
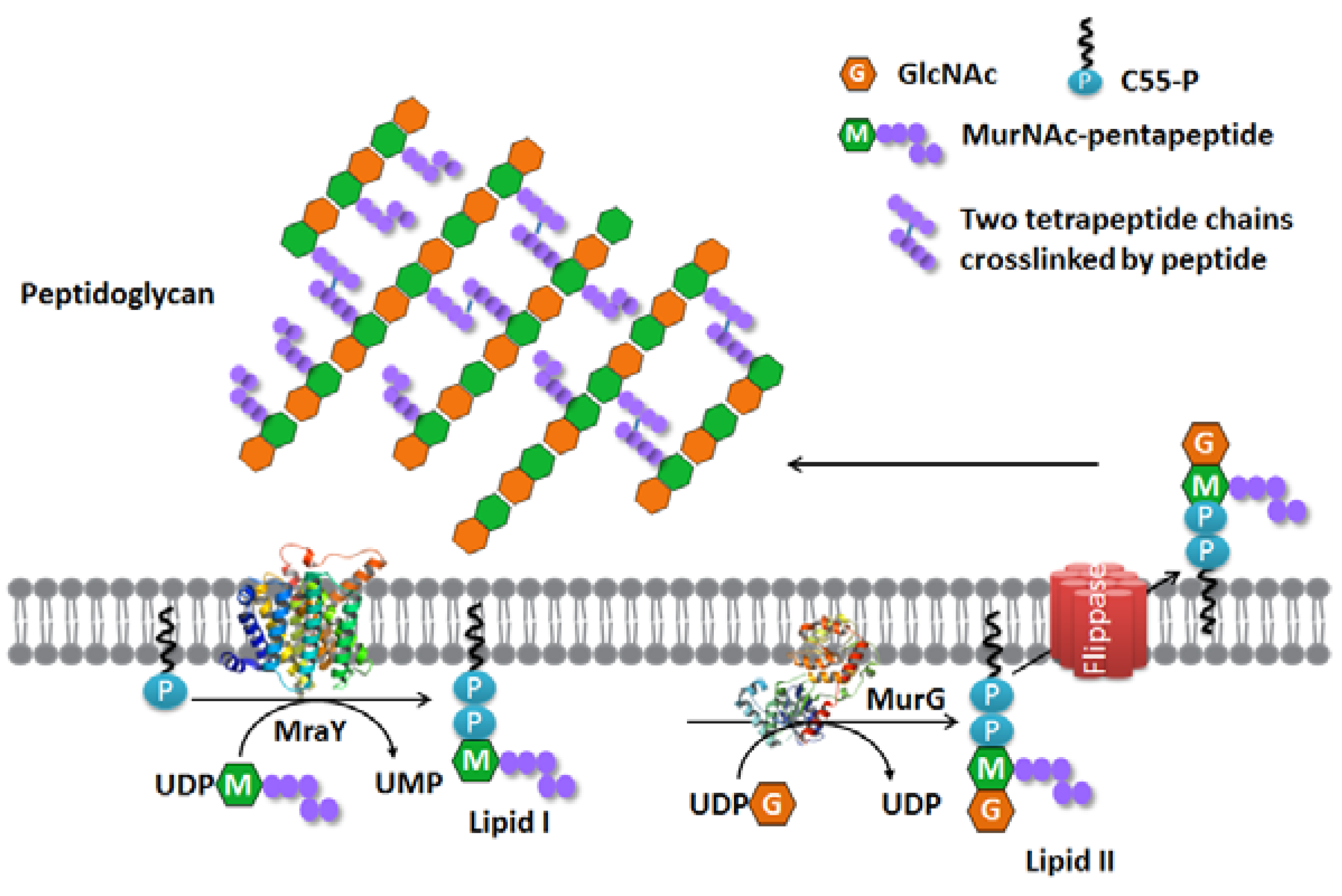
The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
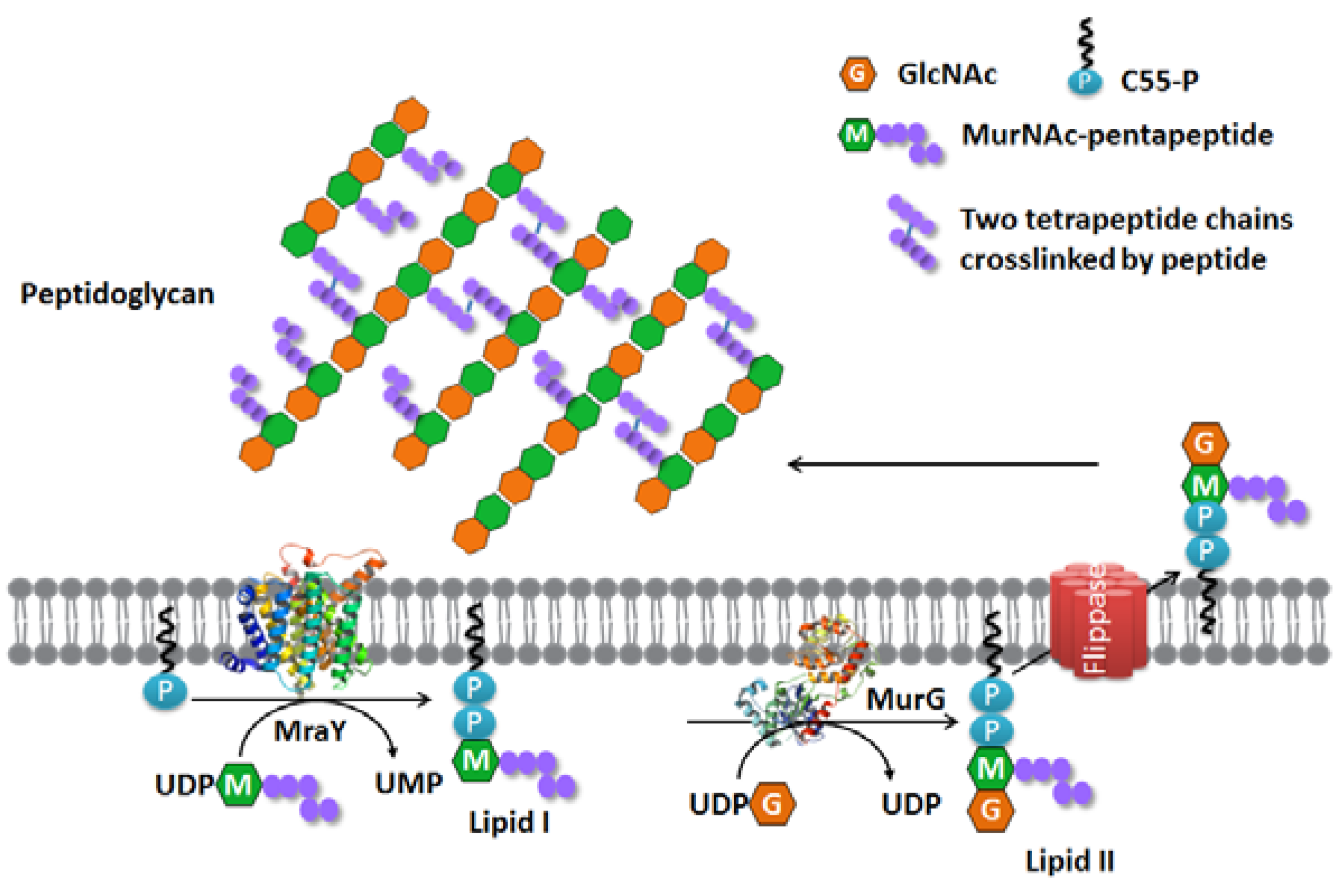
:1. Introduction
2. MraY
2.1. Biochemical Characterization of MraY
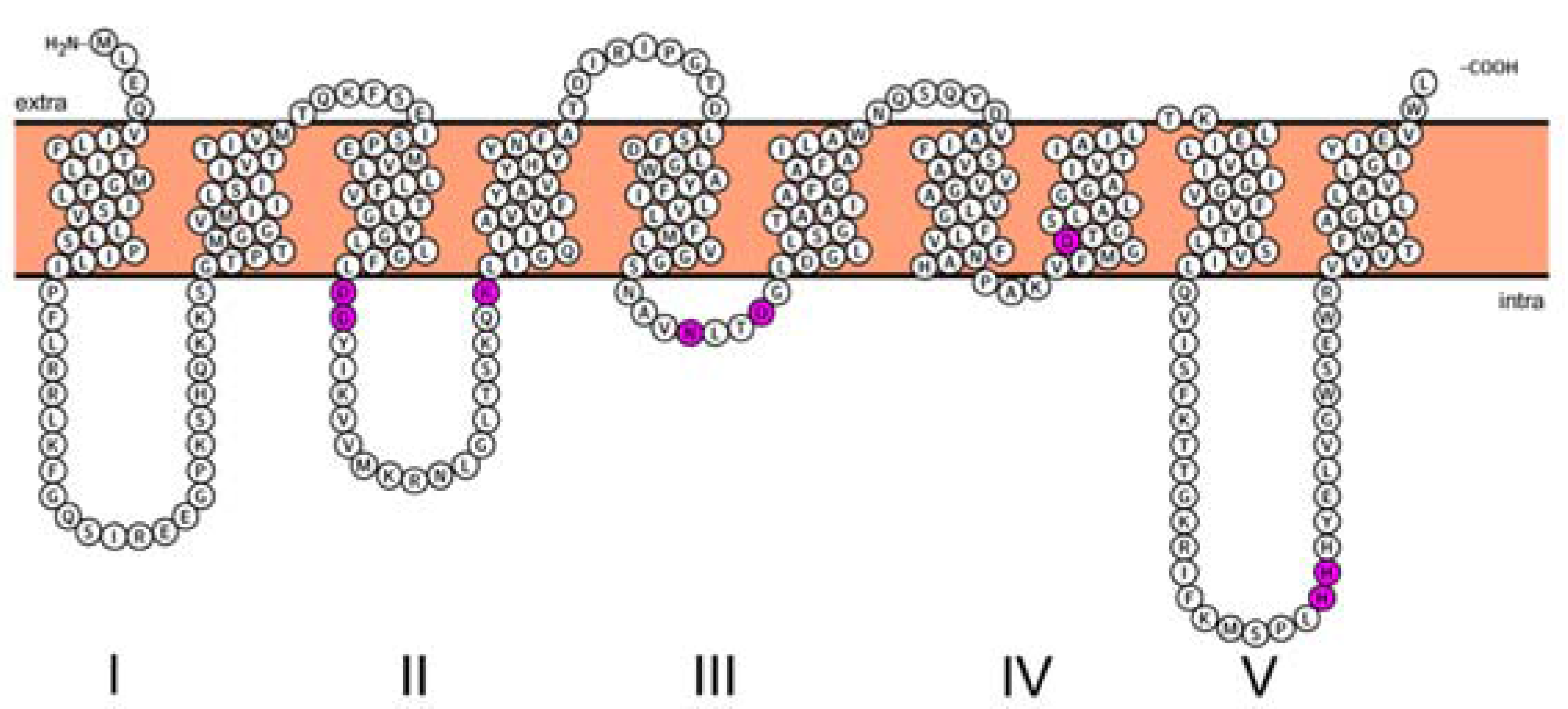
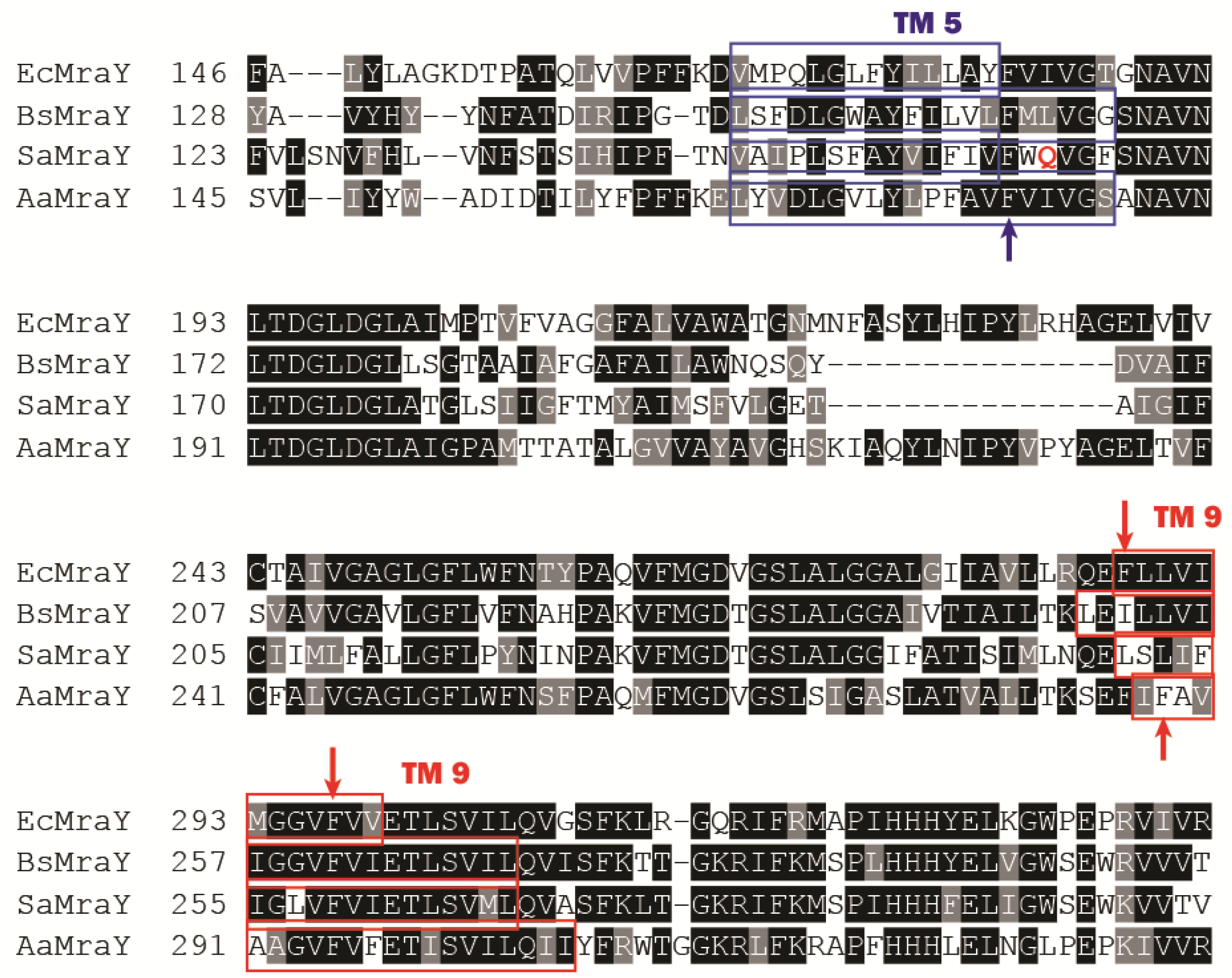
2.2. Structural Characterization of MraY
2.3. MraY Inhibitors and Inhibition Mechanism
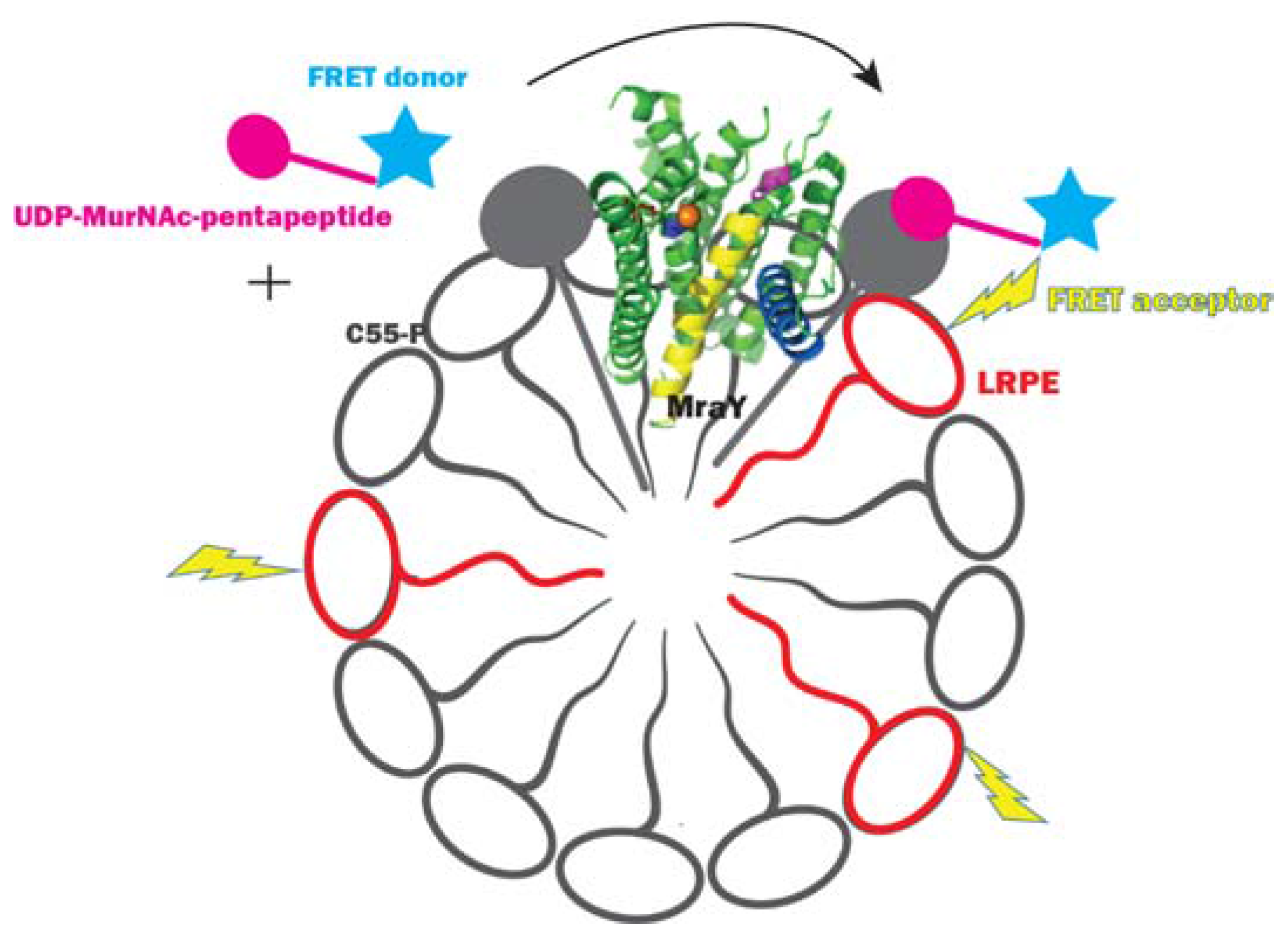
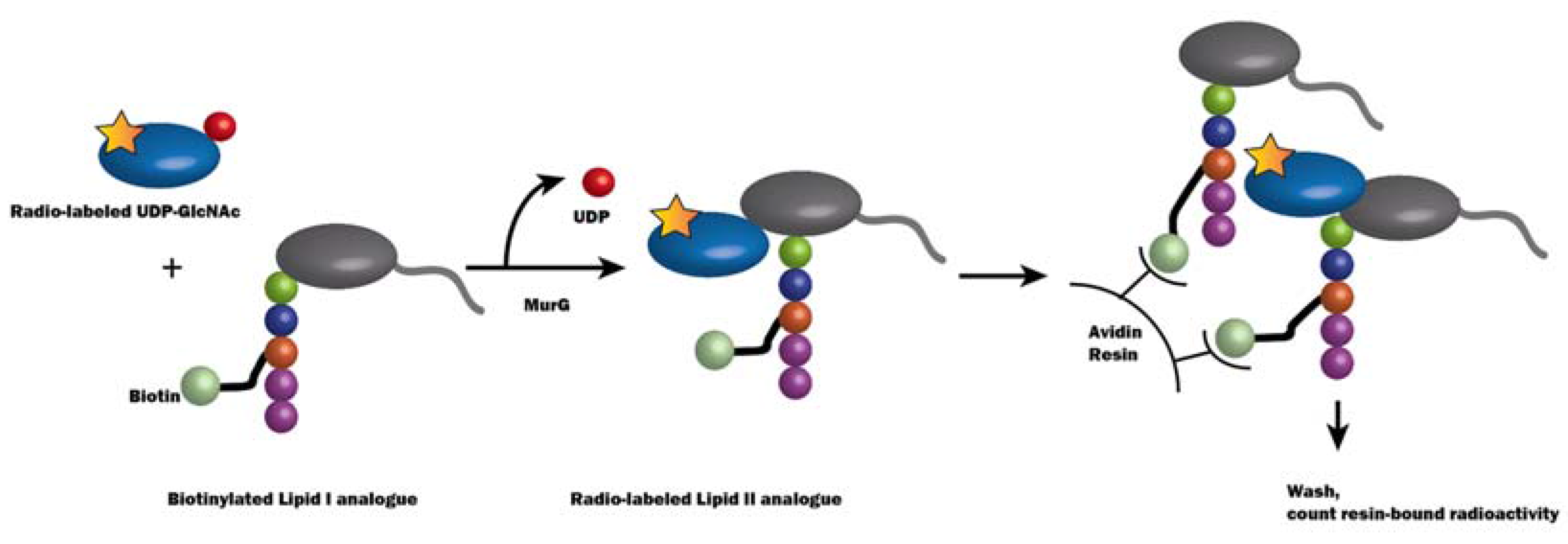
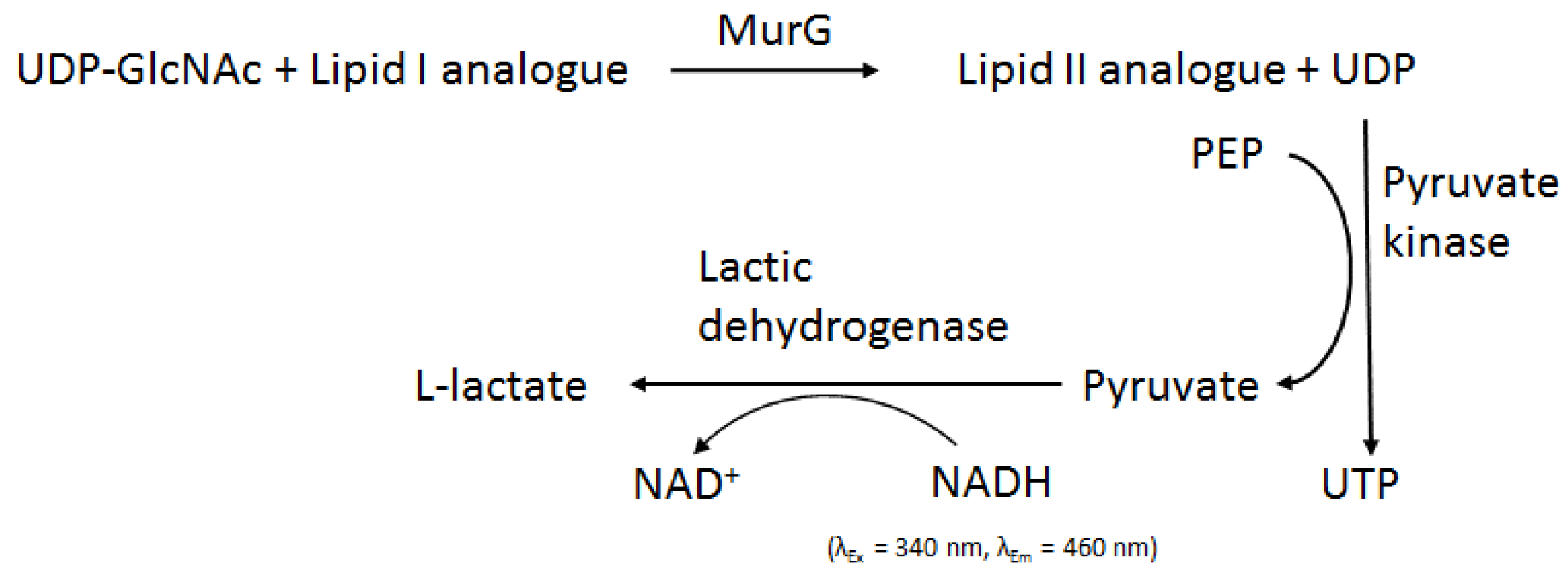
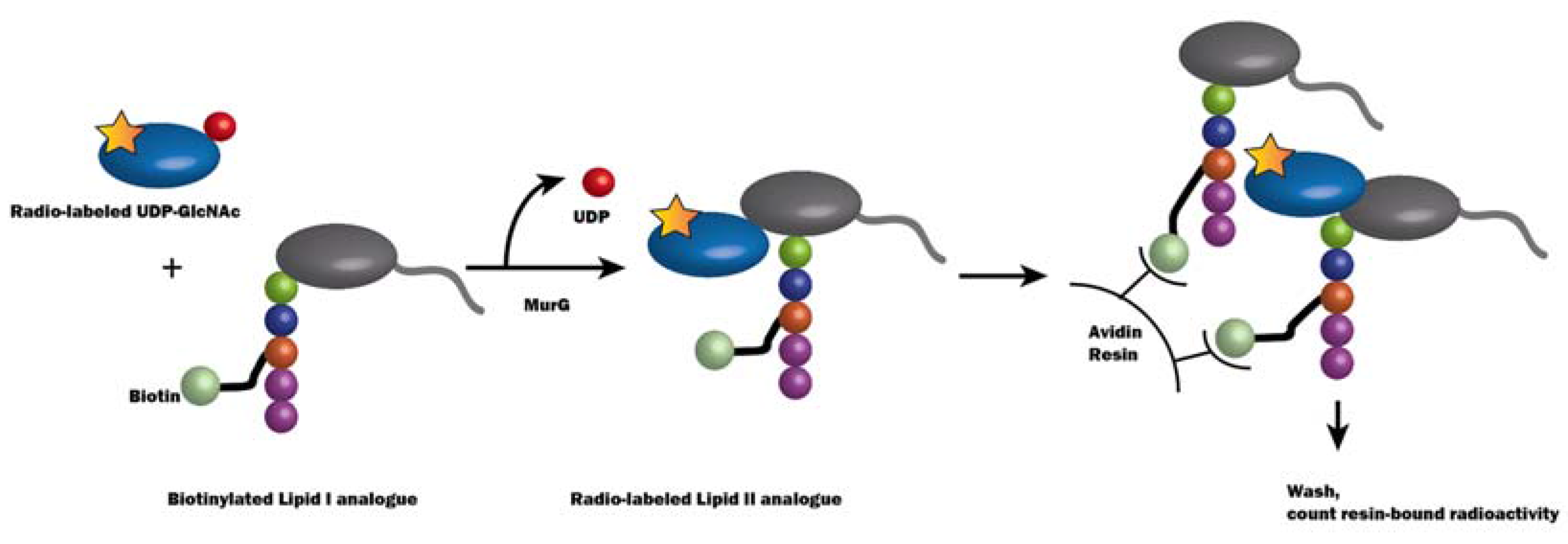
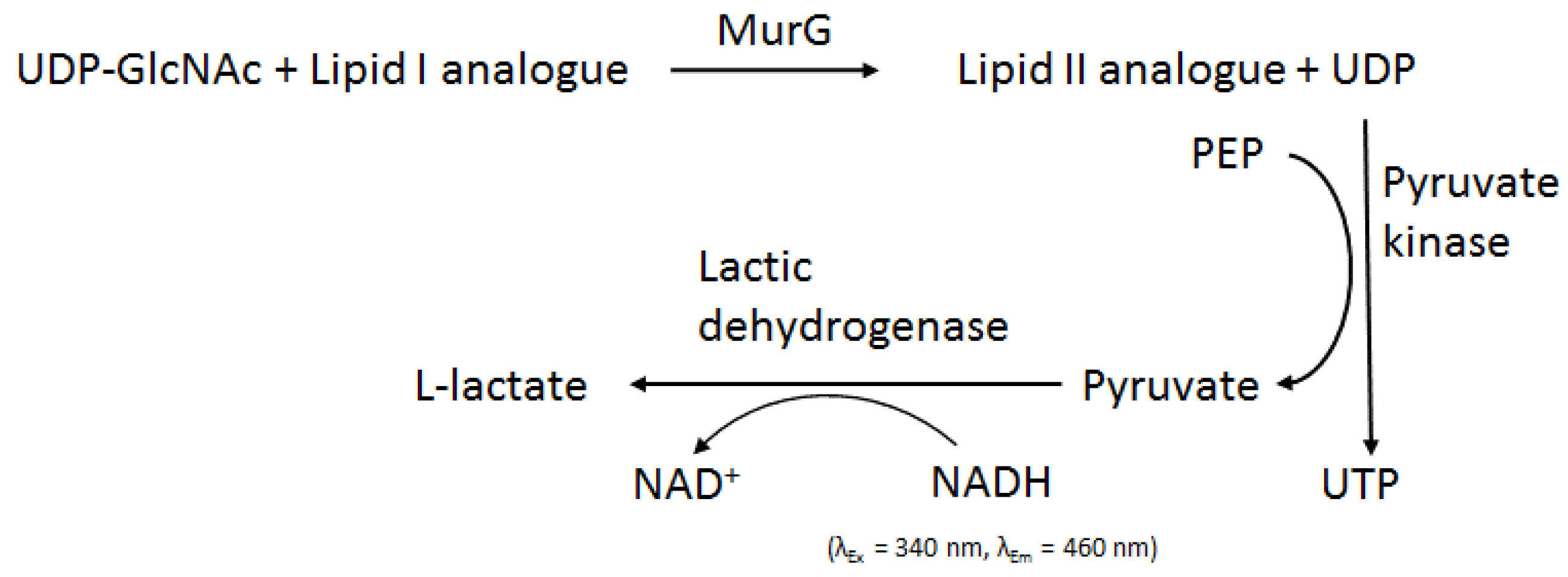
2.3.1. Method Development for MraY Inhibitor Screening
2.3.2. Small Molecules that Inhibit MraY Activity
2.3.3. MraY Inhibition by ΦX174 Protein E
3. MurG
3.1. Biochemical Characterization of MurG
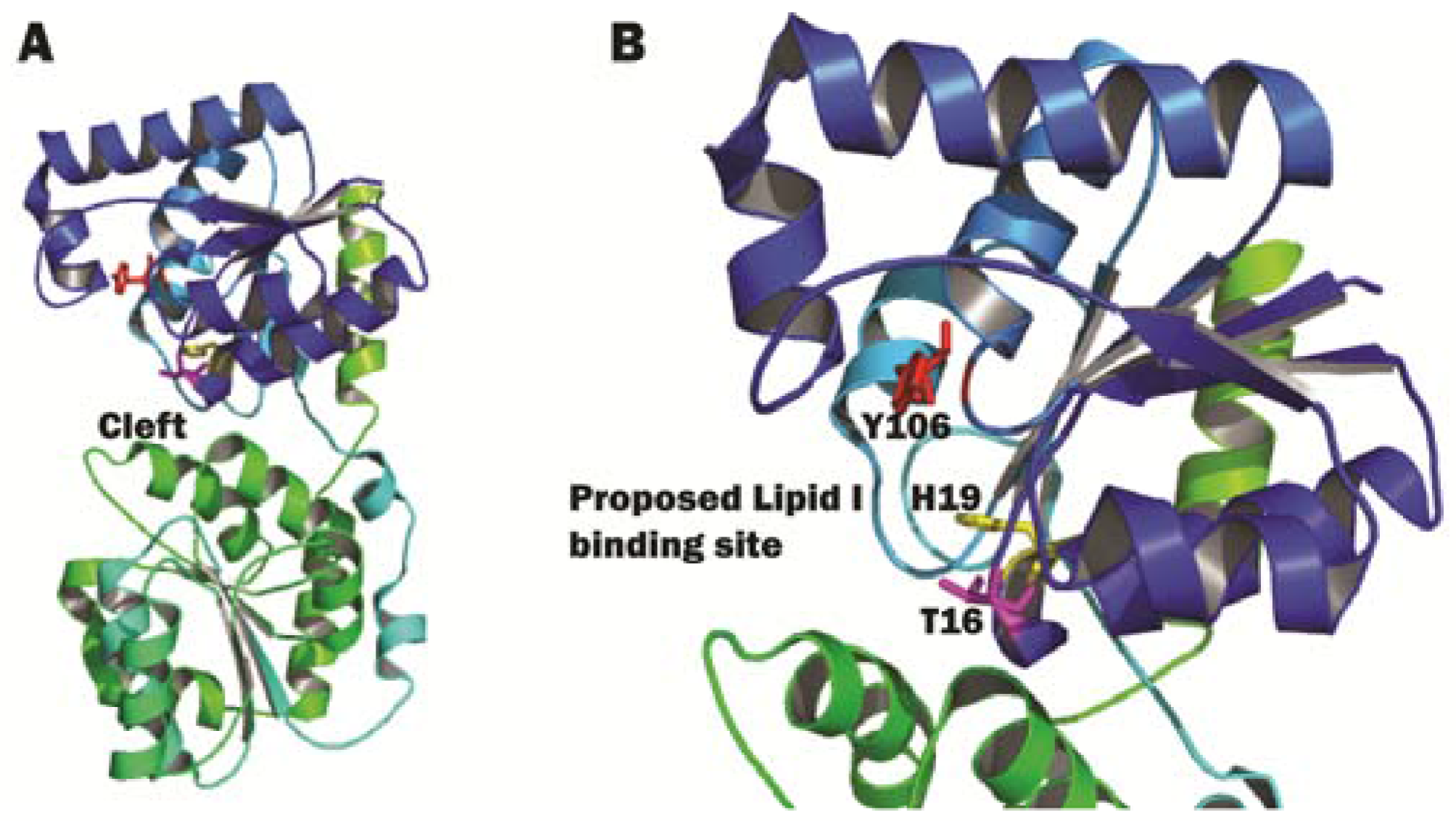
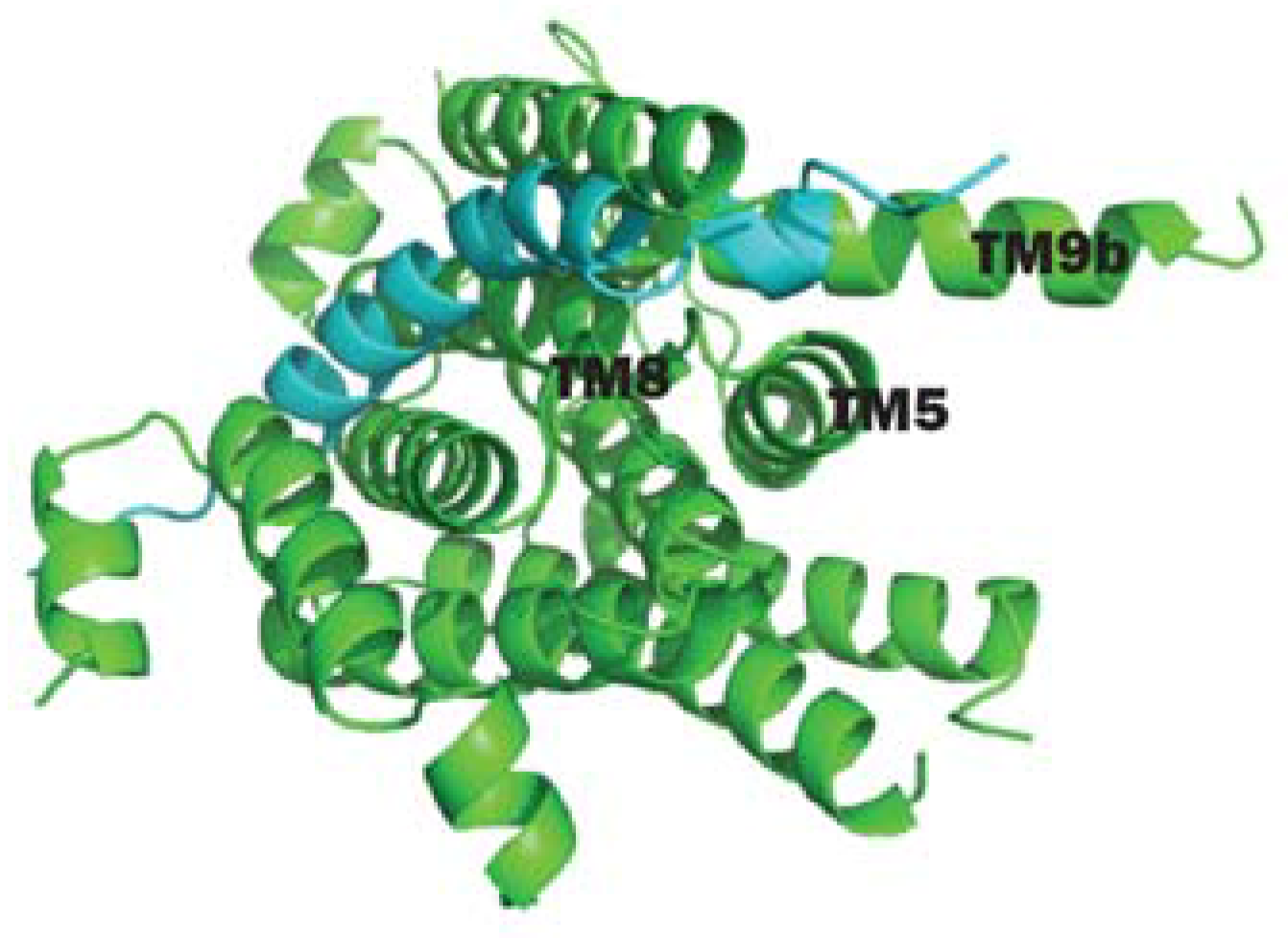
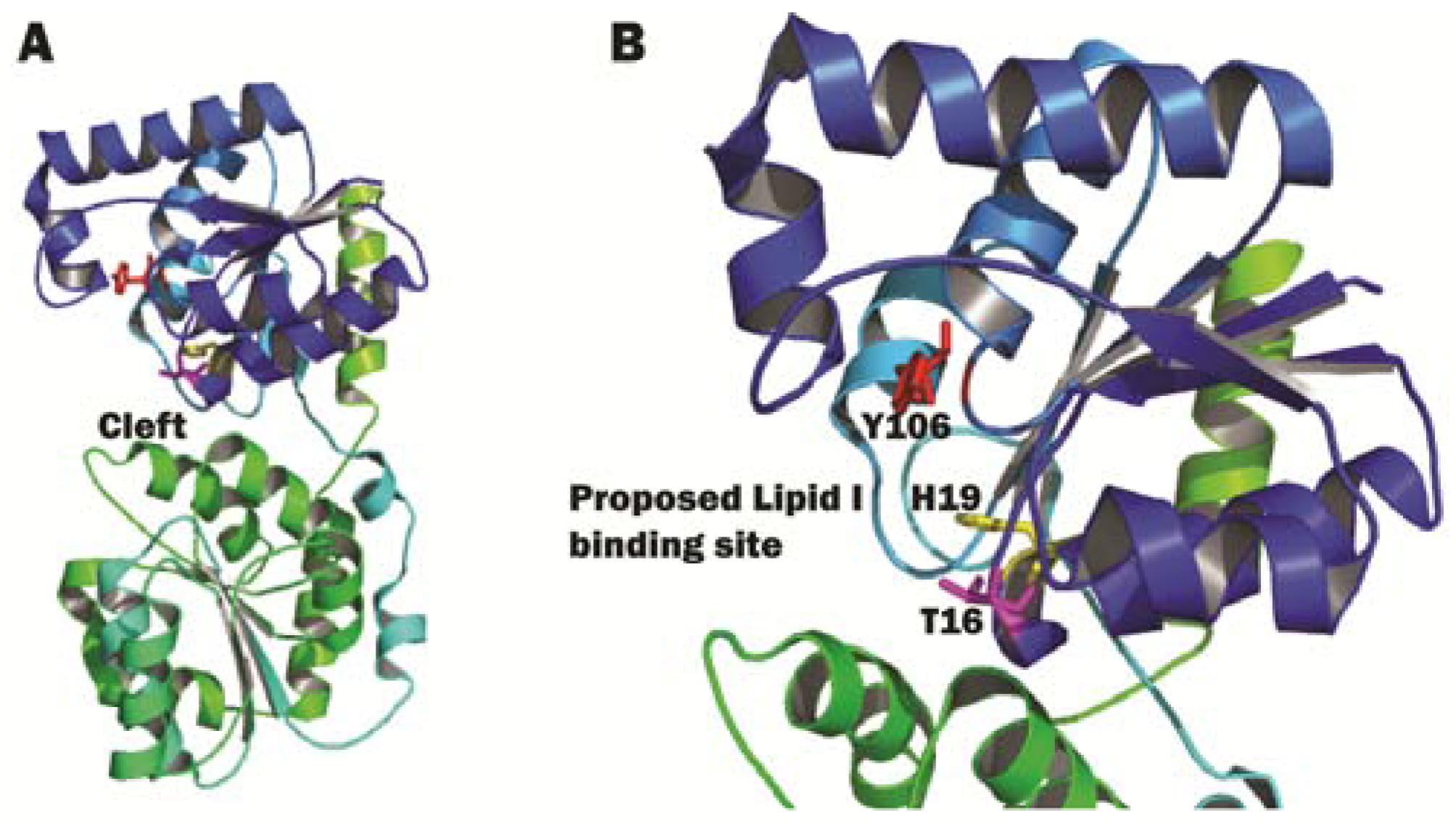
3.2. Structural Characterization of MurG
3.3. MurG Inhibitors
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sobhanifar, S.; King, D.T.; Strynadka, N.C. Fortifying the wall: Synthesis, regulation and degradation of bacterial peptidoglycan. Curr. Opin. Struct. Biol. 2013, 23, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, B.; Mengin-Lecreulx, D.; Bouhss, A. Purification and characterization of the bacterial UDP-GlcNAc: Undecaprenyl-phosphate GlcNAc-1-phosphate transferase WecA. J. Bacteriol. 2008, 190, 7141–7146. [Google Scholar] [CrossRef] [PubMed]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2012, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Rani, C.; Khan, I.A. UDP-GlcNAc pathway: Potential target for inhibitor discovery against M. tuberculosis. Eur. J. Pharm. Sci. 2015, 83, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.L.; Safadi, S.S.; Strynadka, N.C. Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 2012, 81, 451–478. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.T.; Ko, T.P.; Chen, A.P.; Kuo, C.J.; Wang, A.H.; Liang, P.H. Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate: Roles of the metal ion and conserved residues in catalysis. J. Biol. Chem. 2005, 280, 20762–20774. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Chou, C.C.; Hsu, M.F.; Wang, A.H. Proposed carrier lipid-binding site of undecaprenyl pyrophosphate phosphatase from Escherichia coli. J. Biol. Chem. 2014, 289, 18719–18735. [Google Scholar] [CrossRef] [PubMed]
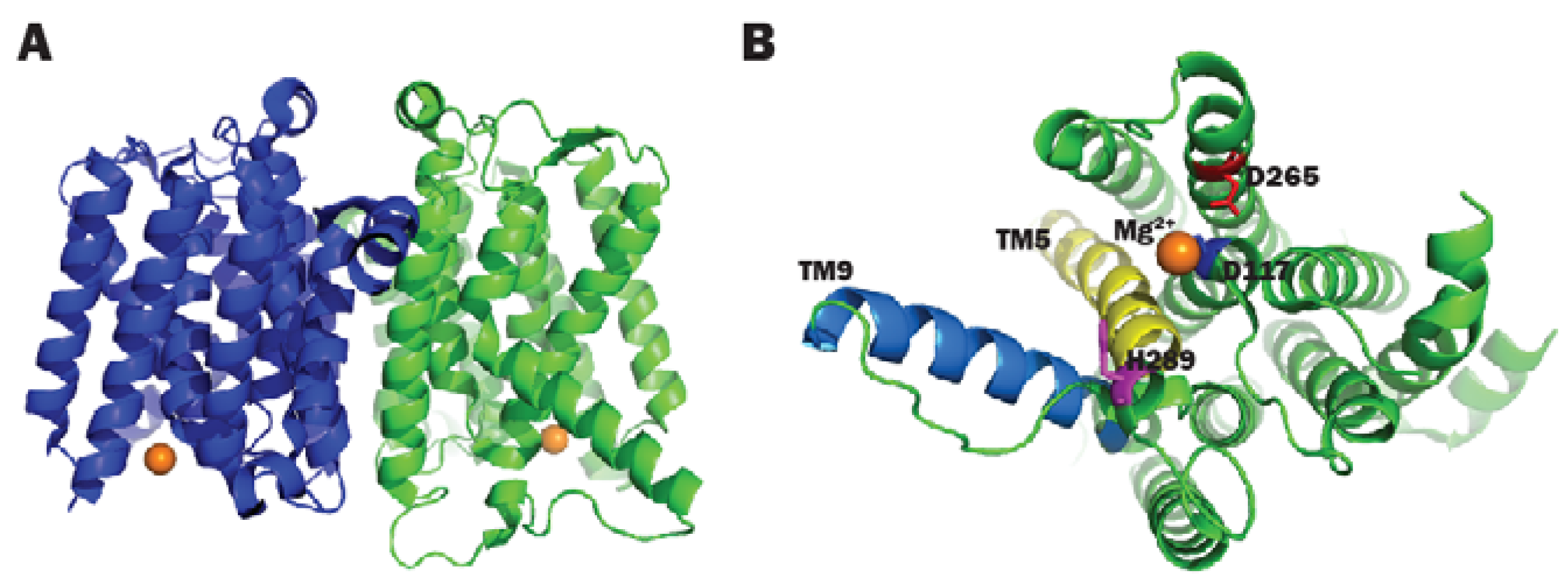
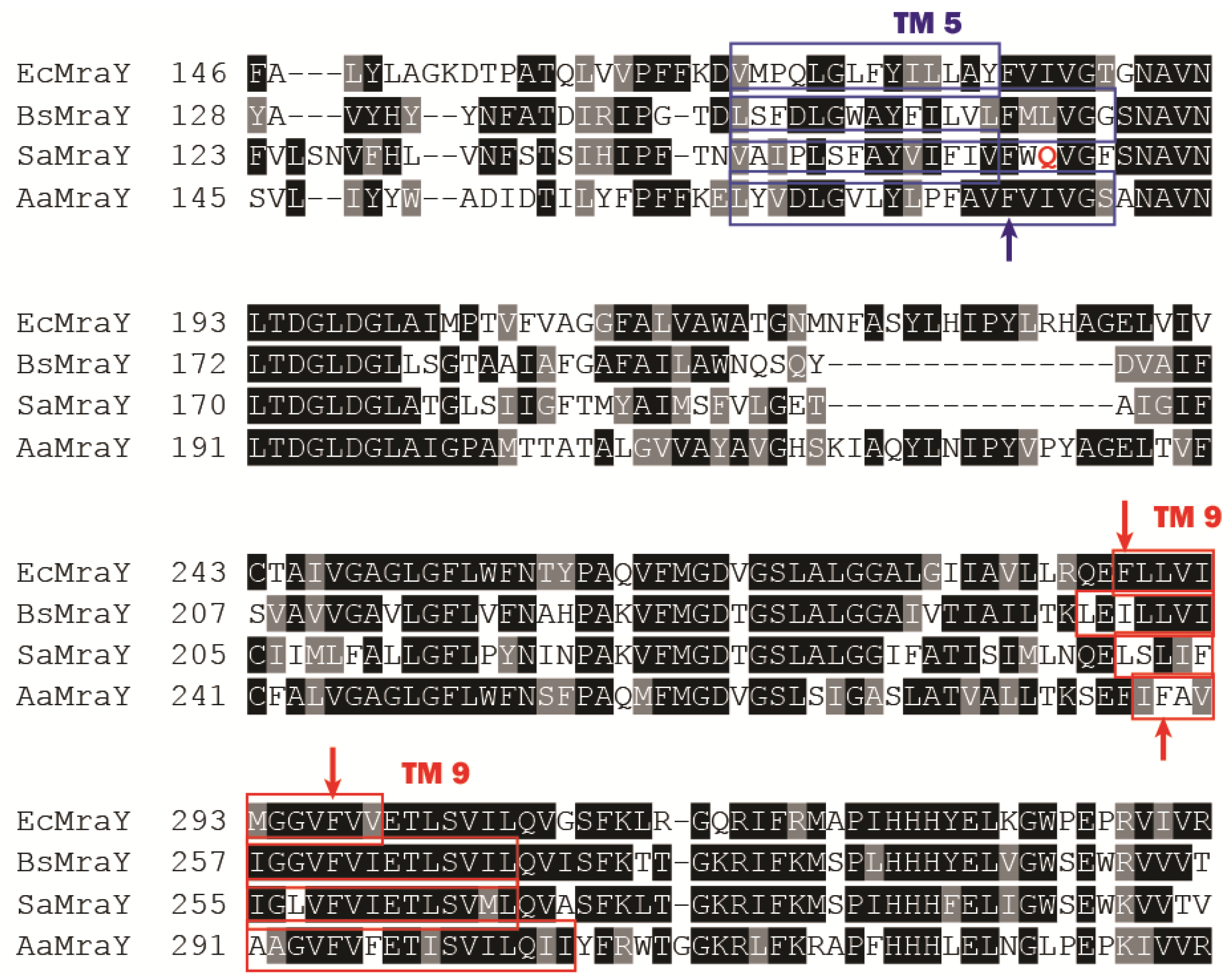
- Chung, B.C.; Zhao, J.; Gillespie, R.A.; Kwon, D.Y.; Guan, Z.; Hong, J.; Zhou, P.; Lee, S.Y. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Sagami, H.; Ogura, K. A novel prenyltransferase from paracoccus denitrificans. Biochem. J. 1986, 233, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Brennan, P.J.; Crick, D.C. Decaprenyl diphosphate synthesis in Mycobacterium tuberculosis. J. Bacteriol. 2004, 186, 7564–7570. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yi, W.; Song, J.K.; Wang, P.G. Current understanding on biosynthesis of microbial polysaccharides. Curr. Top. Med. Chem. 2008, 8, 141–151. [Google Scholar] [PubMed]
- Xia, G.; Maier, L.; Sanchez-Carballo, P.; Li, M.; Otto, M.; Holst, O.; Peschel, A. Glycosylation of wall teichoic acid in Staphylococcus aureus by tarm. J. Biol. Chem. 2010, 285, 13405–13415. [Google Scholar] [CrossRef] [PubMed]
