
Macromolecular Conjugate and Biological Carrier Approaches for the Targeted Delivery of Antibiotics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
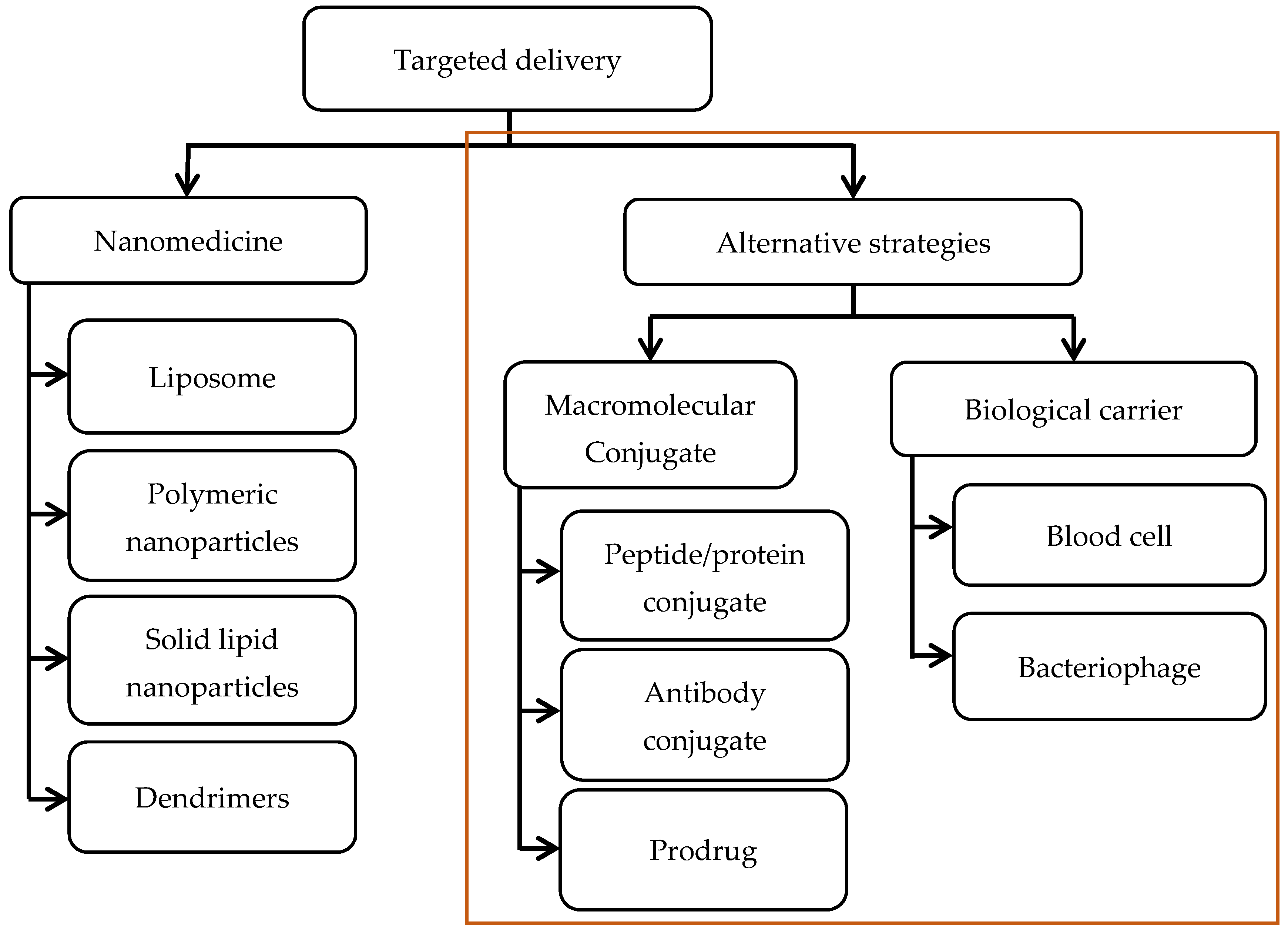
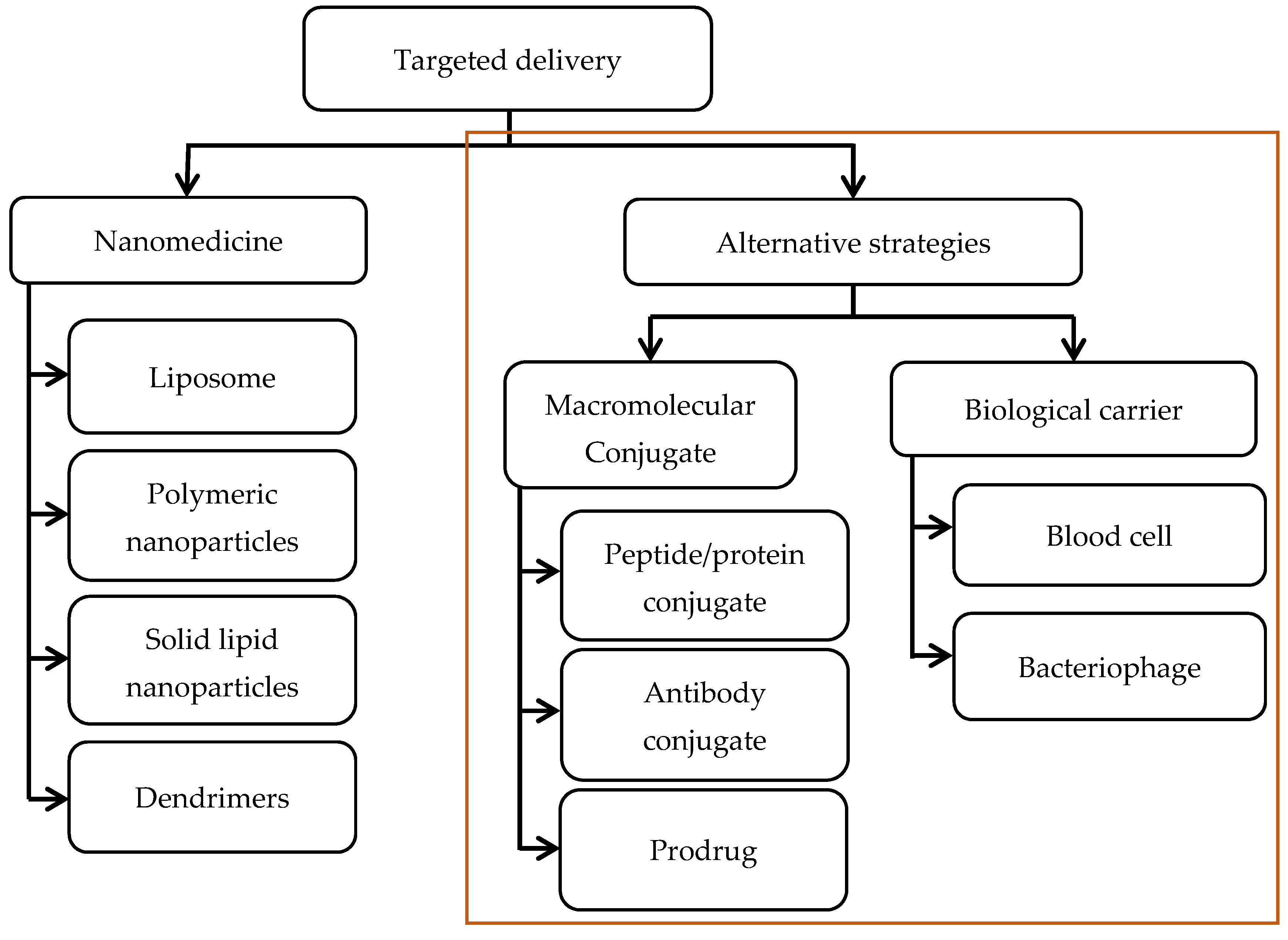
:1. Antibiotic Resistance and the Pressing Need for Alternative Therapeutic Strategies
2. Nanoparticle-Based Strategies for Targeted Delivery: A Brief Update
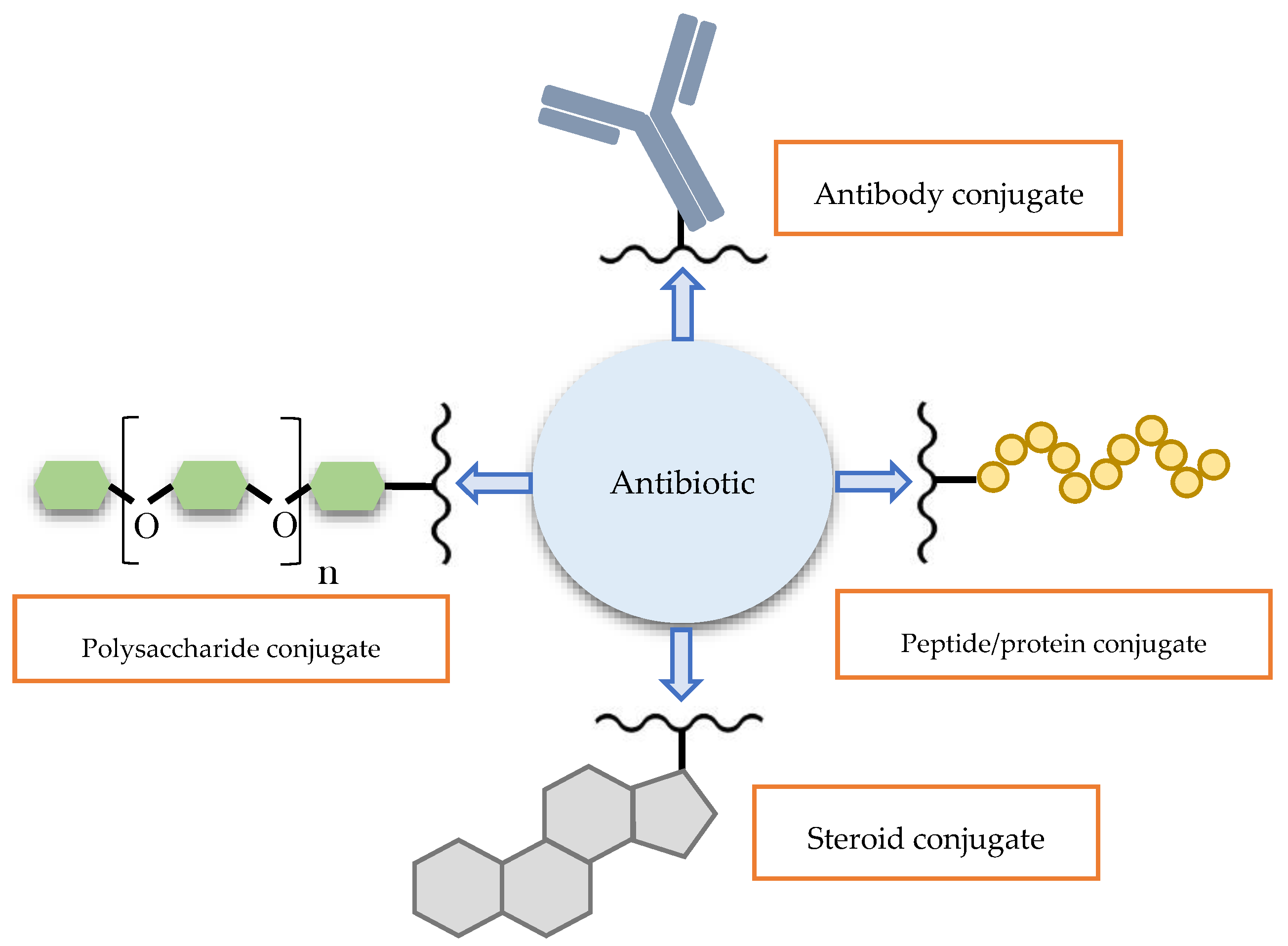
3. Macromolecular Conjugate Strategies
3.1. Conjugate with Peptide/Protein
3.2. Conjugate with Antibody
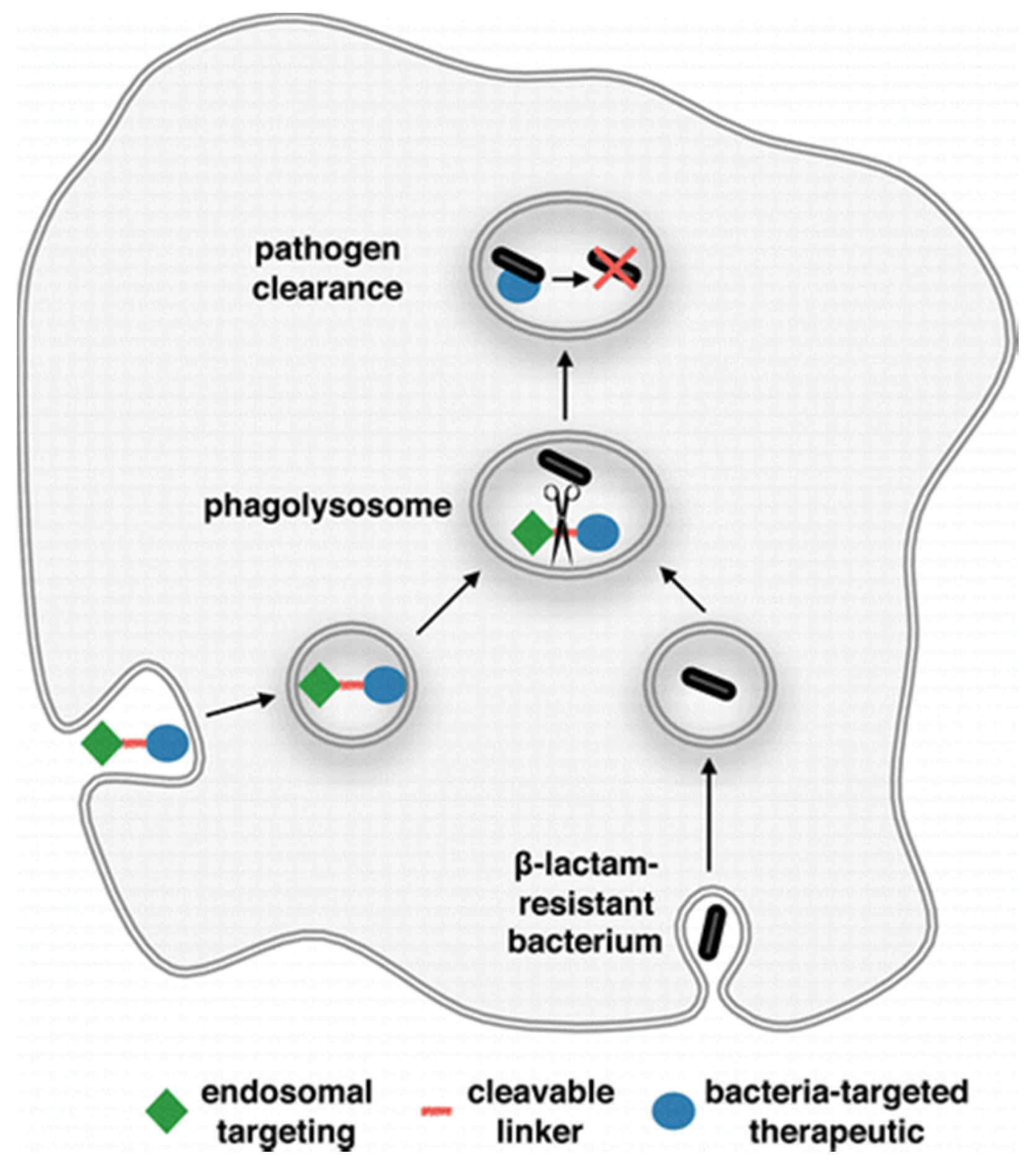
3.3. Prodrug
4. Biological Carrier Strategies
4.1. Blood Cell
4.2. Bacteriophage
5. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gould, I.M.; Bal, A.M. New antibiotic agents in the pipeline and how they can help overcome microbial resistance. Virulence 2013, 4, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Chattopadhyay, M.K.; Grossart, H.-P. The multifaceted roles of antibiotics and antibiotic resistance in nature. Front. Microbiol. 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Van Boeckel, T.P.; Gandra, S.; Ashok, A.; Caudron, Q.; Grenfell, B.T.; Levin, S.A.; Laxminarayan, R. Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet Infect. Dis. 2014, 14, 742–750. [Google Scholar] [CrossRef]
- Luyt, C.-E.; Bréchot, N.; Trouillet, J.-L.; Chastre, J. Antibiotic stewardship in the intensive care unit. Crit. Care 2014, 18, 480. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.G.; Gilbert, D.N.; Spellberg, B. Seven ways to preserve the miracle of antibiotics. Clin. Infect. Dis. 2013, 56, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. The need for new antibiotics. Clin. Microbiol. Infect. 2004, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Lencastre, H.; Oliveira, D.; Tomasz, A. Antibiotic resistant Staphylococcus aureus: A paradigm of adaptive power. Curr. Opin. Microbiol. 2007, 10, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F. Bacteria: An international expert proposal for interim standard definitions for acquired resistance. Microbiology 2011, 18, 268–281. [Google Scholar] [CrossRef]
- McGann, P.; Snesrud, E.; Maybank, R.; Corey, B.; Ong, A.C.; Clifford, R.; Hinkle, M.; Whitman, T.; Lesho, E.; Schaecher, K.E. Escherichia coli Harboring mcr-1 and blaCTX-M on a Novel IncF Plasmid: First report of mcr-1 in the United States. Antimicrob. Agents Chemother. 2016, 60, 4420–4421. [Google Scholar] [CrossRef] [PubMed]
- Conly, J.M. Antimicrobial resistance—Judicious use is the key. Can. J. Infect. Dis. Med. Microbiol. 2004, 15, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. 10 × ′20 Progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Tulkens, P.M. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Baharoglu, Z.; Krin, E.; Mazel, D.; Kulasekara, H.; Bertrand, X. RpoS plays a central role in the SOS induction by sub-lethal aminoglycoside concentrations in vibrio cholerae. PLoS Genet. 2013, 9, e1003421. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, U.; Augustin, H.G. Angiopoietins: A link between angiogenesis and inflammation. Trends Immunol. 2006, 27, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Vitas, A.I.; Díaz, R.; Gamazo, C. Protective effect of liposomal gentamicin against systemic acute murine brucellosis. Chemotherapy 1997, 43, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Diczfalusy, U.; Lund, E.; Lütjohann, D.; Björkhem, I.; Fustik, S.; Goss, C.H.; Lymp, J.; Minic, P.; Quittner, A.L.; Rubenstein, R.C.; et al. Novel pathways for elimination of cholesterol by extrahepatic formation of side-chain oxidized oxysterols. Scand. J. Clin. Lab. Investig. Suppl. 1996, 226, 9–17. [Google Scholar] [CrossRef]
