
Targeting Gene Transcription Prevents Antibiotic Resistance


Abstract

1. Introduction
1.1. Present Challenges from Antimicrobial Resistance and Traditional Pathways to Antibiotics
1.2. Proteins Versus RNA Targets
2. Targeting RNA Function
2.1. RNA Transcription Factors
2.2. A Novel Perspective
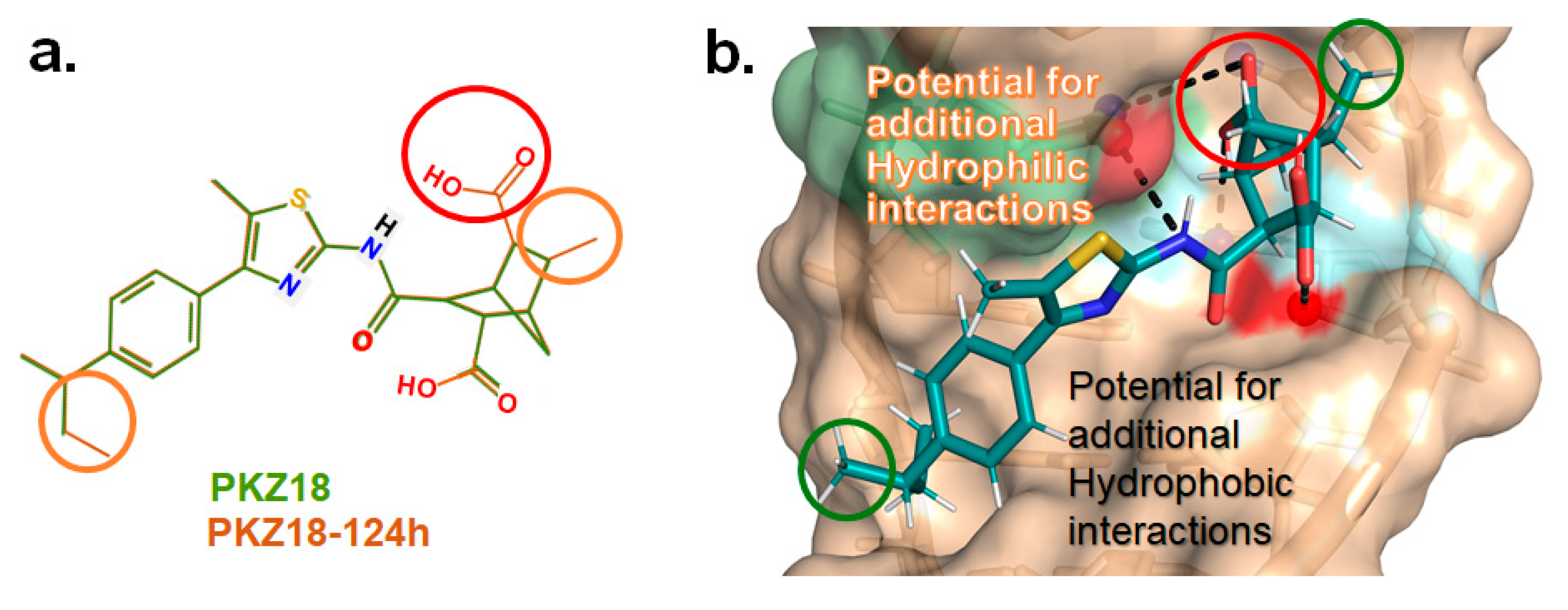
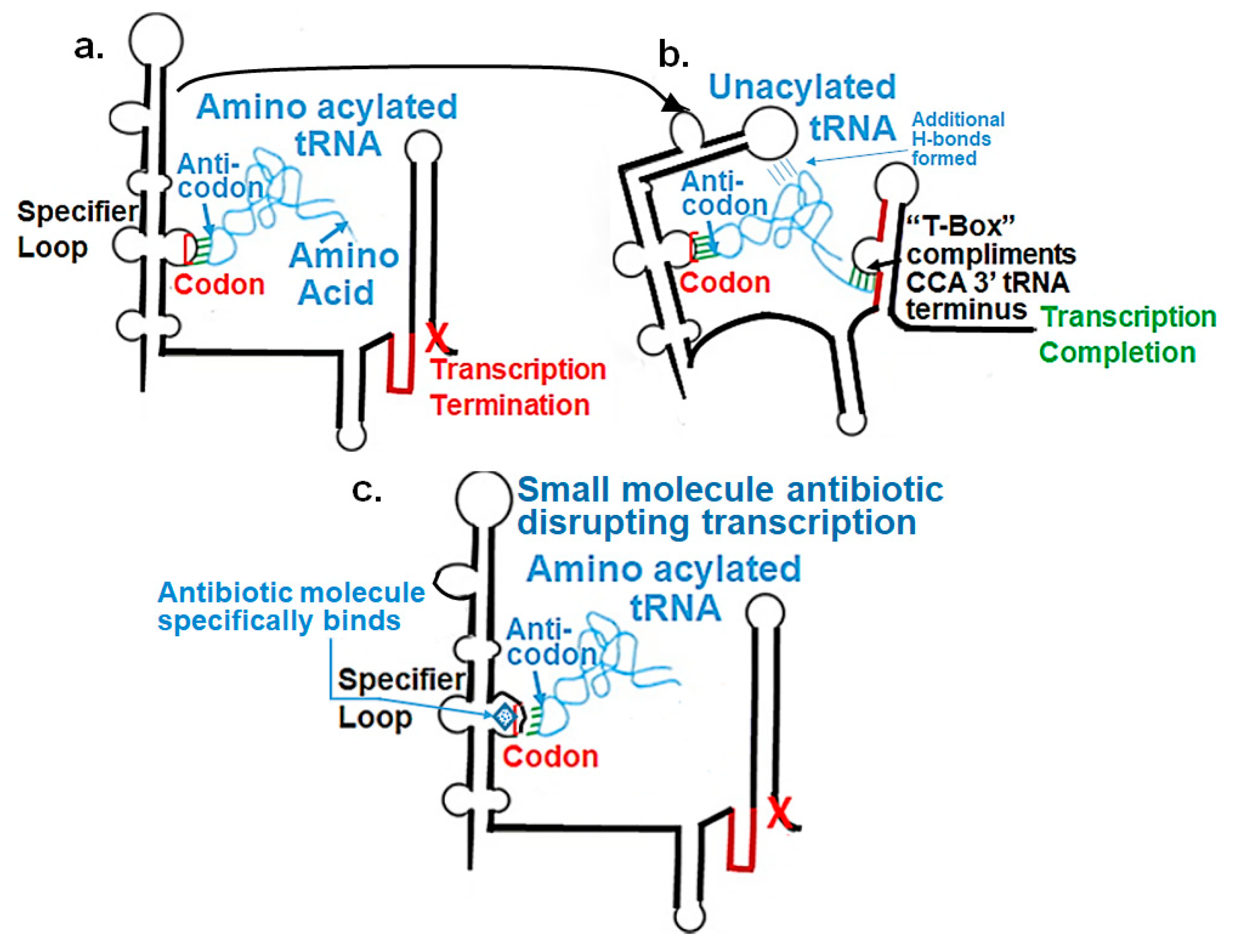
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adefisoye, M.A.; Olaniran, A.O. Antimicrobial resistance expansion in pathogens: A review of current mitigation strategies and advances towards innovative therapy. JAC-Antimicrob. Resist. 2023, 5, dlad127. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention, CDC. Antibiotic Resistance, AR, 2019, Threats Reports Center for Disease Control. Available online: https://www.cdc.gov/antimicrobial-resistance/data-research/facts-stats/index.html (accessed on 17 March 2025).
- Centers for Disease Control and Prevention, CDC. Antibiotic Resistance Threats in the United States, 2021–2022. Center for Disease Control Reports. 2024. Available online: https://www.cdc.gov/antimicrobial-resistance/data-research/threats/update-2022.html (accessed on 17 March 2025).