- Bouhss, A.; Trunkfield, A.E.; Bugg, T.D.; Mengin-Lecreulx, D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008, 32, 208–233. [Google Scholar] [CrossRef] [PubMed]
- Weidenmaier, C.; Lee, J.C. Structure and function of surface polysaccharides of Staphylococcus aureus. Curr. Top. Microbiol. Immunol. 2016. [Google Scholar] [CrossRef]
- Mohammadi, T.; van Dam, V.; Sijbrandi, R.; Vernet, T.; Zapun, A.; Bouhss, A.; Diepeveen-de Bruin, M.; Nguyen-Disteche, M.; de Kruijff, B.; Breukink, E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011, 30, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; Sijbrandi, R.; Lutters, M.; Verheul, J.; Martin, N.I.; den Blaauwen, T.; de Kruijff, B.; Breukink, E. Specificity of the transport of lipid II by FtsW in Escherichia coli. J. Biol. Chem. 2014, 289, 14707–14718. [Google Scholar] [CrossRef] [PubMed]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Kahrstrom, C.T. Bacterial physiology: Flipping out over MurJ. Nat. Rev. Microbiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.J.; Biboy, J.; van’t Veer, I.; Breukink, E.; Vollmer, W. Activities and regulation of peptidoglycan synthases. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Kim, Y.S.; Rojviriya, C.; Ha, S.C.; Kang, B.S.; Kim, Y.G. Crystal structures of bifunctional penicillin-binding protein 4 from listeria monocytogenes. Antimicrob. Agents Chemother. 2013, 57, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.T.; Lai, Y.T.; Huang, C.Y.; Chou, L.Y.; Shih, H.W.; Cheng, W.C.; Wong, C.H.; Ma, C. Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 8824–8829. [Google Scholar] [CrossRef] [PubMed]
- Slusarz, R.; Szulc, M.; Madaj, J. Molecular modeling of gram-positive bacteria peptidoglycan layer, selected glycopeptide antibiotics and vancomycin derivatives modified with sugar moieties. Carbohydr. Res. 2014, 389, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, V.; Akkapeddi, P.; Manjunath, G.B.; Haldar, J. Membrane active vancomycin analogues: A strategy to combat bacterial resistance. J. Med. Chem. 2014, 57, 4558–4568. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Bae, S.I.; Reu, F.J.; Jacobs, B.S.; Lindner, D.J.; Borden, E.C. Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2004, 11, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Worthington, R.J.; Melander, C. Overcoming resistance to beta-lactam antibiotics. J. Org. Chem. 2013, 78, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Hamed, R.B.; Gomez-Castellanos, J.R.; Henry, L.; Ducho, C.; McDonough, M.A.; Schofield, C.J. The enzymes of beta-lactam biosynthesis. Nat. Prod. Rep. 2013, 30, 21–107. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.; Lloyd, A.J.; Roper, D.I. Phospho-murnac-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect. Disord. Drug Targets 2006, 6, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Matsuhashi, M.; Haskin, M.A.; Strominger, J.L. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide: Presumed membrane transport intermediates in cell wall synthesis. Proc. Natl. Acad. Sci. USA 1965, 53, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Wachi, M.; Jung, H.K.; Ishino, F.; Matsuhashi, M. The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide: Undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J. Bacteriol. 1991, 173, 1021–1026. [Google Scholar] [PubMed]
