Phenotypic Resistance to Antibiotics


{kind=link}
Abstract
:1. Introduction
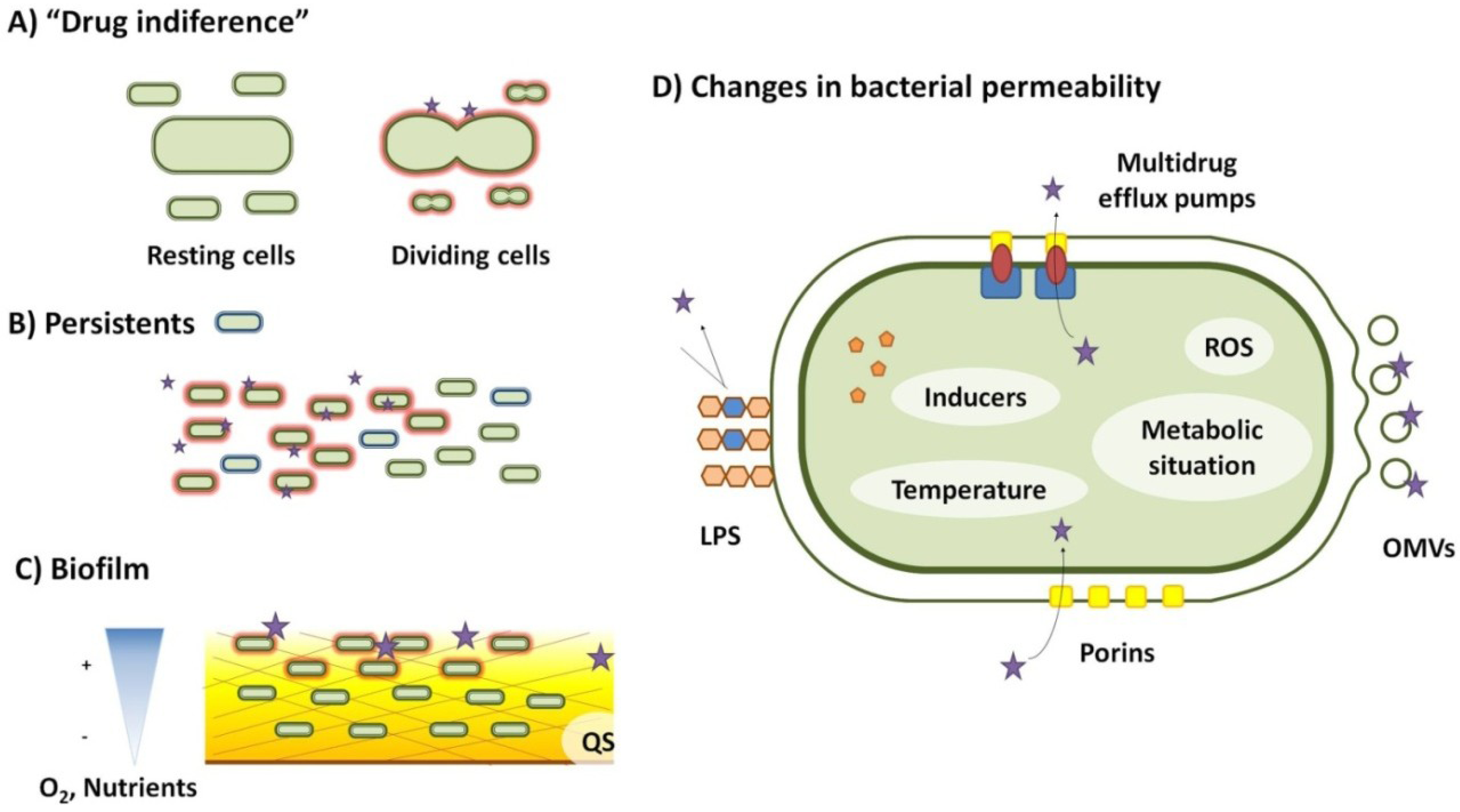
2. Drug Indifference
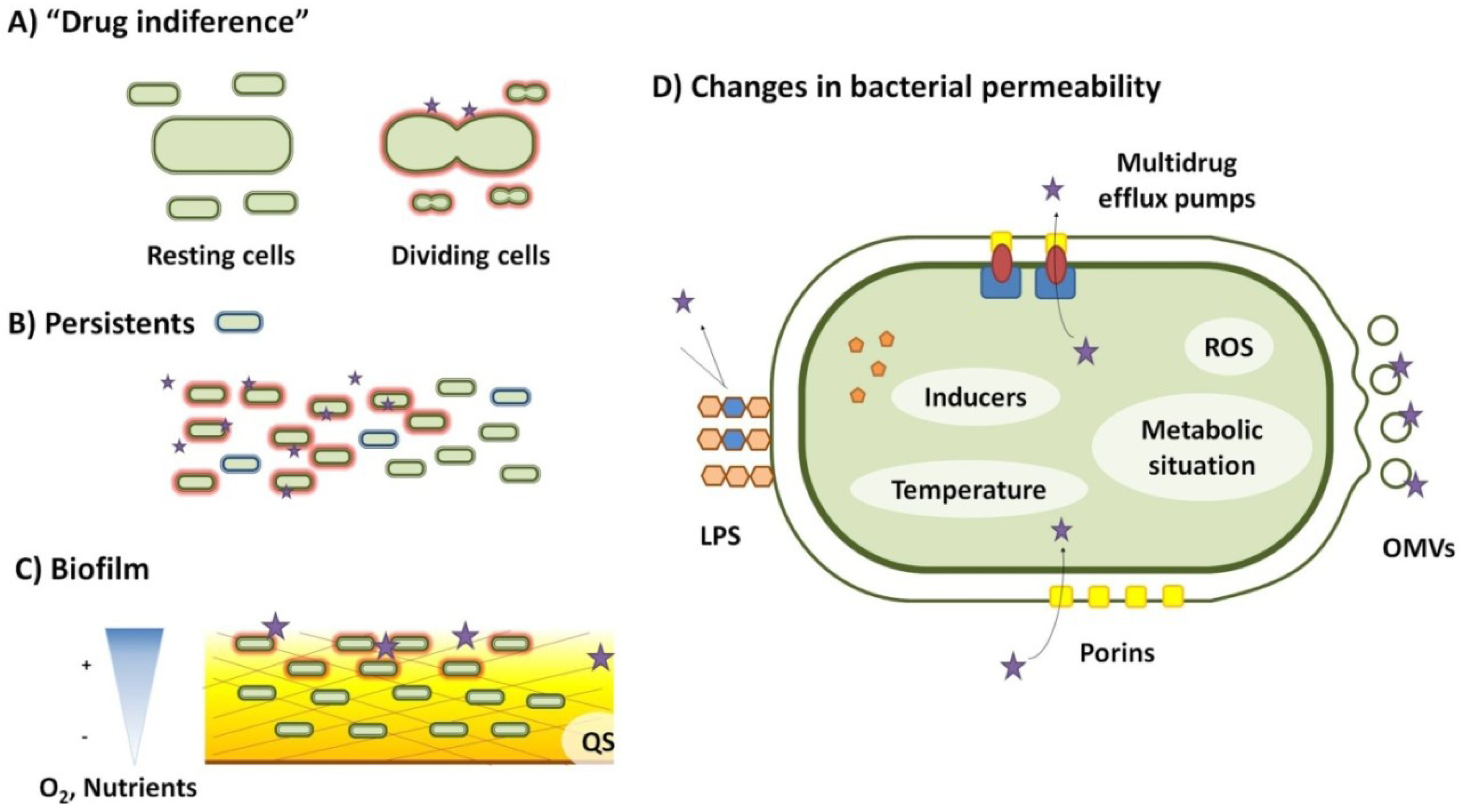
3. Biofilms
4. Transient Changes in Bacterial Permeability to Antibiotics
5. Inoculum Size and Phenotypic Resistance
6. Persistence
7. Concluding Remarks
Acknowledgements
References and Notes
- Baquero, F.; Coque, T.M. Multilevel population genetics in antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 705–706. [Google Scholar] [CrossRef]
- Martinez, J.L.; Fajardo, A.; Garmendia, L.; Hernandez, A.; Linares, J.F.; Martinez-Solano, L.; Sanchez, M.B. A global view of antibiotic resistance. FEMS Microbiol. Rev. 2009, 33, 44–65. [Google Scholar] [CrossRef]
- Martinez, J.L.; Baquero, F. Mutation frequencies and antibiotic resistance. Antimicrob. Agents Chemother. 2000, 44, 1771–1777. [Google Scholar] [CrossRef]
- Boto, L.; Martinez, J.L. Ecological and temporal constraints in the evolution of bacterial genomes. Genes 2011, 2, 804–828. [Google Scholar] [CrossRef]
- Baquero, F.; Alvarez-Ortega, C.; Martinez, J.L. Ecology and evolution of antibiotic resistance. Environ. Microbiol. Reports 2009, 1, 469–476. [Google Scholar] [CrossRef]
- Levin, B.R.; Rozen, D.E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006, 4, 556–562. [Google Scholar] [CrossRef]
- Wiedemann, B.; Pfeifle, D.; Wiegand, I.; Janas, E. beta-Lactamase induction and cell wall recycling in gram-negative bacteria. Drug Resist. Updat. 1998, 1, 223–226. [Google Scholar] [CrossRef]
- Fernandez, L.; Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Kocincova, D.; Lam, J.S.; Martinez, J.L.; Hancock, R.E. Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 110–119. [Google Scholar] [CrossRef]
- Martinez, J.L. The antibiotic resistome: Challenge and opportunity for therapeutic intervention. Future Med. Chem. 2012, 4, 347–359. [Google Scholar] [CrossRef]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.; Martinez, J.L. The intrinsic resistome of Pseudomonas aeruginosa to beta-lactams. Virulence 2011, 2, 144–146. [Google Scholar] [CrossRef]
- Fajardo, A.; Martinez-Martin, N.; Mercadillo, M.; Galan, J.C.; Ghysels, B.; Matthijs, S.; Cornelis, P.; Wiehlmann, L.; Tummler, B.; Baquero, F.; et al. The neglected intrinsic resistome of bacterial pathogens. PLoS One 2008, 3, e1619. [Google Scholar]
- Schurek, K.N.; Marr, A.K.; Taylor, P.K.; Wiegand, I.; Semenec, L.; Khaira, B.K.; Hancock, R.E. Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 4213–4219. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; Khaira, B.K.; Wiegand, I.; Overhage, J.; Hancock, R.E. Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob. Agents Chemother. 2008, 52, 4486–4491. [Google Scholar] [CrossRef]
- Liu, A.; Tran, L.; Becket, E.; Lee, K.; Chinn, L.; Park, E.; Tran, K.; Miller, J.H. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: Generating an antibiotic bar code. Antimicrob. Agents Chemother. 2010, 54, 1393–1403. [Google Scholar] [CrossRef]
- Tamae, C.; Liu, A.; Kim, K.; Sitz, D.; Hong, J.; Becket, E.; Bui, A.; Solaimani, P.; Tran, K.P.; Yang, H.; et al. Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol. 2008, 190, 5981–5988. [Google Scholar] [CrossRef]
- Alvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Hancock, R.E.; Martinez, J.L. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to beta-lactam antibiotics. Antimicrob. Agents Chemother. 2010, 54, 4159–4167. [Google Scholar] [CrossRef]
- Martinez, J.L.; Rojo, F. Metabolic regulation of antibiotic resistance. FEMS Microbiol. Rev. 2011, 35, 768–789. [Google Scholar] [CrossRef]
- Linares, J.F.; Moreno, R.; Fajardo, A.; Martinez-Solano, L.; Escalante, R.; Rojo, F.; Martinez, J.L. The global regulator Crc modulates metabolism, susceptibility to antibiotics and virulence in Pseudomonas aeruginosa. Environ. Microbiol. 2010, 12, 3196–3212. [Google Scholar] [CrossRef]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011, 473, 216–220. [Google Scholar]
- Pethe, K.; Sequeira, P.C.; Agarwalla, S.; Rhee, K.; Kuhen, K.; Phong, W.Y.; Patel, V.; Beer, D.; Walker, J.R.; Duraiswamy, J.; et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 2010, 1, 1–8. [Google Scholar]
- Lee, S.W.; Foley, E.J.; Epstein, J.A. Mode of Action of penicillin: I. Bacterial growth and penicillin activity-Staphylococcus aureus FDA. J. Bacteriol. 1944, 48, 393–399. [Google Scholar]
- Mc Dermott, W. Microbial persistence. Yale J. Biol. Med. 1958, 30, 257–291. [Google Scholar]
- Eagle, H. The effect of the size of the inoculum and the age of the infection on the curative dose of penicillin in experimental infections with Streptococci, Pneumococci, and Treponema pallidum. J. Exp. Med. 1949, 90, 595–607. [Google Scholar] [CrossRef]
- Bull, J.J.; Levin, B.R.; DeRouin, T.; Walker, N.; Bloch, C.A. Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol. 2002, 2, e35. [Google Scholar] [CrossRef]
- Clement, S.; Vaudaux, P.; Francois, P.; Schrenzel, J.; Huggler, E.; Kampf, S.; Chaponnier, C.; Lew, D.; Lacroix, J.S. Evidence of an intracellular reservoir in the nasal mucosa of patients with recurrent Staphylococcus aureus rhinosinusitis. J. Infect. Dis. 2005, 192, 1023–1028. [Google Scholar] [CrossRef]
- Fitoussi, F.; Cohen, R.; Brami, G.; Doit, C.; Brahimi, N.; de la Rocque, F.; Bingen, E. Molecular DNA analysis for differentiation of persistence or relapse from recurrence in treatment failure of Streptococcus pyogenes pharyngitis. Eur. J. Clin. Microbiol. Infect. Dis. 1997, 16, 233–237. [Google Scholar] [CrossRef]
- Toman, K. Bacterial persistence in leprosy. Int. J. Lepr. Other Mycobact. Dis. 1981, 49, 205–217. [Google Scholar]
- McCune, R.M., Jr.; Tompsett, R. Fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. I. The persistence of drug-susceptible tubercle bacilli in the tissues despite prolonged antimicrobial therapy. J. Exp. Med. 1956, 104, 737–762. [Google Scholar] [CrossRef]
- Ginsberg, A.M. Drugs in development for tuberculosis. Drugs 2010, 70, 2201–2214. [Google Scholar]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar]
- Chao, M.C.; Rubin, E.J. Letting sleeping dos lie: Does dormancy play a role in tuberculosis? Annu. Rev. Microbiol. 2010, 64, 293–311. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Kolter, R.; Greenberg, E.P. Microbial sciences: The superficial life of microbes. Nature 2006, 441, 300–302. [Google Scholar] [CrossRef]