- Bardonnet, P.-L.; Faivre, V.; Boullanger, P.; Piffaretti, J.-C.; Falson, F. Pre-formulation of liposomes against Helicobacter pylori: Characterization and interaction with the bacteria. Eur. J. Pharm. Biopharm. 2008, 69, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.J.; Li, J.; Nation, R.L.; Prankerd, R.J.; Boyd, B.J. Interaction of colistin and colistin methanesulfonate with liposomes: Colloidal aspects and implications for formulation. J. Pharm. Sci. 2012, 101, 3347–3359. [Google Scholar] [CrossRef] [PubMed]
- Sangare, L.; Morisset, R.; Ravaoarinoro, M. In vitro anti-chlamydial activities of free and liposomal tetracycline and doxycycline. J. Med. Microbiol. 1999, 48, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Abeylath, S.C.; Turos, E. Drug delivery approaches to overcome bacterial resistance to β-lactam antibiotics. Expert Opin. Drug Deliv. 2008, 5, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Kalluru, R.; Fenaroli, F.; Westmoreland, D.; Ulanova, L.; Maleki, A.; Roos, N.; Paulsen Madsen, M.; Koster, G.; Egge-Jacobsen, W.; Wilson, S.; et al. Poly(lactide-co-glycolide)-rifampicin nanoparticles efficiently clear Mycobacterium bovis BCG infection in macrophages and remain membrane-bound in phago-lysosomes. J. Cell Sci. 2013, 126, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Azhdarzadeh, M.; Lotfipour, F.; Zakeri-Milani, P.; Mohammadi, G.; Valizadeh, H. Anti-bacterial performance of azithromycin nanoparticles as colloidal drug delivery system against different gram-negative and gram-positive bacteria. Adv. Pharm. Bull. 2012, 2, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Lee, B.-Y.; Xue, M.; Thomas, C.R.; Meng, H.; Ferris, D.; Nel, A.E.; Zink, J.I.; Horwitz, M.A. Targeted intracellular delivery of antituberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 2012, 56, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Forestier, F.; Gerrier, P.; Chaumanrd, C.; Quero, A.-M.; Couvreur, P.; Labarre, C. Effect of nanoparticle-bound ampicillin on the survival of Listeria monocytogenes in mouse peritoneal macrophages. J. Antimicrob. Chemother. 1992, 30, 173–179. [Google Scholar] [CrossRef]
- Fattal, E.; Youssef, M.; Couvreur, P.; Andremont, A. Treatment of experimental salmonellosis in mice with ampicillin-bound nanoparticles. Antimicrob. Agents Chemother. 1989, 33, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Varshosaz, J. Stability and antimicrobial effect of amikacin-loaded solid lipid nanoparticles. Int. J. Nanomed. 2010, 6, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, L.; Dong, Z.; Xie, S.; Chen, X.; Lu, M.; Wang, X.; Li, X.; Zhou, W. Preparation and stability study of norfloxacin-loaded solid lipid nanoparticle suspensions. Colloids Surf. B Biointerfaces 2012, 98, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Abul Kalam, M.; Sultana, Y.; Ali, A.; Aqil, M.; Mishra, A.K.; Chuttani, K.; Aljuffali, I.A.; Alshamsan, A. Part II: Enhancement of transcorneal delivery of gatifloxacin by solid lipid nanoparticles in comparison to commercial aqueous eye drops. J. Biomed. Mater. Res. Part A. 2013, 101, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Zhang, S.L.; Zhu, L.Y.; Xie, S.Y.; Dong, Z.; Wang, Y.; Zhou, W.Z. Enhancement