone-like 2,4-and 1,3-diones. Antimicrob. Agents Chemother. 2010, 54, 3011–3014. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Marks, K.R.; Mustaev, A.; Zhao, X.; Chavda, K.; Kerns, R.J.; Drlica, K. Fluoroquinolone and quinazolinedione activities against wild-type and gyrase mutant strains of Mycobacterium smegmatis. Antimicrob. Agents Chemother. 2011, 55, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Al-Trawneh, S.A.; Zahra, J.A.; Kamal, M.R.; El-Abadelah, M.M.; Zani, F.; Incerti, M.; Cavazzoni, A.; Alfieri, R.R.; Petronini, P.G.; Vicini, P. Synthesis and biological evaluation of tetracyclic fluoroquinolones as antibacterial and anticancer agents. Bioorg. Med. Chem. 2010, 18, 5873–5884. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.E.; Osheroff, N. Type II topoisomerases as targets for quinolone antibacterials: Turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001, 7, 337–353. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Grignon, R.; Aslam, N.; Aranha, O.; Sarkar, F.H. Ciprofloxacin inhibits cell growth and synergises the effect of etoposide in hormone resistant prostate cancer cells. Int. J. Oncol. 2002, 21, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.D.; Madafiglio, J.; Gilbert, J.; Marshall, G.M.; Haber, M. Reversal of multidrug resistance-associated protein-mediated drug resistance in cultured human neuroblastoma cells by the quinolone antibiotic difloxacin. Med. Pediatr. Oncol. 2001, 36, 177–180. [Google Scholar] [CrossRef]
- Herold, C.; Ocker, M.; Ganslmayer, M.; Gerauer, H.; Hahn, E.G.; Schuppan, D. Ciprofloxacin induces apoptosis and inhibits proliferation of human colorectal carcinoma cells. Br. J. Cancer 2002, 86, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Hussy, P.; Maass, G.; Tümmler, B.; Grosse, F.; Schomburg, U. Effect of 4-quinolones and novobiocin on calf thymus DNA polymerase alpha primase complex, topoisomerases I and II, and growth of mammalian lymphoblasts. Antimicrob. Agents Chemother. 1986, 29, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Aranha, O.; Grignon, R.; Fernandes, N.; McDonnell, T.J.; Wood, D.P.; Sarkar, F.H. Suppression of human prostate cancer cell growth by ciprofloxacin is associated with cell cycle arrest and apoptosis. Int. J. Oncol. 2003, 22, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, A.J. The expanding role of fluoroquinolones. Am. J. Med. 2002, 113, 45S–54S. [Google Scholar] [CrossRef]
- Simpson, K.J.; Brodie, M.J. Convulsions related to enoxacin. Lancet 1985. [Google Scholar] [CrossRef]
- De Sarro, A.; Zappala, M.; Chimirri, A.; Grasso, S.; De Sarro1, G.B. Quinolones potentiate cefazolin-induced seizures in DBA/2 mice. Antimicrob. Agents Chemother. 1993, 37, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Mandal, E.R.; Das, S.K. Evaluation of antiproliferative activity of enoxacin on a human breast cancer cell line. Int. J. Hum. Genet. 2005, 5, 57–63. [Google Scholar]
- Melo, S.; Villanueva, A.; Moutinho, C.; Davalos, V.; Spizzo, R.; Ivan, C.; Rossi, S. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc. Natl. Acad. Sci. USA 2011, 108, 4394–4399. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Gaur, A.; Jewell, D.A.; Liang, Y.; Ridzon, D.; Moore, J.H.; Chen, C.; Ambros, V.R.; Israel, M.A. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007, 67, 2456–2468. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.; Li, Y.; Zhang, J.; Li, W.; Szulwach, K.E.; Duan, R.; Faghihi, M.A.; Khalil, A.M.; Lu, L.; Paroo, Z.; et al. A small molecule enhances RNA interference and promotes microRNA processing. Nat. Biotechnol. 2008, 26, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Spencer, C.M. Enoxacin: A reappraisal of its clinical efficacy in the treatment of genitourinary tract infections. Drugs 1996, 51, 137–160. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.P.; Arshinoff, S.A.; Mah, F.S. Perspectives on antibiotics for postoperative endophthalmitis prophylaxis: Potential role of moxifloxacin. J. Cataract Refract. Surg. 2007, 33, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Brillault, J.; De Castro, W.V.; Harnois, T.; Kitzis, A.; Olivier, J.C.; Couet, W. P-glycoprotein-mediated transport of moxifloxacin in a Calu-3 lung epithelial cell model. Antimicrob. Agents Chemother. 2009, 53, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, C.R.G.; Caparas, V.L.; Bolinao, J.G. Safety of prophylactic intracameral moxifloxacin 0.5% ophthalmic solution in cataract surgery patients. J. Cataract Refract. Surg. 2007, 33, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Arshinoff, S.A.; Modabber, M. Dose and administration of intracameral moxifloxacin for prophylaxis of postoperative endophthalmitis. J. Cataract Refract. Surg. 2016, 42, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Sobolewska, B.; Hofmann, J.; Spitzer, M.S.; Bartz-Schmidt, K.U.; Szurman, P.; Yoeruek, E. Antiproliferative and cytotoxic properties of moxifloxacin on rat retinal ganglion cells. Curr. Eye Res. 2013, 38, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Miyoshi, T.; Suto, C.; Akura, J.; Inoue, Y. Efficacy and safety of prophylactic intracameral moxifloxacin injection in Japan. J. Cart. Refract. Surg. 2013, 39, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Martin, B.A.; Gootz, T.D.; McGuirk, P.R.; Moynihan, M.; Sutcliffe, J.A.; Osheroff, N. Effects of quinolone derivatives on eukaryotic topoisomerase II. A novel mechanism for enhancement of enzyme-mediated DNA cleavage. J. Biol. Chem. 1991, 266, 14585–14592. [Google Scholar] [PubMed]
- Robinson, M.J.; Martin, B.A.; Gootz, T.D.; McGuirk, P.R.; Osheroff, N. Effects of novel fluoroquinolones on the catalytic activities of eukaryotic topoisomerase II: Influence of the C-8 fluorine group. Antimicrob. Agents Chemother. 1992, 36, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T.; Pili, R.; Qian, D.Z.; Dalrymple, S.L.; Garrison, J.B.; Kyprianou, N.; Björk, A.; Olsson, A.; Leanderson, T. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 2006, 66, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Osanto, S.; van Poppel, H.; Burggraaf, J. Tasquinimod: A novel drug in advanced prostate cancer. Future Oncol. 2013, 9, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Mondal, E.R.; Das, S.K.; Mukherjee, P. Comparative evaluation of antiproliferative activity and induction of apoptosis by some fluoroquinolones with a human non-small cell lung cancer cell line in culture. Asian Pac. J. Cancer Prev. 2004, 5, 196–204. [Google Scholar] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Marinello, J.; Chillemi, G. Type I DNA Topoisomerases. J. Med. Chem. 2017, 60, 2169–2192. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.S.; Fisher, L.M. Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: Selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 1997, 41, 471–474. [Google Scholar] [PubMed]
- Pan, X.S.; Fisher, L.M. DNA gyrase and topoisomerase IV are dual targets of clinafloxacin action in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 1998, 42, 2810–2816. [Google Scholar] [PubMed]
- Fournier, B.; Zhao, X.; Lu, T.; Drlica, K.; Hooper, D.C. Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: Different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 2000, 44, 2160–2165. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, K.N.; Cozzarelli, N.R. Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: Effects on deoxyribonucleic acid replication, transcription, and bacteriophage growth. J. Bacteriol. 1979, 140, 424–435. [Google Scholar] [PubMed]