- Hansen, S.K.; Rainey, P.B.; Haagensen, J.A.; Molin, S. Evolution of species interactions in a biofilm community. Nature 2007, 445, 533–536. [Google Scholar]
- Folkesson, A.; Haagensen, J.A.; Zampaloni, C.; Sternberg, C.; Molin, S. Biofilm induced tolerance towards antimicrobial peptides. PLoS One 2008, 3, e1891. [Google Scholar]
- Fux, C.A.; Costerton, J.W.; Stewart, P.S.; Stoodley, P. Survival strategies of infectious biofilms. Trends Microbiol. 2005, 13, 34–40. [Google Scholar] [CrossRef]
- Hogan, D.; Kolter, R. Why are bacteria refractory to antimicrobials? Curr. Opin. Microbiol. 2002, 5, 472–477. [Google Scholar] [CrossRef]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Drenkard, E.; Ausubel, F.M. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 2002, 416, 740–743. [Google Scholar] [CrossRef]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Hoffman, L.R.; D’Argenio, D.A.; MacCoss, M.J.; Zhang, Z.; Jones, R.A.; Miller, S.I. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 2005, 436, 1171–1175. [Google Scholar] [CrossRef]
- Linares, J.F.; Gustafsson, I.; Baquero, F.; Martinez, J.L. Antibiotics as intermicrobial signaling agents instead of weapons. Proc. Natl. Acad. Sci. USA 2006, 103, 19484–19489. [Google Scholar]
- Suci, P.A.; Mittelman, M.W.; Yu, F.P.; Geesey, G.G. Investigation of ciprofloxacin penetration into Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 1994, 38, 2125–2133. [Google Scholar] [CrossRef]
- Corbin, A.; Pitts, B.; Parker, A.; Stewart, P.S. Antimicrobial penetration and efficacy in an in vitro oral biofilm model. Antimicrob. Agents Chemother. 2011, 55, 3338–3344. [Google Scholar] [CrossRef]
- Singh, R.; Ray, P.; Das, A.; Sharma, M. Penetration of antibiotics through Staphylococcus aureus and Staphylococcus epidermidis biofilms. J. Antimicrob. Chemother. 2010, 65, 1955–1958. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, S.; Wang, Z.; Peng, N.; Yu, J. Synergy of ultrasound microbubbles and vancomycin against Staphylococcus epidermidis biofilm. J. Antimicrob. Chemother. 2013, 68, 816–826. [Google Scholar] [CrossRef]
- Walters, M.C., III; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef]
- Stewart, P.S.; Davison, W.M.; Steenbergen, J.N. Daptomycin rapidly penetrates a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 2009, 53, 3505–3507. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry (Mosc.) 2005, 70, 267–274. [Google Scholar] [CrossRef]
- Sternberg, C.; Christensen, B.B.; Johansen, T.; Toftgaard Nielsen, A.; Andersen, J.B.; Givskov, M.; Molin, S. Distribution of bacterial growth activity in flow-chamber biofilms. Appl. Environ. Microbiol. 1999, 65, 4108–4117. [Google Scholar]
- Huang, C.T.; Yu, F.P.; McFeters, G.A.; Stewart, P.S. Nonuniform spatial patterns of respiratory activity within biofilms during disinfection. Appl. Environ. Microbiol. 1995, 61, 2252–2256. [Google Scholar]
- Rani, S.A.; Pitts, B.; Beyenal, H.; Veluchamy, R.A.; Lewandowski, Z.; Davison, W.M.; Buckingham-Meyer, K.; Stewart, P.S. Spatial patterns of DNA replication, protein synthesis, and oxygen concentration within bacterial biofilms reveal diverse physiological states. J. Bacteriol. 2007, 189, 4223–4233. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar]
- Hoiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar]
- Pamp, S.J.; Gjermansen, M.; Johansen, H.K.; Tolker-Nielsen, T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008, 68, 223–240. [Google Scholar] [CrossRef]
- Nalca, Y.; Jansch, L.; Bredenbruch, F.; Geffers, R.; Buer, J.; Haussler, S. Quorum-sensing antagonistic activities of azithromycin in Pseudomonas aeruginosa PAO1: A global approach. Antimicrob. Agents Chemother. 2006, 50, 1680–1688. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J. Macrolides beyond the conventional antimicrobials: A class of potent immunomodulators. Int. J. Antimicrob. Agents 2008, 31, 12–20. [Google Scholar] [CrossRef]
- Wagner, V.E.; Iglewski, B.H. Pseudomonas aeruginosa Biofilms in CF Infection. Clin. Rev. Allergy Immunol. 2008, 35, 124–134. [Google Scholar] [CrossRef]
- Fernandez, L.; Hancock, R.E. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef]
- Pages, J.-M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef]
- Peterson, A.A.; Hancock, R.E.; McGroarty, E.J. Binding of polycationic antibiotics and polyamines to lipopolysaccharides of Pseudomonas aeruginosa. J. Bacteriol. 1985, 164, 1256–1261. [Google Scholar]
- Macfarlane, E.L.; Kwasnicka, A.; Hancock, R.E. Role of Pseudomonas aeruginosa PhoP-phoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 2000, 146, 2543–2554. [Google Scholar]
- Macfarlane, E.L.; Kwasnicka, A.; Ochs, M.M.; Hancock, R.E. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 1999, 34, 305–316. [Google Scholar] [CrossRef]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E.W. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Fernández, L.; Gooderham, W.J.; Bains, M.; McPhee, J.B.; Wiegand, I.; Hancock, R.E.W. Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob. Agents Chemother. 2010, 54, 3372–3382. [Google Scholar]
- Groisman, E.A.; Kayser, J.; Soncini, F.C. Regulation of polymyxin resistance and adaptation to low-Mg2+ environments. J. Bacteriol. 1997, 179, 7040–7045. [Google Scholar]
- Gellatly, S.L.; Needham, B.; Madera, L.; Trent, M.S.; Hancock, R.E.W. The Pseudomonas aeruginosa PhoP-PhoQ two-component regulatory system is induced upon interaction with epithelial cells and controls cytotoxicity and inflammation. Infect. Immun. 2012, 80, 3122–3131. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Rahmati-Bahram, A.; Magee, J.T.; Jackson, S.K. Temperature-dependent aminoglycoside resistance in Stenotrophomonas (Xanthomonas) maltophilia; alterations in protein and lipopolysaccharide with growth temperature. J. Antimicrob. Chemother. 1996, 37, 665–676. [Google Scholar] [CrossRef]
- Manning, A.J.; Kuehn, M.J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 2011, 11, e258. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Ann. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef]
- Mortimer, P.G.; Piddock, L.J. The accumulation of five antibacterial agents in porin-deficient mutants of Escherichia coli. J. Antimicrob. Chemother. 1993, 32, 195–213. [Google Scholar] [CrossRef]