of antibacterial activity of tilmicosin against Staphylococcus aureus by solid lipid nanoparticles in vitro and in vivo. Vet. J. 2012, 191, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Bargoni, A.; Cavalli, R.; Zara, G.P.; Fundarò, A.; Caputo, O.; Gasco, M.R. Transmucosal transport of tobramycin incorporated in solid lipid nanoparticles (sln) after duodenal administration to rats. Part II—Tissue distribution. Pharmacol. Res. 2001, 43, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Khuller, G.K. Solid lipid particle-based inhalable sustained drug delivery system against experimental tuberculosis. Tuberculosis 2005, 85, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qu, H.; Ma, M.; Xu, Z.; Xu, P.; Fang, Y.; Xu, T. Polyamidoamine (PAMAM) dendrimers as biocompatible carriers of quinolone antimicrobials: An in vitro study. Eur. J. Med. Chem. 2007, 42, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Myc, A.; Silpe, J.E.; Sumit, M.; Wong, P.T.; McCarthy, K.; Desai, A.M.; Thomas, T.P.; Kotlyar, A.; Holl, M.M.B.; et al. Dendrimer-based multivalent vancomycin nanoplatform for targeting the drug-resistant bacterial surface. ACS Nano 2013, 7, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lopina, S.T. Penicillin V-conjugated PEG-PAMAM star polymers. J. Biomater. Sci. Polym. Ed. 2003, 14, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Kotta, K.; Hali, M.; Wykes, S.; Gerard, H.C.; Hudson, A.P.; Whittum-Hudson, J.A.; Kannan, R.M. PAMAM dendrimer-azithromycin conjugate nanodevices for the treatment of Chlamydia trachomatis infections. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Cheng, Y.; Xu, Z.; Xu, P.; Qu, H.; Fang, Y.; Xu, T.; Wen, L. Evaluation of polyamidoamine (PAMAM) dendrimers as drug carriers of anti-bacterial drugs using sulfamethoxazole (SMZ) as a model drug. Eur. J. Med. Chem. 2007, 42, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pornpattananangkul, D.; Hu, C.-M.; Huang, C.-M. Development of nanoparticles for antimicrobial drug delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Beyth, N.; Houri-Haddad, Y.; Domb, A.; Khan, W.; Hazan, R. Alternative antimicrobial approach: nano-antimicrobial materials. Evid.-Based Complement. Altern. Med. 2015, 2015, 246012. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.; Lawlor, A.; Whelan, A.; Regan, F.; Mijangos, C.; Grohens, Y.; Rosenau, F.; Jaeger, K.-E.; Bleve-Zacheo, T.; Zambonin, P.G.; et al. The use of nanoparticles in anti-microbial materials and their characterization. Analyst 2008, 133, 835. [Google Scholar] [CrossRef] [PubMed]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- De Simone, S.; Gallo, A.L.; Paladini, F.; Sannino, A.; Pollini, M. Development of silver nano-coatings on silk sutures as a novel approach against surgical infections. J. Mater. Sci. Mater. Med. 2014, 25, 2205–2214. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Lin, W.Y.; Zainal, Z.; Williams, N.E.; Zhu, K.; Kruzic, A.P.; Smith, R.L.; Rajeshwar, K. Bactericidal activity of TiO2 photocatalyst in aqueous media: Toward a solar-assisted water disinfection system. Environ. Sci. Technol. 1994, 28, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Khan, H.M.; Khan, A.A.; Sultan, A.; Azam, A. Characterization of clinical strains of MSSA, MRSA and MRSE isolated from skin and soft tissue infections and the antibacterial activity of ZnO nanoparticles. World J. Microbiol. Biotechnol. 