- Brandish, P.E.; Kimura, K.I.; Inukai, M.; Southgate, R.; Lonsdale, J.T.; Bugg, T.D. Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: Inhibition of phospho-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob. Agents Chemother. 1996, 40, 1640–1644. [Google Scholar] [PubMed]
- Brandish, P.E.; Burnham, M.K.; Lonsdale, J.T.; Southgate, R.; Inukai, M.; Bugg, T.D. Slow binding inhibition of phospho-N-acetylmuramyl-pentapeptide-translocase (Escherichia coli) by mureidomycin A. J. Biol. Chem. 1996, 271, 7609–7614. [Google Scholar] [PubMed]
- Bouhss, A.; Crouvoisier, M.; Blanot, D.; Mengin-Lecreulx, D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 2004, 279, 29974–29980. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Munch, D.; Schneider, T.; Sahl, H.G.; Bouhss, A.; Ghoshdastider, U.; Wang, J.; Dotsch, V.; Wang, X.; Bernhard, F. Preparative scale cell-free production and quality optimization of MraY homologues in different expression modes. J. Biol. Chem. 2011, 286, 38844–38853. [Google Scholar] [CrossRef]
- Liu, Y.; Rodrigues, J.P.; Bonvin, A.M.; Zaal, E.A.; Berkers, C.R.; Heger, M.; Gawarecka, K.; Swiezewska, E.; Breukink, E.; Egmond, M.R. New insight in the catalytic mechanism of bacterial MraY from enzyme kinetics and docking studies. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, B.; Olatunji, S.; Crouvoisier, M.; El Ghachi, M.; Blanot, D.; Mengin-Lecreulx, D.; Bouhss, A. Catalytic mechanism of MraY and WecA, two paralogues of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily. Biochimie 2016, 127, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Al-Dabbagh, B.; Henry, X.; Ghachi, M.E.; Auger, G.V.; Blanot, D.; Parquet, C.; Mengin-Lecreulx, D.; Bouhss, A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.; Zocher, M.; Müller, D.; Münch, D.; Schneider, T.; Sahl, H.-G.; Scholz, F.; Wachtveitl, J.; Ma, Y.; Proverbio, D.; et al. Characterization of co-translationally formed nanodisc complexes with small multidrug transporters, proteorhodopsin and with the E. coli MraY translocase. BBA—Biomembranes 2012, 1818, 3098–3106. [Google Scholar] [CrossRef] [PubMed]
- Henrich, E.; Dotsch, V.; Bernhard, F. Screening for lipid requirements of membrane proteins by combining cell-free expression with nanodiscs. Methods Enzymol. 2015, 556, 351–369. [Google Scholar] [PubMed]
- Henrich, E.; Ma, Y.; Engels, I.; Munch, D.; Otten, C.; Schneider, T.; Henrichfreise, B.; Sahl, H.G.; Dotsch, V.; Bernhard, F. Lipid requirements for the enzymatic activity of MraY translocases and in vitro reconstitution of the lipid II synthesis pathway. J. Biol. Chem. 2016, 291, 2535–2546. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J.N.; Strominger, J.L. Complex lipid requirements for detergent-solubilized phosphoacetylmuramyl-pentapeptide translocase from micrococcus luteus. Proc. Natl. Acad. Sci. USA 1972, 69, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.J.; Brandish, P.E.; Gilbey, A.M.; Bugg, T.D.H. Phospho-N-acetyl-muramyl-pentapeptide translocase from Escherichia coli: Catalytic role of conserved aspartic acid residues. J. Bacteriol. 2004, 186, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Pless, D.D.; Neuhaus, F.C. Initial membrane reaction in peptidoglycan synthesis. Lipid dependence of phospho-N-acetylmuramyl-pentapeptide translocase (exchange reaction). J. Biol. Chem. 1973, 248, 1568–1576. [Google Scholar] [PubMed]
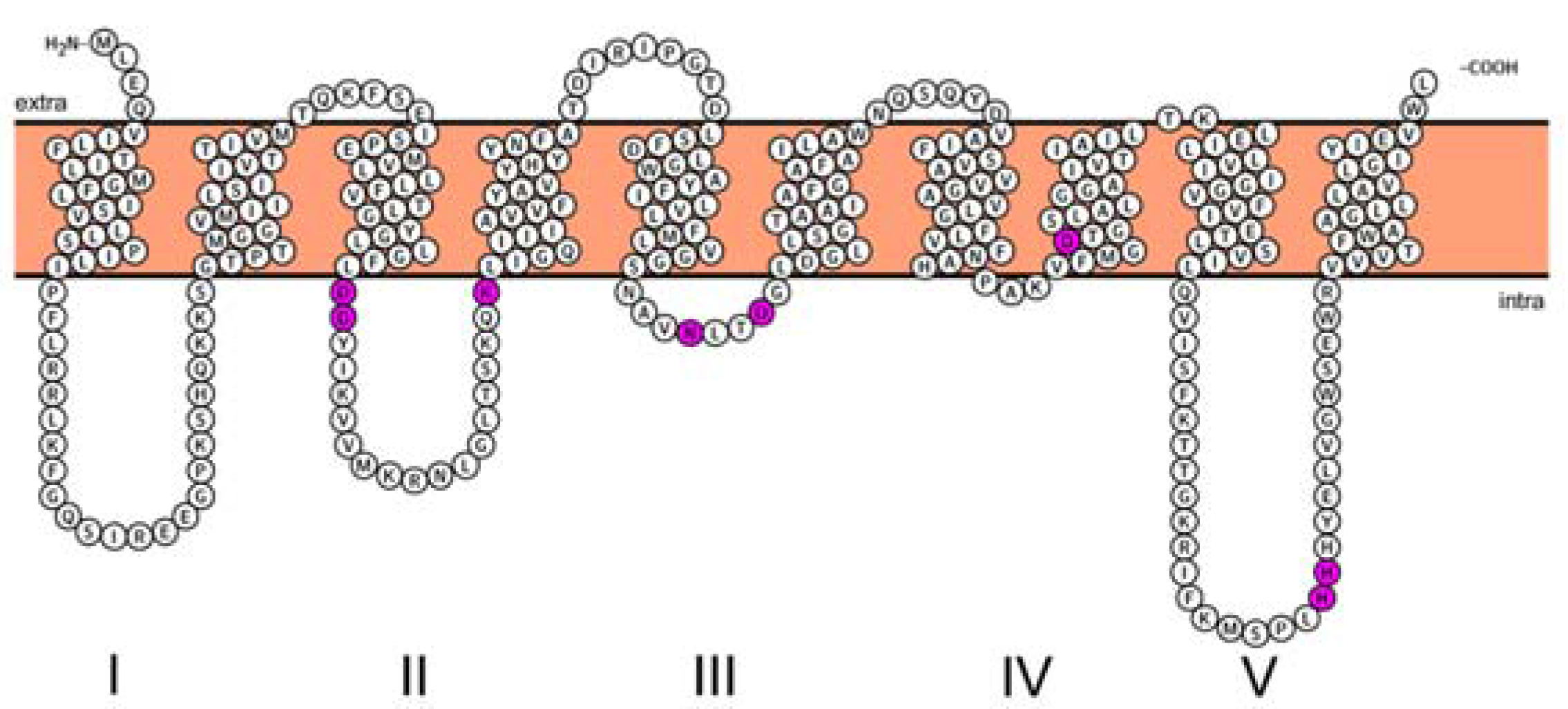