- Cowan, S.W.; Schirmer, T.; Rummel, G.; Steiert, M.; Ghosh, R.; Pauptit, R.A.; Jansonius, J.N.; Rosenbusch, J.P. Crystal structures explain functional properties of two E. coli porins. Nature 1992, 358, 727–733. [Google Scholar] [CrossRef]
- Forst, S.; Delgado, J.; Inouye, M. Phosphorylation of OmpR by the osmosensor EnvZ modulates expression of the ompF and ompC genes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 6052–6056. [Google Scholar] [CrossRef]
- Yoshida, T.; Qin, L.; Egger, L.A.; Inouye, M. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J. Biol Chem 2006, 281, 17114–17123. [Google Scholar] [CrossRef]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A unique mechanism regulating gene expression: Translational inhibition by a complementary RNA transcript (micRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, A.; Blyn, L.B.; Storz, G. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J. Bacteriol. 2004, 186, 6689–6697. [Google Scholar] [CrossRef]
- Takayanagi, K.; Maeda, S.; Mizuno, T. Expression of micF involved in porin synthesis in Escherichia coli: Two distinct cis-acting elements respectively regulate micF expression positively and negatively. FEMS Microbiol. Lett. 1991, 83, 39–44. [Google Scholar] [CrossRef]
- Delihas, N.; Forst, S. MicF: An antisense RNA gene involved in response of Escherichia coli to global stress factors. J. Mol. Biol. 2001, 313, 1–12. [Google Scholar] [CrossRef]
- Pratt, L.A.; Hsing, W.; Gibson, K.E.; Silhavy, T.J. From acids to osmZ: Multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol. Microbiol. 1996, 20, 911–917. [Google Scholar] [CrossRef]
- Chubiz, L.M.; Rao, C.V. Role of the mar-sox-rob regulon in regulating outer membrane porin expression. J. Bacteriol. 2011, 193, 2252–2260. [Google Scholar] [CrossRef]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 2009, 1794, 769–781. [Google Scholar]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef]
- Blair, J.M.; Piddock, L.J. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: An update. Curr. Opin. Microbiol. 2009, 12, 512–519. [Google Scholar] [CrossRef]
- Piddock, L.J. Multidrug-resistance efflux pumps—Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef]
- Martinez, J.L.; Sanchez, M.B.; Martinez-Solano, L.; Hernandez, A.; Garmendia, L.; Fajardo, A.; Alvarez-Ortega, C. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 2009, 33, 430–449. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Paulsen, I.T. Phylogeny of multidrug transporters. Semin. Cell. Dev. Biol. 2001, 12, 205–213. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Chen, J.; Nelson, K.E.; Saier, M.H., Jr. Comparative genomics of microbial drug efflux systems. J. Mol. Microbiol. Biotechnol. 2001, 3, 145–150. [Google Scholar]
- Saier, M.H., Jr.; Paulsen, I.T.; Sliwinski, M.K.; Pao, S.S.; Skurray, R.A.; Nikaido, H. Evolutionary origins of multidrug and drug-specific efflux pumps in bacteria. Faseb. J. 1998, 12, 265–274. [Google Scholar]
- Paulsen, I.T. Multidrug efflux pumps and resistance: Regulation and evolution. Curr. Opin. Microbiol. 2003, 6, 446–451. [Google Scholar] [CrossRef]
- Grkovic, S.; Brown, M.H.; Skurray, R.A. Transcriptional regulation of multidrug efflux pumps in bacteria. Semin. Cell. Dev. Biol. 2001, 12, 225–237. [Google Scholar] [CrossRef]