2012, 28, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Hu, D.; Cheng, E.W.C.; Vargas-Reus, M.A.; Reip, P.; Allaker, R.P. Characterisation of copper oxide nanoparticles for antimicrobial applications. Int. J. Antimicrob. Agents 2009, 33, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Anghel, A.; Grumezescu, A.; Chirea, M.; Grumezescu, V.; Socol, G.; Iordache, F.; Oprea, A.; Anghel, I.; Holban, A. MAPLE fabricated Fe3O4@Cinnamomum verum antimicrobial surfaces for improved gastrostomy tubes. Molecules 2014, 19, 8981–8994. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.S.; Stone, J.W.; Gole, A.; Catherine, J.; Murphy, A.; Tara, L. Sabo-attwood, targeted photothermal lysis of the pathogenic bacteria, Pseudomonas aeruginosa, with gold nanorods. Nano Lett. 2007, 8, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Majdalawieh, A.; Kanan, M.C.; El-Kadri, O.; Kanan, S.M. Recent advances in gold and silver nanoparticles: synthesis and applications. J. Nanosci. Nanotechnol. 2014, 14, 4757–4780. [Google Scholar] [CrossRef] [PubMed]
- Shahverdi, A.R.; Fakhimi, A.; Shahverdi, H.R.; Minaian, S. Synthesis and effect of silver nanoparticles on the antibacterial activity of different antibiotics against Staphylococcus aureus and Escherichia coli. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, A.M.; Balaji, K.; Girilal, M.; Yadav, R.; Kalaichelvan, P.T.; Venketesan, R. Biogenic synthesis of silver nanoparticles and their synergistic effect with antibiotics: A study against gram-positive and gram-negative bacteria. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Okusanya, O.O.; Bhavnani, S.M.; Hammel, J.; Minic, P.; Dupont, L.J.; Forrest, A.; Mulder, G.-J.; Mackinson, C.; Ambrose, P.G.; Gupta, R. Pharmacokinetic and pharmacodynamic evaluation of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infection. Antimicrob. Agents Chemother. 2009, 53, 3847–3854. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, C.; Shao, L. The antimicrobial activity of nanoparticles : Present situation and prospects for the future. Int. J. Nanomed. 2017, 12, 1227–1249. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Soracha Thamphiwatana, P.A.; Zhang, L. Nanoparticle approaches against bacterial infections. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 532–547. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Couvreur, P. Nanocarriers for antibiotics: A promising solution to treat intracellular bacterial infections. Int. J. Antimicrob. Agents. 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Cal, P.M.S.D.; Matos, M.J.; Bernardes, G.J.L. Trends in therapeutic drug conjugates for bacterial diseases: A patent review. Expert Opin. Ther. Pat. 2016, 27, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Groves, E.S.; Aldwin, L.; Winkelhake, J.L.; Nitecki, D.E.; Gauney, S.; Rudolph, A.A. Polymer/Antibiotic Conjugate. Patent WO19,90,015,628 A1, 27 December 1990. Available online: https://www.google.com/patents/WO1990015628A1?cl=frOn (accessed on 27 December 1990).
- Drabick, J.; Lees, A.; Mond, J.; Shah, A.; Walsh, S. Antimicrobial Polymer Conjugates. Patent WO2003082926 A3, 1 April 2004. Available online: https://www.google.com/patents/WO2003082926A3?cl=en (accessed on 1 April 2004).
- Krivan, H.C.; Blomberg, A.L.I. Receptor conjugates for targeting penicillin antibiotics to bacteria. U.S. Patent 5,466,681 A, 14 November 1995. Available online: https://www.google.com/patents/US5466681 (accessed on 14 November 1995).