- Arnoldi, E.; Pan, X.S.; Fisher, L.M. Functional determinants of gate-DNA selection and cleavage by bacterial type II topoisomerases. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Veselkov, D.A.; Crevel, I.M.T.; Pan, X.S.; Fisher, L.M.; Sanderson, M.R. Structure of an “open” clamp type II topoisomerase-DNA complex provides a mechanism for DNA capture and transport. Nucleic Acids Res. 2013, 41, 9911–9923. [Google Scholar] [CrossRef] [PubMed]
- Laponogov, I.; Sohi, M.K.; Veselkov, D.A.; Pan, X.S.; Sawhney, R.; Thompson, A.W.; McAuley, K.E.; Fisher, L.M.; Sanderson, M.R. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009, 16, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by analogues of nalidixic acid: The target of the drugs is DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Palù, G.; Valisena, S.; Ciarrocchi, G.; Gatto, B.; Palumbo, M. Quinolone binding to DNA is mediated by magnesium ions. Proc. Natl. Acad. Sci. USA 1992, 89, 9671–9675. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Mitscher, L.A.; Sharma, P.N.; O’Donnell, T.J.; Chu, D.W.; Cooper, C.S.; Rosen, T.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by quinolone antibacterials: A cooperative drug-DNA binding model. Biochemistry 1989, 28, 3886–3894. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Baranowski, J.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by quinolone antibacterials: Specificity and cooperativity of drug binding to DNA. Biochemistry 1989, 28, 3879–3885. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Kohlbrenner, W.E.; Weigl, D.; Baranowski, J. Mechanism of quinolone inhibition of DNA gyrase. Appearance of unique norfloxacin binding sites in enzyme-DNA complexes. J. Biol. Chem. 1989, 264, 2973–2978. [Google Scholar] [PubMed]
- Yoshida, H.; Bogaki, M.; Nakamura, M.; Yamanaka, L.M.; Nakamura, S. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Williamson, B.H.; Kerns, R.J.; Berger, J.M. Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA. 2016, 113, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Leo, E.; Gould, K.A.; Pan, X.S.; Capranico, G.; Sanderson, M.R.; Palumbo, M.; Fisher, L.M. Novel symmetric and asymmetric DNA scission determinants for Streptococcus pneumoniae topoisomerase IV and gyrase are clustered at the DNA breakage site. J. Biol. Chem. 2005, 280, 14252–14263. [Google Scholar] [CrossRef] [PubMed]
- Kwok, Y.; Zeng, Q.; Hurley, L.H. Structural insight into a quinolone-topoisomerase II-DNA complex. Further evidence for a 2:2 quinobenzoxazine-mg2+ self-assembly model formed in the presence of topoisomerase II. J. Biol. Chem. 1999, 274, 17226–17235. [Google Scholar] [CrossRef] [PubMed]
- Kathiravan, M.K.; Khilare, M.M.; Nikoomanesh, K.; Chothe, A.S.; Jain, K.S. Topoisomerase as target for antibacterial and anticancer drug discovery. J. Enzyme Inhib. Med. Chem. 2018, 28, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F. DNA topoisomerase poisons as antitumor drugs. Annu. Rev. Biochem. 1989, 58, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.V.; Nitiss, J.L. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Investig. 2002, 20, 570–589. [Google Scholar] [CrossRef]
- Goto, T.; Wang, J.C. Yeast DNA topoisomerase II is encoded by a single-copy, essential gene. Cell 1984, 36, 1073–1080. [Google Scholar] [CrossRef]
- Wyckoff, E.; Natalie, D.; Nolan, J.M.; Lee, M.; Hsieh, T. Structure of the Drosophila DNA topoisomerase II gene. Nucleotide sequence and homology among topoisomerases II. J. Mol. Biol. 1989, 205, 1–13. [Google Scholar] [CrossRef]