- Zhou, G.; Shi, Q.S.; Huang, X.M.; Xie, X.B. The Three Bacterial Lines of Defense against Antimicrobial Agents. Int. J. Mol. Sci. 2015, 16, 21711–21733. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lambert, P.A. Bacterial resistance to antibiotics: Modified target sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Viner, C.; Ishak, C.A.; Johnson, J.; Walker, N.J.; Shi, H.; Sjöberg-Herrera, M.K.; Shen, S.Y.; Lardo, S.M.; Adams, D.J.; Ferguson-Smith, A.C.; et al. Modeling methyl-sensitive transcription factor motifs with an expanded epigenetic alphabet. Genome Biol. 2024, 25, 11–57. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cui, H.; Stilgenbauer, M.; Koehler, A.N. Small molecule approaches to target transcription factors. Annu. Rev. Cancer Biol. 2024, 8, 395–415. [Google Scholar]
- Tao, Z.; Wu, Z. Targeting transcription factors in cancer: From “Undruggable” to “Druggable”. Methods Mol. Biol. 2023, 2594, 107–131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barrick, J.E.; Breaker, R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007, 8, R239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Panchal, V.; Brenk, R. Riboswitches as Drug Targets for Antibiotics. Antibiotics 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ellinger, E.; Chauvier, A.; Romero, R.A.; Liu, Y.; Ray, S.; Walter, N.G. Riboswitches as therapeutic targets: Promise of a new era of antibiotics. Expert Opin. Ther. Targets 2023, 27, 433–445. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J.; Dimitrova, N. Transcription regulation by long non-coding RNAs: Mechanisms and disease relevance. Nat. Rev. Mol. Cell Biol. 2024, 25, 396–415. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nazli, A.; He, D.L.; Liao, D.; Khan, M.Z.I.; Huang, C.; He, Y. Strategies and progresses for enhancing targeted antibiotic delivery. Adv. Drug Deliv. Rev. 2022, 189, 114502. [Google Scholar] [CrossRef]
- Henkin, T.M. The T box riboswitch: A novel regulatory RNA that utilizes tRNA as its ligand. Biochim. Biophys. Acta 2014, 1839, 959–963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gutiérrez-Preciado, A.; Henkin, T.M.; Grundy, F.J.; Yanofsky, C.; Merino, E. Biochemical features and functional implications of the RNA-based T-box regulatory mechanism. Microbiol. Mol. Biol. Rev. 2009, 73, 36–61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Green, N.J.; Grundy, F.J.; Henkin, T.M. The T box mechanism: tRNA as a regulatory molecule. FEBS Lett. 2010, 584, 318–324. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frohlich, K.M.; Weintraub, S.F.; Bell, J.T.; Todd, G.C.; Väre, V.Y.P.; Schneider, R.; Kloos, Z.A.; Tabe, E.S.; Cantara, W.A.; Stark, C.J.; et al. Discovery of Small-Molecule Antibiotics against a Unique tRNA-Mediated Regulation of Transcription in Gram-Positive Bacteria. ChemMedChem 2019, 14, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Väre, V.Y.P.; Schneider, R.F.; Kim, H.; Lasek-Nesselquist, E.; McDonough, K.A.; Agris, P.F. Small-molecule antibiotics inhibiting tRNA-regulated gene expression is a viable strategy for targeting Gram-positive bacteria. Antimicrob. Agents Chemother. 