- Schiffman, E.; Altman, L.C. Formyl-Methionyl Chemotatic Peptide Antibiotic Conjugates Useful in Treating Infections. U.S. Patent 4,427,660 A, 24 January 1984. Available online: https://www.google.com/patents/US4427660 (accessed on 24 January 1984).
- Warden, G.D.; Mason, A.D.; Pruitt, B.A.; Pruitt, B.A., Jr. Suppression of leukocyte chemotaxis in vitro by chemotherapeutic agents used in the management of thermal injuries. Ann. Surg. 1975, 181, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Morita, H.; Yoo, Y.-C.; Kim, W.-S.; Kumura, H.; Shimazaki, K. Detection of bovine lactoferrin binding protein on jurkat human lymphoblastic T cell line. J. Vet. Med. Sci. 2004, 66, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Schryvers, A.B.; Morris, L.J. Identification and characterization of the transferrin receptor from Neisseria meningitidis. Mol. Microbiol. 1988, 2, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.R.; Tatsumoto, S.; Hajime, O.; van Immerseel, F.; Raspoet, R.; Miyata, T. A novel antibiotic-delivery system by using ovotransferrin as targeting molecule. Eur. J. Pharm. Sci. 2015, 66, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, F.; Leboffe, L.; Pitari, G.; Ippoliti, R.; Antonini, G. Physiological roles of ovotransferrin. Biochim. Biophys. Acta-Gen. Subj. 2012, 1820, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.R.; Sugimoto, Y.; Aoki, T. Ovotransferrin antimicrobial peptide (OTAP-92) kills bacteria through a membrane damage mechanism. Biochim. Biophys. Acta-Gen. Subj. 2000, 1523, 196–205. [Google Scholar] [CrossRef]
- Hai, J.; Serradji, N.; Mouton, L.; Redeker, V.; Cornu, D.; El Hage Chahine, J.M.; Verbeke, P.; Hémadi, M. Targeted delivery of amoxicillin to C. trachomatis by the transferrin iron acquisition pathway. PLoS ONE 2016, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, Y.M.; Belland, R.J. The chlamydial developmental cycle. FEMS Microbiol. Rev. 2005, 29, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Mpiga, P.; Ravaoarinoro, M. Chlamydia trachomatis persistence: An update. Microbiol. Res. 2006, 161, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.P.; Shi, J.; Kelley, S.O. Peptide targeting of an antibiotic prodrug toward phagosome-entrapped mycobacteria. ACS Infect. Dis. 2016, 1, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.R.; Parsons, L.M.; Pavelka, M.S. Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 2005, 151, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.P.; Kelley, S.O. Maximizing the therapeutic window of an antimicrobial drug by imparting mitochondrial sequestration in human cells. J. Am. Chem. Soc. 2011, 133, 3260–3263. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Stover, P.J. Chapter 1 folate-mediated one-carbon metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar] [CrossRef]
- Surolia, N. Receptor-mediated targeting of toxins to intraerythrocytic parasite Plasmodium falciparum. Adv. Drug Deliv. Rev. 2000, 41, 163–170. [Google Scholar] [CrossRef]
- Wawrzynczak, E.J. Systemic immunotoxin therapy of cancer: Advances and prospects. Br. J. Cancer 1991, 64, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Haldar, K.; Henderson, C.L.; Cross, G.A.M. Identification of the parasite transferrin receptor of Plasmodium falciparum-infected erythrocytes and its acylation via 1,2-diacyl-sn-glycerol. Biochemistry 1986, 83, 8565–8569. [Google Scholar] [CrossRef]
- Geller, B.L.; Deere, J.D.; Stein, D.A.; Kroeker, A.D.; Moulton, H.M.; Iversen, P.L. Inhibition of gene expression in Escherichia coli by antisense phosphorodiamidate morpholino oligomers. Antimicrob. Agents Chemother. 2003, 47, 3233–3239. [Google Scholar] [CrossRef] [PubMed]
- Geller, B.L.; Deere, J.; Tilley, L.; Iversen, P.L. Antisense phosphorodiamidate morpholino oligomer inhibits viability of Escherichia coli in pure culture and in mouse peritonitis. J. Antimicrob. Chemother. 2005, 55, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Tilley, L.D.; Hine, O.S.; Kellogg, J.A.; Hassinger, J.N.; Weller, D.D.; Iversen, P.L.; Geller, B.L. Gene-specific effects of antisense phosphorodiamidate morpholino oligomer-peptide conjugates on Escherichia coli and Salmonella enterica serovar typhimurium in pure culture and in tissue culture. Antimicrob. Agents Chemother. 2006, 50, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Mellbye, B.L.; Weller, D.D.; Hassinger, J.N.; Reeves, M.D.; Lovejoy, C.E.; Iversen, P.L.; Geller, B.L. Cationic phosphorodiamidate morpholino oligomers efficiently prevent growth of Escherichia coli in vitro and in vivo. J. Antimicrob. Chemother. 2009, 65, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Mellbye, B.L.; Puckett, S.E.; Tilley, L.D.; Iversen, P.L.; Geller, B.L. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob. Agents Chemother. 2009, 53, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.E.; Marshall-Batty, K.R.; Brinster, L.R.; Zarember, K.A.; Shaw, P.A.; Mellbye, B.L.; Iversen, P.L.; Holland, S.M.; Geller, B.L. Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J. Infect. Dis. 2010, 201, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Tan, M.-W. Antibody–antibiotic conjugates: A novel therapeutic platform against bacterial infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Flygare, J.; Hazenbos, W.; Lehar, S.M.; Mariathasan, S.; Morisaki, J.H.; Pillow, T.H.; Staben, L.; Vandlen, R.; Koefoed, K. Anti-Wall Teichoic Antibodies and Conjugates. Patent WO2,014,193,722 A1, 4 December 2014. Available online: https://www.google.com/patents/WO2014193722A1?cl=en (accessed on 4 December 2014).
- Brown, E.J.; Flygare, J.; Hazenbos, W.; Lehar, S.M.; Mariathasan, S.; MORISAKI, J.H.; Pillow, T.H.; Staben, L.; Vandlen, R.; Koefoed, K. Anti-Wall Teichoic Antibodies and Conjugates. U.S. Patent 20,150,366,985 A1, 24 December 2015. Available online: https://www.google.com/patents/US20150366985 (accessed on 24 December 2015).