- Nitiss, J.L. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta Gene Struct. Expr. 1998, 1400, 63–81. [Google Scholar] [CrossRef]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIβ and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Mandell, G.L.; Coleman, E. Uptake, transport, and delivery of antimicrobial agents by human polymorphonuclear neutrophils. Antimicrob. Agents Chemother. 2001, 45, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Carryn, S.; Van Bambeke, F.; Mingeot-Leclercq, M.P.; Tulkens, P.M. Comparative intracellular (THP-1 macrophage) and extracellular activities of beta-lactams, azithromycin, gentamicin, and fluoroquinolones against Listeria monocytogenes at clinically relevant concentrations. Antimicrob. Agents Chemother. 2002, 46, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Rodriguez, A.C.; Osheroff, N. Human topoisomerase IIalpha rapidly relaxes positively supercoiled DNA: Implications for enzyme action ahead of replication forks. J. Biol. Chem. 2005, 280, 39337–39345. [Google Scholar] [CrossRef] [PubMed]
- D’arpa, P.; Beardmore, C.; Liu, L.F. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990, 50, 6919–6924. [Google Scholar] [PubMed]
- Andoh, T.; Ishida, R. Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta 1998, 1400, 155–171. [Google Scholar] [CrossRef]
- Hsiung, Y.; Elsea, S.H.; Osheroff, N.; Nitiss, J.L. A mutation in yeast TOP2 homologous to a quinolone-resistant mutation in bacteria. Mutation of the amino acid homologous to Ser83 of Escherichia coli gyrA alters sensitivity to eukaryotic topoisomerase inhibitors. J. Biol. Chem. 1995, 270, 20359–20364. [Google Scholar] [CrossRef] [PubMed]
- Gruger, T.; Nitiss, J.L.; Maxwell, A.; Zechiedrich, E.L.; Heisig, P.; Seeber, S.; Pommier, Y.; Strumberg, D. A mutation in Escherichia coli DNA gyrase conferring quinolone resistance results in sensitivity to drugs targeting eukaryotic topoisomerase II. Antimicrob. Agents Chemother. 2004, 48, 4495–4504. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, B.; Brooks, K.; Pavey, S. Defective cell cycle checkpoints as targets for anti-cancer therapies. Front. Pharmacol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Schaad, U.B. Will fluoroquinolones ever be recommended for common infections in children? Pediatr. Infect. Dis. J. 2007, 26, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Kohlbrenner, W.E.; Wideburg, N.; Weigl, D.; Saldivar, A.; Chu, D.T. Induction of calf thymus topoisomerase II-mediated DNA breakage by the antibacterial isothiazoloquinolones A-65281 and A-65282. Antimicrob. Agents Chemother. 1992, 36, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Permana, P.A.; Snapka, R.M.; Shen, L.L.; Chu, D.T.; Clement, J.J.; Plattner, J.J. Quinobenoxazines: A class of novel antitumor quinolones and potent mammalian DNA topoisomerase II catalytic inhibitors. Biochemistry 1994, 33, 11333–11339. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Tsuzuki, Y.; Shibamori, K.; Tashima, M.; Kajikawa, F.; Sato, Y. Synthesis and structure-activity relationships of novel 7-substituted antitumor agents. Part 1. J. Med. Chem. 2002, 45, 5564–5575. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, Y.; Tomita, K.; Shibamori, K.I.; Sato, Y.; Kashimoto, S.; Chiba, K. Synthesis and structure-activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 2. J. Med. Chem. 2004, 47, 2097–2109. [Google Scholar] [CrossRef] [PubMed]
- Golub, A.G.; Yakovenko, O.Y.; Bdzhola, V.G.; Sapelkin, V.M.; Zien, P.; Yarmoluk, S.M. Evaluation of 3-carboxy-4(1H)-quinolones as inhibitors of human protein kinase CK2. J. Med. Chem. 2006, 49, 6443–6450. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Tauber, S.C.; Nau, R. Immunomodulatory properties of antibiotics. Curr. Mol. Pharmacol. 2008, 1, 68–79. [Google Scholar] [PubMed]