2021, 65, 01247-20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Seyler, T.M.; Moore, C.; Kim, H.; Ramachandran, S.; Agris, P.F. A new promising anti-infective agent inhibits biofilm growth by targeting simultaneously a conserved RNA function that controls multiple genes. Antibiotics 2021, 10, 41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agarwal, R.; Rajeshaw, T.R.; Smith, J.C. Comparative Assessment of Pose Prediction Accuracy in RNA-Ligand Docking. J. Chem. Inf. Model. 2023, 63, 7444–7452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferré-D’Amaré, A. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature 2013, 500, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.C.; Ke, A. Structural determinants for geometry and information decoding of tRNA by T box leader RNA. Structure 2013, 21, 2025–2032. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cera, E.D. Mechanisms of ligand binding. Biophys. Rev. 2020, 1, 011303. [Google Scholar] [CrossRef]
- Wang, J.; Henkin, T.M.; Nikonowicz, E.P. NMR structure and dynamics of the Specifier Loop domain from the Bacillus subtilis tyrS T box leader RNA. Nucleic Acids Res. 2010, 38, 3388–3398. [Google Scholar]
- Wang, J.; Nikonowicz, E.P. Solution structure of the K-turn and Specifier Loop domains from the Bacillus subtilis tyrS T-box leader RNA. J. Mol. Biol. 2011, 22, 99–117. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
{kind=link}
| Bacteria | MIC 1 (MBC) [µg/mL] |
|---|---|
| Gram-positive | |
| B. subtilis (BGSC 1A1) | 64 (64) |
| B. cereus (ATCC 7064) | 64 (64) |
| S. aureus (ATCC 29213) | 64 (N/O) |
| E. faecalis (ATCC 29212) | 32 (N/D) 2 |
| S. pyogenes (clinical isolate) | 16 (16) |
| S. agalactiae (clinical isolate) | 32 (32) |
| S. pneumoniae (clinical isolate) | 16 (16) |
| S. mutans (clinical isolate) | 125 (125) |
| MRSA (clinical isolate, pleural fluid) | 64 (N/O) |
| MRSA (clinical isolate, blood) | 64 (N/O) |
| Gram-negative | |
| E. coli (ATCC 25922) | N/O (N/D) |
| P. aeruginosa (ATCC 27853) | N/O (N/D) |
| K. pneumoniae (clinical isolate) | N/O (N/D) |
| PKZ18 Family | 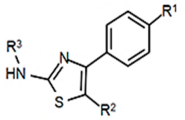 | MIC (µg/mL) |
|---|---|---|
| PKZ18 |  | 64 |
| PKZ18-21 | 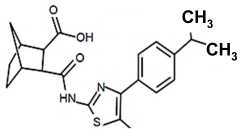 | 64 |
| PKZ18-22 | 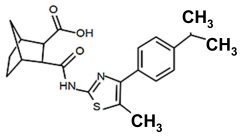 | 32 |
| PKZ18-52 | 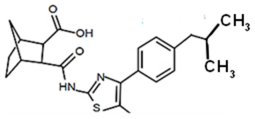 | 32 |
| PKZ18-53 |  | 32 |
| PKZ18-54 | 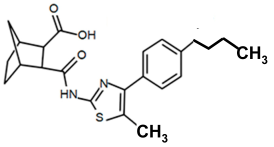 | 128 |
| PKZ18-55 |  | >256 |
| PKZ18-56 | 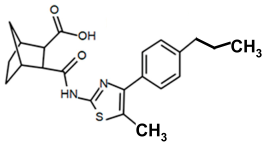 | >256 |
| PKZ18-57 | 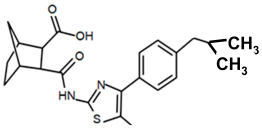 | >256 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agris, P.F. Targeting Gene Transcription Prevents Antibiotic Resistance. Antibiotics 2025, 14, 345. https://doi.org/10.3390/antibiotics14040345
Agris PF. Targeting Gene Transcription Prevents Antibiotic Resistance. Antibiotics. 2025; 14(4):345. https://doi.org/10.3390/antibiotics14040345
Chicago/Turabian StyleAgris, Paul F. 2025. "Targeting Gene Transcription Prevents Antibiotic Resistance" Antibiotics 14, no. 4: 345. https://doi.org/10.3390/antibiotics14040345
APA StyleAgris, P. F. (2025). Targeting Gene Transcription Prevents Antibiotic Resistance. Antibiotics, 14(4), 345. https://doi.org/10.3390/antibiotics14040345