- Kaiser, P.; Regoes, R.R.; Dolowschiak, T.; Wotzka, S.Y.; Lengefeld, J.; Slack, E.; Grant, A.J.; Ackermann, M.; Hardt, W.-D. Cecum lymph node dendritic cells harbor slow-growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol. 2014, 12, e1001793. [Google Scholar] [CrossRef] [PubMed]
- Böttger, E.; Multhoff, G.; Kun, J.F.J.; Esen, M.; Mahmud, H. Plasmodium falciparum-Infected erythrocytes induce granzyme B by NK cells through expression of Host-Hsp70. PLoS ONE 2012, 7, e33774. [Google Scholar] [CrossRef] [PubMed]
- Mavoungou, E.; Luty, A.J.F.; Kremsner, P.G. Natural killer (NK) cell-mediated cytolysis of Plasmodium falciparum-infected human red blood cells in vitro. Eur. Cytokine Netw. 2003, 14, 134–142. [Google Scholar] [PubMed]
- Kapelski, S.; de Almeida, M.; Fischer, R.; Barth, S.; Fendel, R. Antimalarial activity of granzyme B and its targeted delivery by a granzyme B-single-chain Fv fusion protein. Antimicrob. Agents Chemother. 2015, 59, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Garcia, Y.; Puentes, A.; Curtidor, H.; Cifuentes, G.; Reyes, C.; Barreto, J.; Moreno, A.; Patarroyo, M.E. Identifying merozoite surface protein 4 and merozoite surface protein 7 Plasmodium falciparum protein family members specifically binding to human erythrocytes suggests a new malarial Parasite-Redundant survival mechanism. J. Med. Chem. 2007, 50, 5665–5675. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, D.; Bong, R.; Stewart, B.H. Improved oral drug delivery: Solubility limitations overcome by the use of prodrugs. Adv. Drug Deliv. Rev. 1996, 19, 115–130. [Google Scholar] [CrossRef]
- Dahan, A.; Duvdevani, R.; Shapiro, I.; Elmann, A.; Finkelstein, E.; Hoffman, A. The oral absorption of phospholipid prodrugs: In vivo and in vitro mechanistic investigation of trafficking of a lecithin-valproic acid conjugate following oral administration. J. Control. Release 2008, 126, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, P.B.; Ramos, D.C.C.; Cotrim, P.C.; Ferreira, E.I. Synthesis and in vitro evaluation of potential anti-leishmanial targeted drugs of pyrimethamine. J. Pharm. Sci. 2003, 92, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Bhaduri, A.N.; Das, P.K. Sugar receptor mediated drug delivery to macrophages in the therapy of experimental visceral leishmaniasis. Biochem. Biophys. Res. Commun. 1990, 166, 404–410. [Google Scholar] [CrossRef]
- Takakura, Y.; Atsumi, R.; Hashida, M.; Sezaki, H. Development of a novel polymeric prodrug of mitomycin C, mitomycin C-dextran conjugate with anionic charge. II. Disposition and pharmacokinetics following intravenous and intramuscular administration. Int. J. Pharm. 1987, 37, 145–154. [Google Scholar] [CrossRef]
- Banerjee, G.; Nandi, G.; Mahato, S.B.; Pakrashi, A.; Basu, M.K. Drug delivery system: Targeting of pentamidines to specific sites using sugar grafted liposomes. J. Antimicrob. Chemother. 1996, 38, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Roseeuw, E.; Coessens, V.; Balazuc, A.M.; Lagranderie, M.; Chavarot, P.; Pessina, A.; Neri, M.G.; Schacht, E.; Marchal, G.; Domurado, D. Synthesis, degradation, and antimicrobial properties of targeted macromolecular prodrugs of norfloxacin. Antimicrob. Agents Chemother. 2003, 47, 3435–3441. [Google Scholar] [CrossRef] [PubMed]
- Abed, N.; Saïd-Hassane, F.; Zouhiri, F.; Mougin, J.; Nicolas, V.; Desmaële, D.; Gref, R.; Couvreur, P. An efficient system for intracellular delivery of beta-lactam antibiotics to overcome bacterial resistance. Sci. Rep. 2015, 5, 13500. [Google Scholar] [CrossRef] [PubMed]
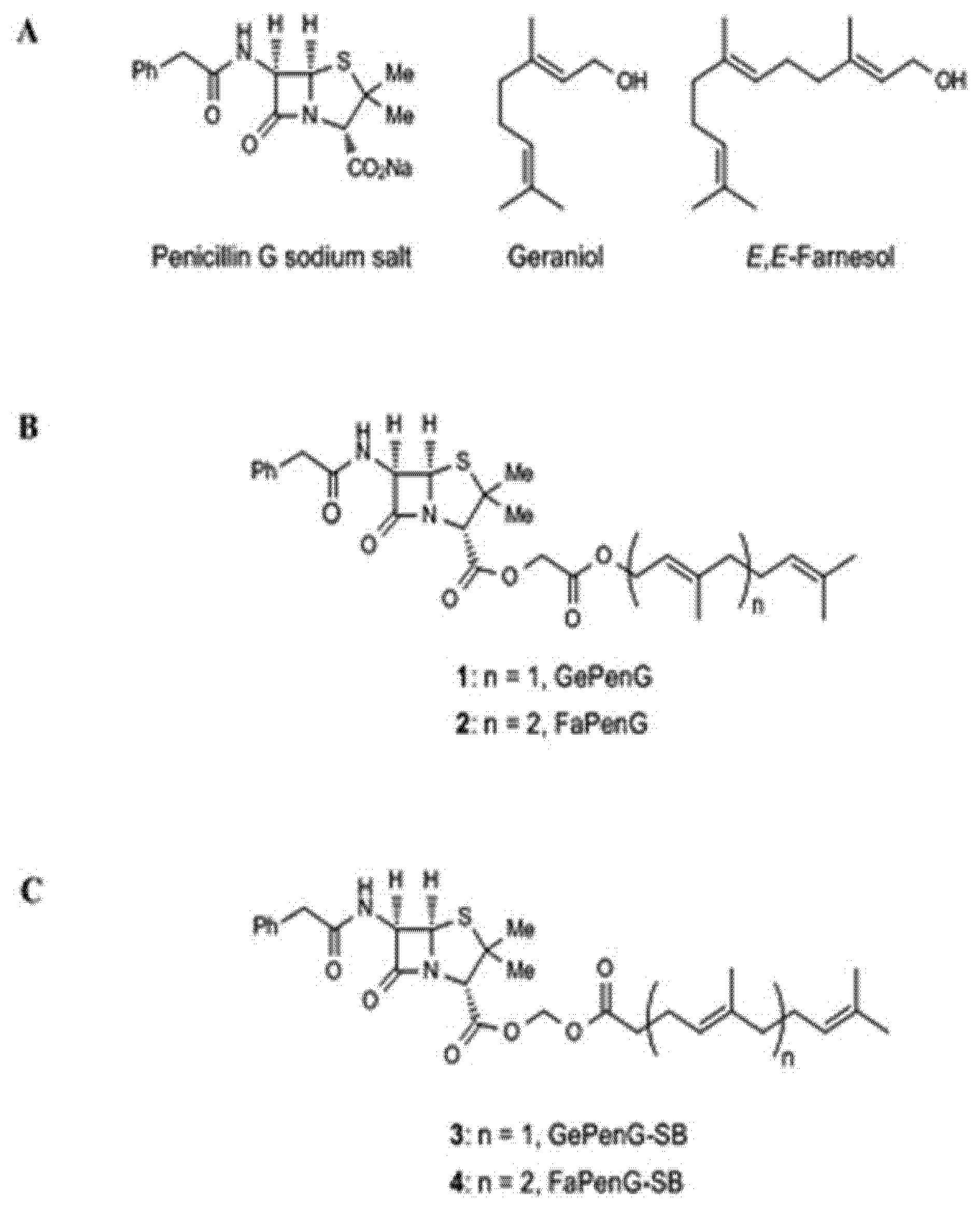
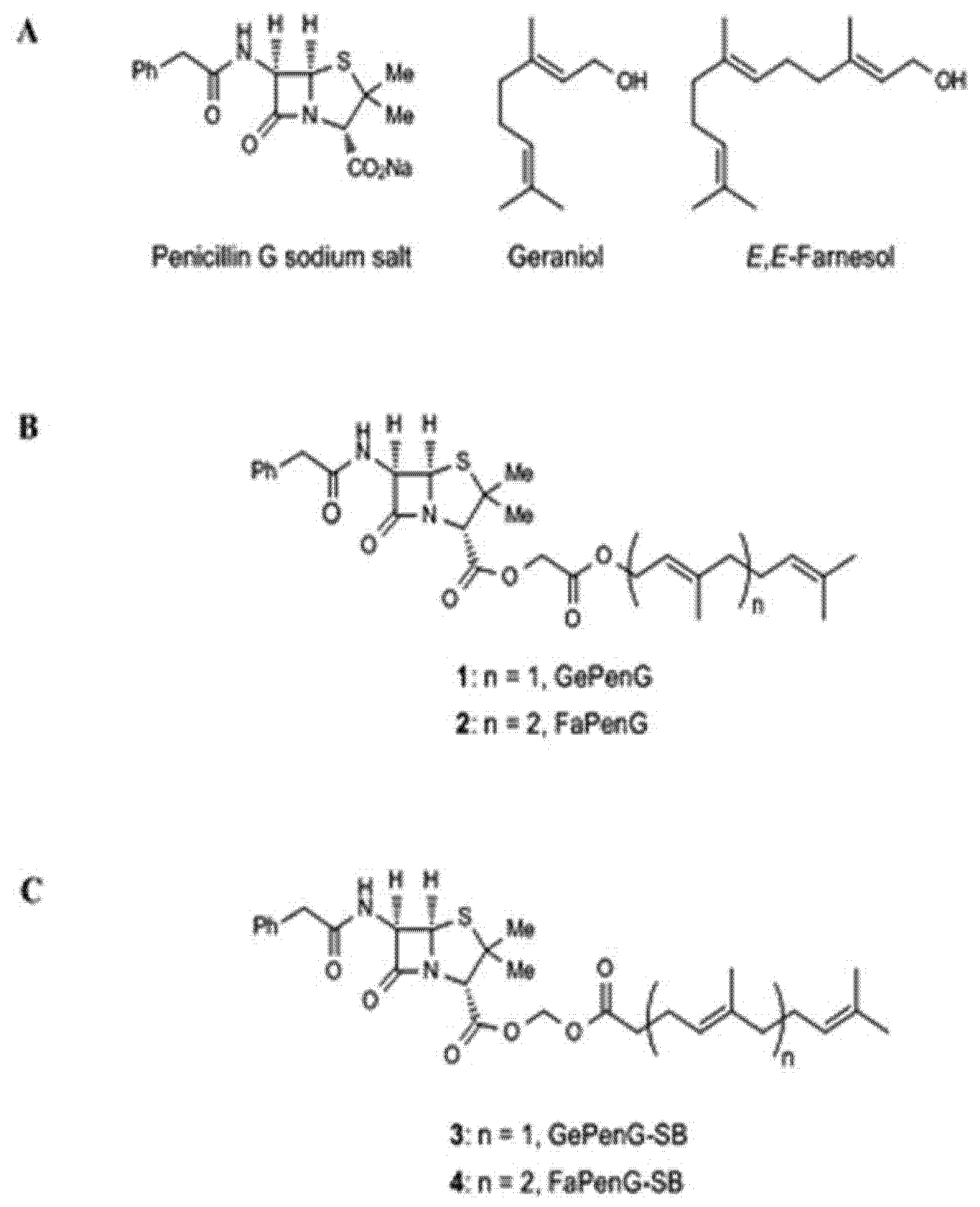
- Sémiramoth, N.; di Meo, C.; Zouhiri, F.; Saïd-Hassane, F.; Valetti, S.; Gorges, R.; Nicolas, V.; Poupaert, J.H.; Chollet-Martin, S.; Desmaële, D.; et al. Self-assembled squalenoylated penicillin bioconjugates: An original approach for the treatment of intracellular infections. ACS Nano 2012, 6, 3820–3831. [Google Scholar] [CrossRef] [PubMed]
- Albayati, Z.A.F.; Sunkara, M.; Schmidt-Malan, S.M.; Karau, M.J.; Morris, A.J.; Steckelberg, J.M.; Patel, R.; Breen, P.J.; Smeltzer, M.S.; Taylor, K.G.; et al. Novel bone-targeting agent for enhanced delivery of vancomycin to bone. Antimicrob. Agents Chemother. 2016, 60, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- Massias, L.; Dubois, C.; de Lentdecker, P.; Brodaty, O.; Fischler, M.; Farinotti, R. Penetration of vancomycin in uninfected sternal bone. Antimicrob. Agents Chemother. 1992, 36, 2539–2541. [Google Scholar] [CrossRef] [PubMed]
- Nasim, S.; Vartak, A.P.; Pierce, W.M.; Taylor, K.G.; Smith, N.; Crooks, P.A. 3-O-Phosphate ester conjugates of 17-β-O-{1-[2-carboxy-(2-hydroxy-4-methoxy-3-carboxamido)anilido]ethyl}1,3,5(10)-estra-triene as novel bone-targeting agents. Bioorg. Med. Chem. Lett. 2010, 20, 7450–7453. [Google Scholar] [CrossRef] [PubMed]