- Kanoh, S.; Rubin, B.K. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 2010, 23, 590–615. [Google Scholar] [CrossRef] [PubMed]
- Guchhait, G.; Altieri, A.; Gorityala, B.; Yang, X.; Findlay, B.; Zhanel, G.G.; Mookherjee, N.; Schweizer, F. Amphiphilic tobramycins with immunomodulatory properties. Angew. Chem. Int. Ed. Engl. 2015, 54, 6278–6282. [Google Scholar] [CrossRef] [PubMed]
- Roche, Y.; Fay, M.; Gougerot-Pocidalo, M.A. Effects of quinolones on interleukin 1 production in vitro by human monocytes. Immunopharmacology 1987, 13, 99–109. [Google Scholar] [CrossRef]
- Riesbeck, K.; Andersson, J.; Gullberg, M.; Forsgren, A. Fluorinated 4-quinolones induce hyperproduction of interleukin 2. Proc. Natl. Acad. Sci. USA 1989, 86, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Riesbeck, K. Immunomodulating activity of quinolones: Review. J. Chemother. 2002, 14, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A.; Shalit, I. Immunomodulatory effects of quinolones. Lancet Infect. Dis. 2003, 3, 359–371. [Google Scholar] [CrossRef]
- Nau, R.; Eiffert, H. Modulation of release of proinflammatory bacterial compounds by antibacterials: Potential impact on course of inflammation and outcome in sepsis and meningitis. Clin. Microbiol. Rev. 2002, 15, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Eiffert, H. Minimizing the release of proinflammatory and toxic bacterial products within the host: A promising approach to improve outcome in life-threatening infections. FEMS Immunol. Med. Microbiol. 2005, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Laing, K.J.; Secombes, C.J. Chemokines. Dev. Comp. Immunol. 2004, 28, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, D.; Schulze, C.; Oesser, S.; Dalhoff, A.; Sack, M. Impact of different classes of antimicrobial agents on plasma endotoxin activity. Arch. Surg. 1996. [Google Scholar] [CrossRef]
- Gollapudi, S.V.; Chuah, S.K.; Harvey, T.; Thadepalli, H.D.; Thadepalli, H. In vivo effects of rufloxacin and ciprofloxacin on T-cell subsets and tumor necrosis factor production in mice infected with Bacteroides fragilis. Antimicrob. Agents Chemother. 1993, 37, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Thadepalli, H.; Gollapudi, S.V.; Chuah, S.K. Therapeutic evaluation of difloxacin (A-56619) and A-56620 for experimentally induced Bacteroides fragilis-associated intra-abdominal abscess. Antimicrob. Agents Chemother. 1986, 30, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Thadepalli, H.; Hajji, M.; Perumal, V.K.; Chuah, S.K.; Gollapudi, S. Evaluation of temafloxacin in a rat model of intra-abdominal abscess. J. Antimicrob. Chemother. 1992, 29, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Thadepalli, H.; Reddy, U.; Chuah, S.K.; Thadepalli, F.; Malilay, C.; Polzer, R.J.; Hanna, N.; Esfandiari, A.; Brown, P.; Gollapudi, S. In vivo efficacy of trovafloxacin (CP-99,217), a new quinolone, in experimental intra-abdominal abscesses caused by Bacteroides fragilis and Escherichia coli. Antimicrob. Agents Chemother. 1997, 41, 583–586. [Google Scholar] [PubMed]
- Thadepalli, H.; Chuah, S.K.; Reddy, U.; Hanna, N.; Clark, R.; Polzer, R.J.; Gollapudi, S. Efficacy of trovafloxacin for treatment of experimental Bacteroides infection in young and senescent mice. Antimicrob. Agents Chemother. 1997, 41, 1933–1936. [Google Scholar] [PubMed]
- King, A.; May, J.; French, G.; Phillips, I. Comparative in vitro activity of gemifloxacin. J. Antimicrob. Chemother. 2000, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Riesbeck, K.; Sigvardsson, M.; Leanderson, T.; Forsgren, A. Superinduction of cytokine gene transcription by ciprofloxacin. J. Immunol. 1994, 153, 343–352. [Google Scholar] [PubMed]