- Bouhss, A.; Mengin-Lecreulx, D.; le Beller, D.; van Heijenoort, J. Topological analysis of the MraY protein catalysing the first membrane step of peptidoglycan synthesis. Mol. Microbiol. 1999, 34, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.H.; Muller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Heydanek, M.G., Jr.; Struve, W.G.; Neuhaus, F.C. On the initial stage in peptidoglycan synthesis. 3. Kinetics and uncoupling of phospho-N-acetylmuramyl-pentapeptide translocase (uridine 5’-phosphate). Biochemistry 1969, 8, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J.N.; Strominger, J.L. Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J. Bacteriol. 1972, 112, 1306–1309. [Google Scholar] [PubMed]
- Maillard, A.P.; Biarrotte-Sorin, S.; Villet, R.; Mesnage, S.; Bouhss, A.; Sougakoff, W.; Mayer, C.; Arthur, M. Structure-based site-directed mutagenesis of the UDP-MurNAc-pentapeptide-binding cavity of the FemX alanyl transferase from Weissella viridescens. J. Bacteriol. 2005, 187, 3833–3838. [Google Scholar] [CrossRef] [PubMed]
- Price, N.P.; Momany, F.A. Modeling bacterial UDP-HexNAc: Polyprenol-P HexNAc-1-P transferases. Glycobiology 2005, 15, 29R–42R. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Kitich, A.; Gober, J.W. Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol. Microbiol. 2010, 76, 616–633. [Google Scholar] [CrossRef] [PubMed]
- Stachyra, T.; Dini, C.; Ferrari, P.; Bouhss, A.; van Heijenoort, J.; Mengin-Lecreulx, D.; Blanot, D.; Biton, J.; Le Beller, D. Fluorescence detection-based functional assay for high-throughput screening for MraY. Antimicrob. Agents Chemother. 2004, 48, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Mihalyi, A.; Jamshidi, S.; Slikas, J.; Bugg, T.D.H. Identification of novel inhibitors of phospho-murnac-pentapeptide translocase MraY from library screening: Isoquinoline alkaloid michellamine B and xanthene dye phloxine B. Bioorg. Med. Chem. 2014, 22, 4566–4571. [Google Scholar] [CrossRef] [PubMed]
- Howard, N.I.; Bugg, T.D. Synthesis and activity of 5′-uridinyl dipeptide analogues mimicking the amino terminal peptide chain of nucleoside antibiotic mureidomycin A. Bioorg. Med. Chem. 2003, 11, 3083–3099. [Google Scholar] [CrossRef]
- Mihalyi, A. Screening for novel inhibitors of phospho-murnac-pentapeptide translocase MraY. Thesis Univ. Warwick 2014, 22, 4566–4571. [Google Scholar]
- Branstrom, A.A.; Midha, S.; Longley, C.B.; Han, K.; Baizman, E.R.; Axelrod, H.R. Assay for identification of inhibitors for bacterial MraY translocase or MurG transferase. Anal. Biochem. 2000, 280, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, S.; Kumar, V.P.; Chandrakala, B.; Jha, R.K.; Solapure, S.M.; de Sousa, S.M. Scintillation proximity assay for inhibitors of Escherichia coli MurG and, optionally, MraY. Antimicrob. Agents Chemother. 2005, 49, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
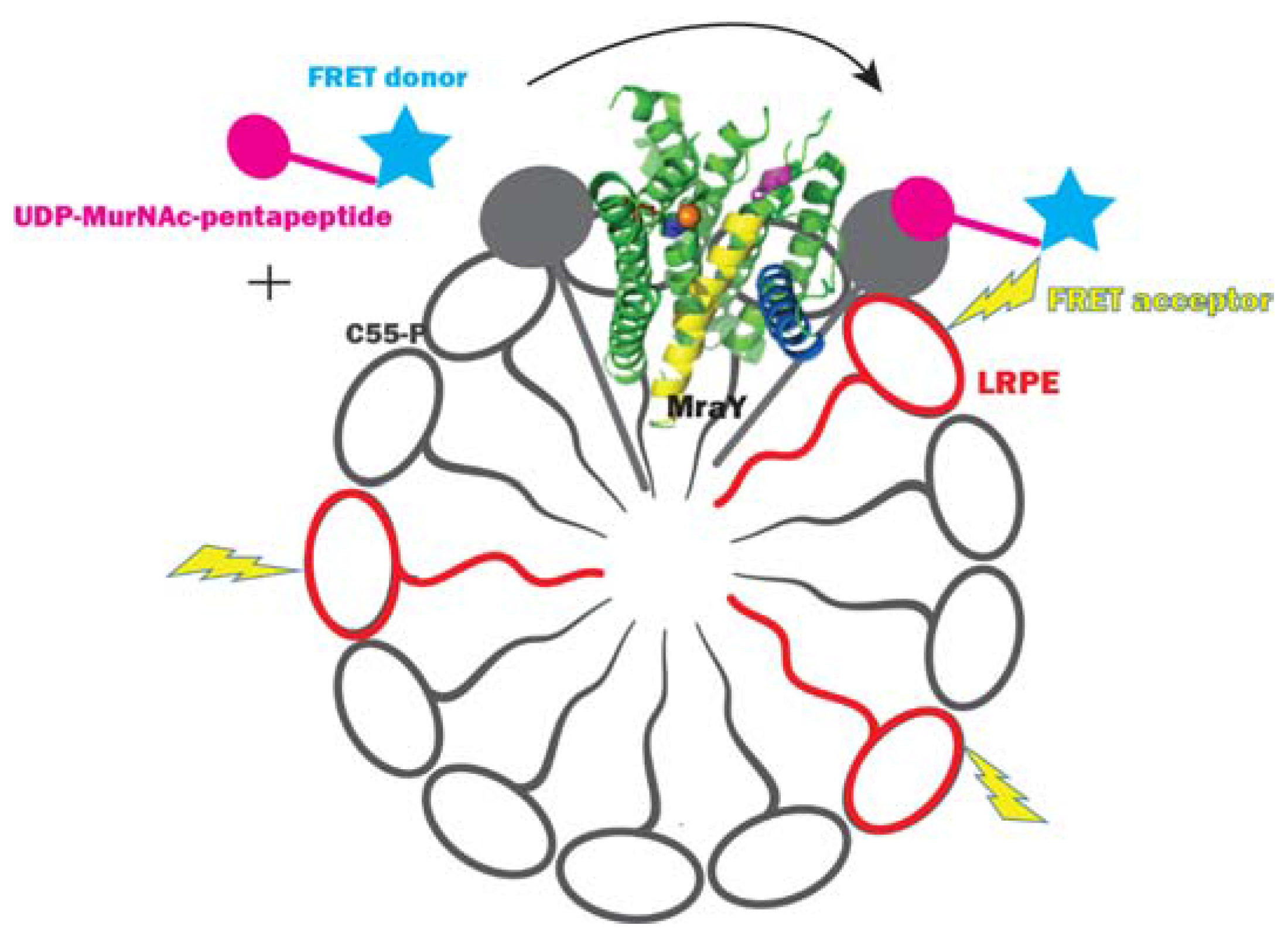
- Shapiro, A.B.; Jahic, H.; Gao, N.; Hajec, L.; Rivin, O. A high-throughput, homogeneous, fluorescence resonance energy transfer-based assay for phospho-N-acetylmuramoyl-pentapeptide translocase (MraY). J. Biomol. Screen. 2012, 17, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Solapure, S.M.; Raphael, P.; Gayathri, C.N.; Barde, S.P.; Chandrakala, B.; Das, K.S.; de Sousa, S.M. Development of a microplate-based scintillation proximity assay for MraY using a modified substrate. J. Biomol. Screen. 2005, 10, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hyland, S.A.; Anderson, M.S. A high-throughput solid-phase extraction assay capable of measuring diverse polyprenyl phosphate: Sugar-1-phosphate transferases as exemplified by the WecA, MraY, and MurG proteins. Anal. Biochem. 2003, 317, 156–165. [Google Scholar] [CrossRef]