- Rybak, M.J. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin. Infect. Dis. 2006, 42, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Zarrin, A.; Foroozesh, M.; Mohammadi-Samani, S. Applications of carrier erythrocytes in delivery of biopharmaceuticals. J. Control. Release 2007, 118, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Alpar, H.O.; Irwin, W.J. Some unique applications of erythrocytes as carrier systems. Adv. Biosci. 1987, 67, 1–9. [Google Scholar]
- Lynch, W.E.; Sartiano, G.P.; Ghaffar, A. Erythrocytes as carriers of chemotherapeutic agents for targeting the reticuloendothelial system. Am. J. Hematol. 1980, 9, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Valbonesi, M.; Bruni, R.; Florio, G.; Zanella, A.; Bunkens, H. Cellular contamination of plasma collected with various apheresis systems. Transfus. Apher. Sci. 2001, 24, 91–94. [Google Scholar] [CrossRef]
- Lisovskaya, I.L.; Shcherbachenko, I.M.; Volkova, R.I.; Ataullakhanov, F.I. Clotrimazole enhances lysis of human erythrocytes induced by t-BHP. Chem. Biol. Interact. 2009, 180, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Schrier, S.L.; Junga, I. Entry and distribution of chlorpromazine and vinblastine into human erythrocytes during endocytosis. Proc. Soc. Exp. Biol. Med. 1981, 168, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.S.; Weiner, M.; Wagner, K.; Martelo, O.J. Incorporation of inositol hexaphosphate into red blood cells mediated by dimethyl sulfoxide. Life Sci. 1983, 32, 2763–2768. [Google Scholar] [CrossRef]
- Mitchell, D.H.; James, G.T.; Kruse, C.A. Bioactivity of electric field-pulsed human recombinant interleukin-2 and its encapsulation into erythrocyte carriers. Biotechnol. Appl. Biochem. 1990, 12, 264–275. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Ye, J.; Wang, Y.; Liu, Q.; Chung, H.S.; Kwon, Y.M.; Shin, M.C.; Lee, K.; Yang, V.C. Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. J. Control. Release 2014, 176, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Tajerzadeh, H.; Dehpour, A.-R.; Rouini, M.-R.; Ejtemaee-Mehr, S. In vitro characterization of human intact erythrocytes loaded by enalaprilat. Drug Deliv. 2001, 8, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.E.; Bain, M.D.; Fairbanks, L.D.; Webster, A.D.; Chalmers, R.A. In vitro and in vivo studies with human carrier erythrocytes loaded with polyethylene glycol-conjugated and native adenosine deaminase. Br. J. Haematol. 2000, 109, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Bourgeaux, V.; Lanao, J.M.; Bax, B.E.; Godfrin, Y. Drug-loaded erythrocytes: On the road toward marketing approval. Drug Des. Dev. Ther. 2016, 10, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Talwar, N.; Jain, N.K. Erythrocytes as carriers of primaquine-preparation: Characterization and evaluation. J. Control. Release 1992, 20, 133–141. [Google Scholar] [CrossRef]
- Zocchi, E.; Tonetti, M.; Polvani, C.; Guida, L.; Benatti, U.; de Flora, A. In vivo liver and lung targeting of adriamycin encapsulated in glutaraldehyde-treated murine erythrocytes. Biotechnol. Appl. Biochem. 1988, 10, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Deloach, J.; Peters, S.; Pinkard, O.; Glew, R.; Ihler, G. Effect of glutaraldehyde treatment on enzyme-loaded erythrocytes. Biochim. Biophys. Acta 1977, 496, 507–515. [Google Scholar] [CrossRef]
- Eichler, H.G.; Gasic, S.; Bauer, K.; Korn, A.; Bacher, S. In vivo clearance of antibody-sensitized human drug carrier erythrocytes. Clin. Pharmacol. Ther. 1986, 40, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Jain, S.K.; Dixit, V.K. Magnetically guided rat erythrocytes bearing isoniazid: Preparation, characterization, and evaluation. Drug Dev. Ind. Pharm. 1997, 23, 999–1006. [Google Scholar] [CrossRef]
- Hamidi, M.; Tajerzadeh, H. Carrier erythrocytes: An overview. Drug Deliv. 2003, 10, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, L.E. Immunobiology of Mycobacterium avium infection. Eur. J. Clin. Microbiol. Infect. Dis. 1994, 13, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Brandi, G.; Malatesta, M.; Serafini, S.; Pierigè, F.; Celeste, A.G.; Schiavano, G.F.; Gazzanelli, G.; Magnani, M. Effect of listeriolysin O-loaded erythrocytes on Mycobacterium avium replication within macrophages. J. Antimicrob. Chemother. 2004, 53, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Baistrocchi, S.R.; Lee, M.J.; Lehoux, M.; Ralph, B.; Snarr, B.D.; Robitaille, R.; Sheppard, D.C. Posaconazole-loaded leukocytes as a novel treatment strategy targeting invasive pulmonary aspergillosis. J. Infect. Dis. 2016, 215, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Tochigi, N.; Okubo, Y.; Ando, T.; Wakayama, M.; Shinozaki, M.; Gocho, K.; Hata, Y.; Ishiwatari, T.; Nemoto, T.; Shibuya, K. Histopathological implications of Aspergillus infection in lung. Mediat. Inflamm. 2013, 2013, 809798. [Google Scholar] [CrossRef] [PubMed]