- Hernandez, A.; Ruiz, F.M.; Romero, A.; Martinez, J.L. The binding of triclosan to SmeT, the repressor of the multidrug efflux pump SmeDEF, induces antibiotic resistance in Stenotrophomonas maltophilia. PLoS Pathog. 2011, 7, e1002103. [Google Scholar]
- Ma, D.; Cook, D.N.; Alberti, M.; Pon, N.G.; Nikaido, H.; Hearst, J.E. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol. Microbiol. 1995, 16, 45–55. [Google Scholar]
- Lin, J.; Cagliero, C.; Guo, B.; Barton, Y.W.; Maurel, M.C.; Payot, S.; Zhang, Q. Bile salts modulate expression of the CmeABC multidrug efflux pump in Campylobacter jejuni. J. Bacteriol. 2005, 187, 7417–7424. [Google Scholar] [CrossRef]
- Nikaido, E.; Yamaguchi, A.; Nishino, K. AcrAB multidrug efflux pump regulation in Salmonella enterica serovar Typhimurium by RamA in response to environmental signals. J. Biol. Chem. 2008, 283, 24245–24253. [Google Scholar] [CrossRef]
- Lee, E.H.; Shafer, W.M. The farAB-encoded efflux pump mediates resistance of gonococci to long-chained antibacterial fatty acids. Mol. Microbiol. 1999, 33, 839–845. [Google Scholar] [CrossRef]
- Shafer, W.M.; Qu, X.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/ division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar]
- Poole, K. Stress responses as determinants of antimicrobial resistance in Gram-negative bacteria. Trends Microbiol. 2012, 20, 227–234. [Google Scholar] [CrossRef]
- Miller, P.F.; Sulavik, M.C. Overlaps and parallels in the regulation of intrinsic multiple-antibiotic resistance in Escherichia coli. Mol. Microbiol. 1996, 21, 441–448. [Google Scholar] [CrossRef]
- Chen, H.; Hu, J.; Chen, P.R.; Lan, L.; Li, Z.; Hicks, L.M.; Dinner, A.R.; He, C. The Pseudomonas aeruginosa multidrug efflux regulator MexR uses an oxidation-sensing mechanism. Proc. Natl. Acad. Sci. USA 2008, 105, 13586–13591. [Google Scholar]
- Fraud, S.; Poole, K. Oxidative stress induction of the MexXY multidrug efflux genes and promotion of aminoglycoside resistance development in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2011, 55, 1068–1074. [Google Scholar] [CrossRef]
- Fetar, H.; Gilmour, C.; Klinoski, R.; Daigle, D.M.; Dean, C.R.; Poole, K. mexEF-oprN multidrug efflux operon of Pseudomonas aeruginosa: Regulation by the MexT activator in response to nitrosative stress and chloramphenicol. Antimicrob. Agents Chemother. 2011, 55, 508–514. [Google Scholar] [CrossRef]
- Chico-Calero, I.; Suarez, M.; Gonzalez-Zorn, B.; Scortti, M.; Slaghuis, J.; Goebel, W.; Vazquez-Boland, J.A. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. USA 2002, 99, 431–436. [Google Scholar] [CrossRef]
- Kahan, F.M.; Kahan, J.S.; Cassidy, P.J.; Kropp, H. The mechanism of action of fosfomycin (phosphonomycin). Ann. NY Acad. Sci. 1974, 235, 364–386. [Google Scholar] [CrossRef]
- Ripio, M.T.; Brehm, K.; Lara, M.; Suarez, M.; Vazquez-Boland, J.A. Glucose-1-phosphate utilization by Listeria monocytogenes is PrfA dependent and coordinately expressed with virulence factors. J. Bacteriol. 1997, 179, 7174–7180. [Google Scholar]