- Zawadzke, L.E.; Wu, P.; Cook, L.; Fan, L.; Casperson, M.; Kishnani, M.; Calambur, D.; Hofstead, S.J.; Padmanabha, R. Targeting the MraY and MurG bacterial enzymes for antimicrobial therapeutic intervention. Anal. Biochem. 2003, 314, 243–252. [Google Scholar] [CrossRef]
- Dini, C.; Collette, P.; Drochon, N.; Guillot, J.C.; Lemoine, G.; Mauvais, P.; Aszodi, J. Synthesis of the nucleoside moiety of liposidomycins: Elucidation of the pharmacophore of this family of MraY inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 1839–1843. [Google Scholar] [CrossRef]
- Isono, F.; Katayama, T.; Inukai, M.; Haneishi, T. Mureidomycins A–D, novel peptidylnucleoside antibiotics with spheroplast forming activity. III. Biological properties. J. Antibiot. 1989, 42, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Muramatsu, Y.; Minami, E.; Masuda, K.; Sakaida, Y.; Endo, S.; Suzuki, T.; Ishida, O.; Takatsu, T.; Miyakoshi, S.; et al. A novel assay of bacterial peptidoglycan synthesis for natural product screening. J. Antibiot. 2009, 62, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rodolis, M.T.; Mihalyi, A.; Ducho, C.; Eitel, K.; Gust, B.; Goss, R.J.; Bugg, T.D. Mechanism of action of the uridyl peptide antibiotics: An unexpected link to a protein-protein interaction site in translocase MraY. Chem. Commun. 2014, 50, 13023–13025. [Google Scholar] [CrossRef] [PubMed]
- Rodolis, M.T.; Mihalyi, A.; O’Reilly, A.; Slikas, J.; Roper, D.I.; Hancock, R.E.W.; Bugg, T.D.H. Identification of a novel inhibition site in translocase MraY based upon the site of interaction with lysis protein E from bacteriophage ΦX174. ChemBioChem 2014, 15, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Tanino, T.; Al-Dabbagh, B.; Mengin-Lecreulx, D.; Bouhss, A.; Oyama, H.; Ichikawa, S.; Matsuda, A. Mechanistic analysis of muraymycin analogues: A guide to the design of MraY inhibitors. J. Med. Chem. 2011, 54, 8421–8439. [Google Scholar] [CrossRef] [PubMed]
- Takeoka, Y.; Tanino, T.; Sekiguchi, M.; Yonezawa, S.; Sakagami, M.; Takahashi, F.; Togame, H.; Tanaka, Y.; Takemoto, H.; Ichikawa, S.; et al. Expansion of antibacterial spectrum of muraymycins toward Pseudomonas aeruginosa. ACS Med. Chem. Lett. 2014, 5, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Wiegmann, D.; Koppermann, S.; Wirth, M.; Niro, G.; Leyerer, K.; Ducho, C. Muraymycin nucleoside-peptide antibiotics: Uridine-derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J. Org. Chem. 2016, 12, 769–795. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.C.; Mashalidis, E.H.; Tanino, T.; Kim, M.; Matsuda, A.; Hong, J.; Ichikawa, S.; Lee, S.Y. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 2016, 533, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Dini, C.; Didier-Laurent, S.; Drochon, N.; Feteanu, S.; Guillot, J.C.; Monti, F.; Uridat, E.; Zhang, J.; Aszodi, J. Synthesis of sub-micromolar inhibitors of MraY by exploring the region originally occupied by the diazepanone ring in the liposidomycin structure. Bioorg. Med. Chem. Lett. 2002, 12, 1209–1213. [Google Scholar] [CrossRef]