- Riesbeck, K.; Forsgren, A. Commentary on ciprofloxacin-dependent superinduction of IL-2 synthesis and thymidine uptake. Transplantation 1998, 1282–1283. [Google Scholar]
- Abdel-Aziz, M.; Park, S.E.; Abuo-Rahma, G.D.; Sayed, M.A.; Kwon, Y. Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: Synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur. J. Med. Chem. 2013, 69, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F.M. Amphiphilic modulation of glycosylated antitumor ether lipids results in a potent triamino scaffold against epithelial cancer cell lines and BT474 cancer stem cells. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F. Design, synthesis and antitumor properties of glycosylated antitumor ether lipid (GAEL)-chlorambucil-hybrids. Chem. Phys. Lipids 2016, 194, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Replacing D-glucosamine with Its L-enantiomer in glycosylated antitumor ether lipids (GAELs) retains cytotoxic effects against epithelial cancer cells and cancer stem cells. J. Med. Chem. 2017, 60, 2142–2147. [Google Scholar] [CrossRef] [PubMed]
- Silva, H.; Valério Barra, C.; França da Costa, C.; de Almeida, M.V.; César, E.T.; Silveira, J.N.; Garnier-Suillerot, A.; Silva de Paula, F.C.; Pereira-Maia, E.C.; Fontes, A.P.S. Impact of the carbon chain length of novel platinum complexes derived from N-alkyl-propanediamines on their cytotoxic activity and cellular uptake. J. Inorg. Biochem. 2008, 102, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, R.; Campos, J.M.; Conejo-García, A.; Cruz-López, O.; Báñez-Coronel, M.; Rodríguez-González, A.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Symmetrical bis-quinolinium compounds: New human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 2005, 48, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Next generation topoisomerase I inhibitors: Rationale and biomarker strategies. Biochem. Pharmacol. 2008, 75, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: Mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Azéma, J.; Guidetti, B.; Dewelle, J.; Le Calve, B.; Mijatovic, T.; Korolyov, A.; Vaysse, J.; Malet-Martino, M.; Martino, R.; Kiss, R. 7-((4-Substituted)piperazin-1-yl) derivatives of ciprofloxacin: Synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg. Med. Chem. 2009, 17, 5396–5407. [Google Scholar] [CrossRef] [PubMed]
- Shalit, I.; Kletter, Y.; Weiss, K.; Gruss, T.; Fabian, I. Enhanced hematopoiesis in sublethally irradiated mice treated with various quinolones. Eur. J. Haematol. 2009, 58, 92–98. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.W.; Das, G.; Devadas, S. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ball, P.; Mandell, L.; Niki, Y.; Tillotson, G. Comparative tolerability of the newer fluoroquinolone antibacterials. Drug Saf. 1999, 21, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.D.; Graham, D.J.; McCloskey, C.A. Temafloxacin syndrome: Review of 95 cases. Clin. Infect. Dis. 1994, 18, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Stahlmann, R. Clinical toxicological aspects of fluoroquinolones. Toxicol. Lett. 2002, 127, 269–277. [Google Scholar] [CrossRef]
- Ball, P. Quinolone-induced QT interval prolongation: A not-so-unexpected class effect. J. Antimicrob. Chemother. 2000, 45, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Uivarosi, V. Metal complexes of quinolone antibiotics and their applications: An update. Molecules 2013, 18, 11153–11197. [Google Scholar] [CrossRef] [PubMed]
- Turel, I. The interactions of metal ions with quinolone antibacterial agents. Coord. Chem. Rev. 2002, 232, 27–47. [Google Scholar] [CrossRef]