- Moreno, R.; Marzi, S.; Romby, P.; Rojo, F. The Crc global regulator binds to an unpaired A-rich motif at the Pseudomonas putida alkS mRNA coding sequence and inhibits translation initiation. Nucleic Acids Res. 2009, 37, 7678–7690. [Google Scholar] [CrossRef]
- Moreno, R.; Martinez-Gomariz, M.; Yuste, L.; Gil, C.; Rojo, F. The Pseudomonas putida Crc global regulator controls the hierarchical assimilation of amino acids in a complete medium: Evidence from proteomic and genomic analyses. Proteomics 2009, 9, 2910–2928. [Google Scholar] [CrossRef]
- Morales, G.; Linares, J.F.; Beloso, A.; Albar, J.P.; Martinez, J.L.; Rojo, F. The Pseudomonas putida Crc global regulator controls the expression of genes from several chromosomal catabolic pathways for aromatic compounds. J. Bacteriol. 2004, 186, 1337–1344. [Google Scholar] [CrossRef]
- MacGregor, C.H.; Wolff, J.A.; Arora, S.K.; Phibbs, P.V., Jr. Cloning of a catabolite repression control (crc) gene from Pseudomonas aeruginosa, expression of the gene in Escherichia coli, and identification of the gene product in Pseudomonas aeruginosa. J. Bacteriol. 1991, 173, 7204–7212. [Google Scholar]
- Brook, I. Inoculum effect. Rev. Infect. Dis. 1989, 11, 361–368. [Google Scholar] [CrossRef]
- Soriano, F.; Ponte, C.; Santamaria, M.; Jimenez-Arriero, M. Relevance of the inoculum effect of antibiotics in the outcome of experimental infections caused by Escherichia coli. J. Antimicrob. Chemother. 1990, 25, 621–627. [Google Scholar] [CrossRef]
- Reguera, J.A.; Baquero, F.; Perez-Diaz, J.C.; Martinez, J.L. Factors determining resistance to beta-lactam combined with beta-lactamase inhibitors in Escherichia coli. J. Antimicrob. Chemother. 1991, 27, 569–575. [Google Scholar]
- Reguera, J.A.; Baquero, F.; Perez-Diaz, J.C.; Martinez, J.L. Synergistic effect of dosage and bacterial inoculum in TEM-1 mediated antibiotic resistance. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 778–779. [Google Scholar] [CrossRef]
- Martinez, J.L.; Blazquez, J.; Baquero, F. Non-canonical mechanisms of antibiotic resistance. Eur. J. Clin. Microbiol. Infect. Dis. 1994, 13, 1015–1022. [Google Scholar] [CrossRef]
- Martinez, J.L.; Blazquez, J.; Vicente, M.F.; Martinez-Ferrer, M.; Reguera, J.A.; Culebras, E.; Baquero, F. Influence of gene dosing on antibiotic resistance mediated by inactivating enzymes. J. Chemother. 1989, 1, 265–266. [Google Scholar]
- Udekwu, K.I.; Parrish, N.; Ankomah, P.; Baquero, F.; Levin, B.R. Functional relationship between bacterial cell density and the efficacy of antibiotics. J. Antimicrob. Chemother. 2009, 63, 745–757. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Levin, B.R. Microbiology: Noninherited resistance to antibiotics. Science 2004, 305, 1578–1579. [Google Scholar]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Kussell, E.; Kishony, R.; Balaban, N.Q.; Leibler, S. Bacterial persistence: A model of survival in changing environments. Genetics 2005, 169, 1807–1814. [Google Scholar] [CrossRef]
- Balaban, N.Q. Persistence: Mechanisms for triggering and enhancing phenotypic variability. Curr. Opin. Genet. Dev. 2011, 21, 768–775. [Google Scholar] [CrossRef]