- Dini, C.; Drochon, N.; Feteanu, S.; Guillot, J.C.; Peixoto, C.; Aszodi, J. Synthesis of analogues of the O-β-d-ribofuranosyl nucleoside moiety of liposidomycins. Part 1: Contribution of the amino group and the uracil moiety upon the inhibition of MraY. Bioorg. Med. Chem. Lett. 2001, 11, 529–531. [Google Scholar] [CrossRef]
- Dini, C.; Drochon, N.; Guillot, J.C.; Mauvais, P.; Walter, P.; Aszodi, J. Synthesis of analogues of the O-β-d-ribofuranosyl nucleoside moiety of liposidomycins. Part 2: Role of the hydroxyl groups upon the inhibition of MraY. Bioorg. Med. Chem. Lett. 2001, 11, 533–536. [Google Scholar] [CrossRef]
- Fer, M.J.; Olatunji, S.; Bouhss, A.; Calvet-Vitale, S.; Gravier-Pelletier, C. Toward analogues of MraY natural inhibitors: Synthesis of 5′-triazole-substituted-aminoribosyl uridines through a Cu-catalyzed azide-alkyne cycloaddition. J. Org. Chem. 2013, 78, 10088–10105. [Google Scholar] [CrossRef] [PubMed]
- Fer, M.J.; Bouhss, A.; Patrao, M.; le Corre, L.; Pietrancosta, N.; Amoroso, A.; Joris, B.; Mengin-Lecreulx, D.; Calvet-Vitale, S.; Gravier-Pelletier, C. 5′-methylene-triazole-substituted-aminoribosyl uridines as MraY inhibitors: Synthesis, biological evaluation and molecular modeling. Org. Biomol. Chem. 2015, 13, 7193–7222. [Google Scholar] [CrossRef] [PubMed]
- Unterreitmeier, S.; Fuchs, A.; Schaffler, T.; Heym, R.G.; Frishman, D.; Langosch, D. Phenylalanine promotes interaction of transmembrane domains via GxxxG motifs. J. Mol. Biol. 2007, 374, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, A.; Keenan, R.W.; Elbein, A.D. Mechanism of action of tunicamycin on the UDP-GlcNAc: Dolichyl-phosphate GlcNAc-1-phosphate transferase. Biochemistry 1979, 18, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses P-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in L1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Blasi, U.; Halfmann, G.; Szostak, M.; Wanner, G.; Lubitz, W. PhiX174 protein e-mediated lysis of Escherichia coli. Biochimie 1990, 72, 191–200. [Google Scholar] [CrossRef]
- Bernhardt, T.G.; Roof, W.D.; Young, R. Genetic evidence that the bacteriophage PhiX174 lysis protein inhibits cell wall synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4297–4302. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, G.; Gotz, F.; Lubitz, W. Expression of bacteriophage PhiX174 lysis gene E in Staphylococcus carnosus TM300. FEMS Microbiol. Lett. 1993, 108, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, T.G.; Struck, D.K.; Young, R. The lysis protein E of PhiX174 is a specific inhibitor of the MraY-catalyzed step in peptidoglycan synthesis. J. Biol. Chem. 2001, 276, 6093–6097. [Google Scholar] [CrossRef] [PubMed]
- Mendel, S.; Holbourn, J.M.; Schouten, J.A.; Bugg, T.D.H. Interaction of the transmembrane domain of lysis protein E from bacteriophage PhiX174 with bacterial translocase MraY and peptidyl-prolyl isomerase SlyD. Microbiology 2006, 152, 2959–2967. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Clemons, W.M., Jr. Minimal requirements for inhibition of MraY by lysis protein E from bacteriophage PhiX174. Mol. Microbiol. 2012, 85, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Struck, D.K.; Bernhardt, T.G.; Young, R. Genetic analysis of MraY inhibition by the PhiX174 protein E. Genetics 2008, 180, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J.; Egmond, M.R.; Breukink, E.; Utrecht University, Utrecht, the Netherlands. Docking studies of protein E and bacterial MraY. Unpublished work. 2016. [Google Scholar]