- Paterson, P.J.; Seaton, S.; Prentice, H.G.; Kibbler, C.C. Treatment failure in invasive aspergillosis: Susceptibility of deep tissue isolates following treatment with amphotericin B. J. Antimicrob. Chemother. 2003, 5252, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Strieter, R.M.; Moore, T.A.; Tsai, W.C.; Lira, S.A.; Standiford, T.J. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J. Immunol. 1999, 163, 6086–6094. [Google Scholar] [PubMed]
- Lin, L.; Ibrahim, A.S.; Baquir, B.; Palosaari, A.; Spellberg, B. Luminescent-activated transfected killer cells to monitor leukocyte trafficking during systemic bacterial and fungal infection. J. Infect. Dis. 2012, 205, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, V.; Deresinski, S. Posaconazole: A broad-spectrum triazole antifungal agent. Clin. Infect. Dis. 2007, 45, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically engineered phages: A review of advances over the last. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; de Roock, S.; Opal, S.M. A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 2014, 5, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Vaks, L.; Benhar, I. In vivo characteristics of targeted drug-carrying filamentous bacteriophage nanomedicines. J. Nanobiotechnol. 2011, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Van Houten, N.E.; Zwick, M.B.; Menendez, A.; Scott, J.K. Filamentous phage as an immunogenic carrier to elicit focused antibody responses against a synthetic peptide. Vaccine 2006, 24, 4188–4200. [Google Scholar] [CrossRef] [PubMed]
- Turton, J.A.; Andrews, C.M.; Havard, A.C.; Williams, T.C. Studies on the haemotoxicity of chloramphenicol succinate in the Dunkin Hartley guinea pig. Int. J. Exp. Pathol. 2002, 83, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Yacoby, I.; Shamis, M.; Bar, H.; Shabat, D.; Benhar, I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob. Agents Chemother. 2006, 50, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; di Giorgio, M.L. Toxicogenomics to improve comprehension of the mechanisms underlying responses of in vitro and in vivo systems to nanomaterials: A review. Curr. Genom. 2008, 9, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Wang, H.; Yan, L.; Wang, X.; Pei, R.; Yan, T.; Zhao, Y.; Guo, X. Cytotoxicity of carbon nanomaterials: Single-wall nanotube, multi-wall nanotube, and fullerene. Environ. Sci. Technol. 2005, 39, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Shvedova, A.; Castranova, V.; Kisin, E.; Schwegler-Berry, D.; Murray, A.; Gandelsman, V.; Maynard, A.; Baron, P. Exposure to carbon nanotube material: Assessment of nanotube cytotoxicity using human keratinocyte cells. J. Toxicol. Environ. Heal. Part A 2003, 66, 1909–1926. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.H.; Borm, P.J.A. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef]
- Lei, R.; Wu, C.; Yang, B.; Ma, H.; Shi, C.; Wang, Q.; Wang, Q.; Yuan, Y.; Liao, M. Integrated metabolomic analysis of the nano-sized copper particle-induced hepatotoxicity and nephrotoxicity in rats: A rapid in vivo screening method for nanotoxicity. Toxicol. Appl. Pharmacol. 2008, 232, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Stryjewski, M.E.; Potgieter, P.D.; Li, Y.-P.; Barriere, S.L.; Churukian, A.; Kingsley, J.; Corey, G.R. TD-1792 versus vancomycin for treatment of complicated skin and skin structure infections. Antimicrob. Agents Chemother. 2012, 56, 5476–5483. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tram, N.D.T.; Ee, P.L.R. Macromolecular Conjugate and Biological Carrier Approaches for the Targeted Delivery of Antibiotics. Antibiotics 2017, 6, 14. https://doi.org/10.3390/antibiotics6030014
Tram NDT, Ee PLR. Macromolecular Conjugate and Biological Carrier Approaches for the Targeted Delivery of Antibiotics. Antibiotics. 2017; 6(3):14. https://doi.org/10.3390/antibiotics6030014
Chicago/Turabian StyleTram, Nhan Dai Thien, and Pui Lai Rachel Ee. 2017. "Macromolecular Conjugate and Biological Carrier Approaches for the Targeted Delivery of Antibiotics" Antibiotics 6, no. 3: 14. https://doi.org/10.3390/antibiotics6030014
APA StyleTram, N. D. T., & Ee, P. L. R. (2017). Macromolecular Conjugate and Biological Carrier Approaches for the Targeted Delivery of Antibiotics. Antibiotics, 6(3), 14. https://doi.org/10.3390/antibiotics6030014