- Hansen, S.; Lewis, K.; Vulic, M. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 2008, 52, 2718–2726. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Gefen, O.; Gabay, C.; Mumcuoglu, M.; Engel, G.; Balaban, N.Q. Single-cell protein induction dynamics reveals a period of vulnerability to antibiotics in persister bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 6145–6149. [Google Scholar]
- Korch, S.B.; Henderson, T.A.; Hill, T.M. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 2003, 50, 1199–1213. [Google Scholar] [CrossRef]
- Rotem, E.; Loinger, A.; Ronin, I.; Levin-Reisman, I.; Gabay, C.; Shoresh, N.; Biham, O.; Balaban, N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. USA 2010, 107, 12541–12546. [Google Scholar] [CrossRef]
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180. [Google Scholar] [CrossRef]
- Dorr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef]
- Helaine, S.; Thompson, J.A.; Watson, K.G.; Liu, M.; Boyle, C.; Holden, D.W. Dynamics of intracellular bacterial replication at the single cell level. Proc. Natl. Acad. Sci. USA 2010, 107, 3746–3751. [Google Scholar]
- Moker, N.; Dean, C.R.; Tao, J. Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol. 2010, 192, 1946–1955. [Google Scholar] [CrossRef]
- Hentzer, M.; Wu, H.; Andersen, J.B.; Riedel, K.; Rasmussen, T.B.; Bagge, N.; Kumar, N.; Schembri, M.A.; Song, Z.; Kristoffersen, P.; et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003, 22, 3803–3815. [Google Scholar] [CrossRef]
- Wu, H.; Song, Z.; Hentzer, M.; Andersen, J.B.; Molin, S.; Givskov, M.; Hoiby, N. Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J. Antimicrob. Chemother. 2004, 53, 1054–1061. [Google Scholar] [CrossRef]
- Pan, J.; Bahar, A.A.; Syed, H.; Ren, D. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PLoS One 2012, 7, e45778. [Google Scholar]
- Kim, J.S.; Heo, P.; Yang, T.J.; Lee, K.S.; Cho, D.H.; Kim, B.T.; Suh, J.H.; Lim, H.J.; Shin, D.; Kim, S.K.; et al. Selective killing of bacterial persisters by a single chemical compound without affecting normal antibiotic-sensitive cells. Antimicrob. Agents Chemother. 2011, 55, 5380–5383. [Google Scholar] [CrossRef]
- Grant, S.S.; Kaufmann, B.B.; Chand, N.S.; Haseley, N.; Hung, D.T. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc. Natl. Acad. Sci. USA 2012, 109, 12147–12152. [Google Scholar]
- Lomovskaya, O.; Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic—A vision for applied use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Martinez, J.L.; Rojo, F.; Vila, J. Are nonlethal targets useful for developing novel antimicrobials? Future Microbiol. 2011, 6, 605–607. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Corona, F.; Martinez, J.L. Phenotypic Resistance to Antibiotics. Antibiotics 2013, 2, 237-255. https://doi.org/10.3390/antibiotics2020237
Corona F, Martinez JL. Phenotypic Resistance to Antibiotics. Antibiotics. 2013; 2(2):237-255. https://doi.org/10.3390/antibiotics2020237
Chicago/Turabian StyleCorona, Fernando, and Jose L. Martinez. 2013. "Phenotypic Resistance to Antibiotics" Antibiotics 2, no. 2: 237-255. https://doi.org/10.3390/antibiotics2020237
APA StyleCorona, F., & Martinez, J. L. (2013). Phenotypic Resistance to Antibiotics. Antibiotics, 2(2), 237-255. https://doi.org/10.3390/antibiotics2020237