- Mohammadi, T.; Karczmarek, A.; Crouvoisier, M.; Bouhss, A.; Mengin-Lecreulx, D.; den Blaauwen, T. The essential peptidoglycan glycosyltransferase MurG forms a complex with proteins involved in lateral envelope growth as well as with proteins involved in cell division in Escherichia coli. Mol. Microbiol. 2007, 65, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Salmond, G.P.; Lutkenhaus, J.F.; Donachie, W.D. Identification of new genes in a cell envelope-cell division gene cluster of Escherichia coli: Cell envelope gene MurG. J. Bacteriol. 1980, 144, 438–440. [Google Scholar] [PubMed]
- Mengin-Lecreulx, D.; Texier, L.; Rousseau, M.; van Heijenoort, J. The MurG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J. Bacteriol. 1991, 173, 4625–4636. [Google Scholar] [PubMed]
- Chen, L.; Men, H.; Ha, S.; Ye, X.Y.; Brunner, L.; Hu, Y.; Walker, S. Intrinsic lipid preferences and kinetic mechanism of Escherichia coli MurG. Biochemistry 2002, 41, 6824–6833. [Google Scholar] [CrossRef] [PubMed]
- Bupp, K.; van Heijenoort, J. The final step of peptidoglycan subunit assembly in Escherichia coli occurs in the cytoplasm. J. Bacteriol. 1993, 175, 1841–1843. [Google Scholar] [PubMed]
- Crouvoisier, M.; Mengin-Lecreulx, D.; van Heijenoort, J. UDP-N-acetylglucosamine: N-acetylmuramoyl-(pentapeptide) pyrophosphoryl undecaprenol N-acetylglucosamine transferase from Escherichia coli: Overproduction, solubilization, and purification. FEBS Lett. 1999, 449, 289–292. [Google Scholar] [CrossRef]
- Ha, S.; Chang, E.; Lo, M.-C.; Men, H.; Park, P.; Ge, M.; Walker, S. The kinetic characterization of Escherichia coli MurG using synthetic substrate analogues. J. Am. Chem. Soc. 1999, 121, 8415–8426. [Google Scholar] [CrossRef]
- Epand, R.M. Role of membrane lipids in modulating the activity of membrane-bound enzyme. In The Structure of Biological Membranes, 2nd ed.; Yeagle, P.L., Ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Van den Brink-van der Laan, E.; Boots, J.W.; Spelbrink, R.E.; Kool, G.M.; Breukink, E.; Killian, J.A.; de Kruijff, B. Membrane interaction of the glycosyltransferase MurG: A special role for cardiolipin. J. Bacteriol. 2003, 185, 3773–3779. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ritter, T.K.; Sadamoto, R.; Sears, P.S.; Wu, M.; Wong, C.H. Acceptor specificity and inhibition of the bacterial cell-wall glycosyltransferase MurG. ChemBioChem 2003, 4, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Walker, D.; Shi, Y.; Walker, S. The 1.9 Å crystal structure of Escherichia coli MurG, a membrane-associated glycosyltransferase involved in peptidoglycan biosynthesis. Protein Sci. 2000, 9, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Brakoulias, A.; Jackson, R.M. Towards a structural classification of phosphate binding sites in protein-nucleotide complexes: An automated all-against-all structural comparison using geometric matching. Proteins 2004, 56, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.L.; Edman, M.; Sjostrom, M.; Wieslander, A. Recognition of fold and sugar linkage for glycosyltransferases by multivariate sequence analysis. J. Biol. Chem. 2004, 279, 38683–38692. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, L.; Ha, S.; Gross, B.; Falcone, B.; Walker, D.; Mokhtarzadeh, M.; Walker, S. Crystal structure of the MurG: UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc. Natl. Acad. Sci. USA 2003, 100, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Öhrlein, R. Glycosyltransferase-catalyzed synthesis of non-natural oligosaccharides. In Biocatalysis—From Discovery to Application; Fessner, W.-D., Archelas, A., Demirjian, D.C., Furstoss, R., Griengl, H., Jaeger, K.E., Morís-Varas, E., Öhrlein, R., Reetz, M.T., Reymond, J.L., et al., Eds.; Springer: Berlin, Germany, 1999; Volume 200, pp. 227–254. [Google Scholar]
- Crouvoisier, M.; Auger, G.; Blanot, D.; Mengin-Lecreulx, D. Role of the amino acid invariants in the active site of MurG as evaluated by site-directed mutagenesis. Biochimie 2007, 89, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Vial, S.C.; Dedi, N.; Westcott, J.; Scally, S.; Bugg, T.D.; Charlton, P.A.; Cheetham, G.M. Crystal structure of the Pseudomonas aeruginosa MurG: UDP-GlcNAc substrate complex. Protein Pept. Lett. 2013, 20, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Helm, J.S.; Hu, Y.; Chen, L.; Gross, B.; Walker, S. Identification of active-site inhibitors of MurG using a generalizable, high-throughput glycosyltransferase screen. J. Am. Chem. Soc. 2003, 125, 11168–11169. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Helm, J.S.; Chen, L.; Ginsberg, C.; Gross, B.; Kraybill, B.; Tiyanont, K.; Fang, X.; Wu, T.; Walker, S. Identification of selective inhibitors for the glycosyltransferase MurG via high-throughput screening. Chem. Biol. 2004, 11, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Auger, G.; van Heijenoort, J.; Mengin-Lecreulx, D.; Blanot, D. A MurG assay which utilises a synthetic analogue of lipid I. FEMS Microbiol. Lett. 2003, 219, 115–119. [Google Scholar] [CrossRef]
- Terrak, M.; Ghosh, T.K.; van Heijenoort, J.; van Beeumen, J.; Lampilas, M.; Aszodi, J.; Ayala, J.A.; Ghuysen, J.M.; Nguyen-Disteche, M. The catalytic, glycosyl transferase and acyl transferase modules of the cell wall peptidoglycan-polymerizing penicillin-binding protein 1b of Escherichia coli. Mol. Microbiol. 1999, 34, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Mann, P.A.; Muller, A.; Xiao, L.; Pereira, P.M.; Yang, C.; Ho Lee, S.; Wang, H.; Trzeciak, J.; Schneeweis, J.; Dos Santos, M.M.; et al. Murgocil is a highly bioactive staphylococcal-specific inhibitor of the peptidoglycan glycosyltransferase enzyme MurG. ACS Chem. Biol. 2013, 8, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Mitachi, K.; Siricilla, S.; Klaic, L.; Clemons, W.M., Jr.; Kurosu, M. Chemoenzymatic syntheses of water-soluble lipid I fluorescent probes. Tetrahedron Lett. 2015, 56, 3441–3446. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Breukink, E. The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets. Antibiotics 2016, 5, 28. https://doi.org/10.3390/antibiotics5030028
Liu Y, Breukink E. The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets. Antibiotics. 2016; 5(3):28. https://doi.org/10.3390/antibiotics5030028
Chicago/Turabian StyleLiu, Yao, and Eefjan Breukink. 2016. "The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets" Antibiotics 5, no. 3: 28. https://doi.org/10.3390/antibiotics5030028
APA StyleLiu, Y., & Breukink, E. (2016). The Membrane Steps of Bacterial Cell Wall Synthesis as Antibiotic Targets. Antibiotics, 5(3), 28. https://doi.org/10.3390/antibiotics5030028