- Shimada, J.; Shiba, K.; Oguma, T.; Miwa, H.; Yoshimura, Y.; Nishikawa, T.; Okabayashi, Y.; Kitagawa, T.; Yamamoto, S. Effect of antacid on absorption of the quinolone lomefloxacin. Antimicrob. Agents Chemother. 1992, 36, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.C.; Ambrose, P.G. Antimicrobial safety: Focus on fluoroquinolones. Clin. Infect. Dis. 2005, 41, S144–S157. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J.V. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Crozat, E.; Philippe, N.; Lenski, R.E.; Geiselmann, J.; Schneider, D. Long-term experimental evolution in Escherichia coli. XII. DNA topology as a key target of selection. Genetics 2005, 169, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Riesbeck, K.; Gullberg, M.; Forsgren, A. Evidence that the antibiotic ciprofloxacin counteracts cyclosporine-dependent suppression of cytokine production. Transplantation 1994, 57, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- O’Shea, R.; Moser, H.E. Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generations | Microbiologic Activity | Administration and Characteristics | Indications |
|---|---|---|---|
| First generation Nalidixic acid, Cinoxacin (Discontinued), Flumequine | Enterobacteriaceae | Oral administration. Low serum and tissue drug concentrations. Narrow gram-negative coverage | Uncomplicated urinary tract infections Not for use in systemic infections |
| Second generation Class I Lomefloxacin (Discontinued), Norfloxacin, Enoxacin Class II Ofloxacin Ciprofloxacin | Enterobacteriaceae. Enterobacteriaceae, atypical pathogens; Pseudomonas aeruginosa (ciprofloxacin only), Pneumoccoci | Oral administration. Low serum and tissue drug concentrations. Improved gram-negative coverage, limited gram-positive coverage. Oral and intravenous administration. Higher serum, tissue, and intracellular drug concentrations, coverage of atypical pathogens | Uncomplicated urinary tract infections. Not for use in systemic infections. Complicated urinary tract and catheter-related infections. Gastroenteritis with severe diarrhea, prostatitis, nosocomial infections, sexually transmitted diseases |
| Third generation Levofloxacin, Sparfloxacin (Discontinued) Gatifloxacin (Discontinued) | Enterobacteriaceae, atypical pathogens, streptococci. Pneumoccoci MIC: 0.25–0.5 μg/mL | Oral and intravenous administration, similar to class II second-generation but with modest streptococcal coverage. Increased hepatic metabolism (sparfloxacin) | Similar indications as for second-generation. Community-acquired pneumonia in hospitalized patients or if atypical pathogens are strongly suspected |
| Fourth generation Trovafloxacin (Discontinued) Moxifloxacin Gemifloxacin | Enterobacteriaceae, P. aeruginosa, atypical pathogens, MSSA, streptococci, anaerobes, Pneumoccoci | Oral and intravenous administration. Similar to third-generation, but with improved gram-positive and anaerobic coverages | Consider for treatment of intra-abdominal infections |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idowu, T.; Schweizer, F. Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities. Antibiotics 2017, 6, 26. https://doi.org/10.3390/antibiotics6040026
Idowu T, Schweizer F. Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities. Antibiotics. 2017; 6(4):26. https://doi.org/10.3390/antibiotics6040026
Chicago/Turabian StyleIdowu, Temilolu, and Frank Schweizer. 2017. "Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities" Antibiotics 6, no. 4: 26. https://doi.org/10.3390/antibiotics6040026
APA StyleIdowu, T., & Schweizer, F. (2017). Ubiquitous Nature of Fluoroquinolones: The Oscillation between Antibacterial and Anticancer Activities. Antibiotics, 6(4), 26. https://doi.org/10.3390/antibiotics6040026