

Multiplex Real-Time Polymerase Chain Reaction and Recombinase Polymerase Amplification: Methods for Quick and Cost-Effective Detection of Vancomycin-Resistant Enterococci (VRE)
 , , and
, , and
Abstract
1. Introduction
2. Results
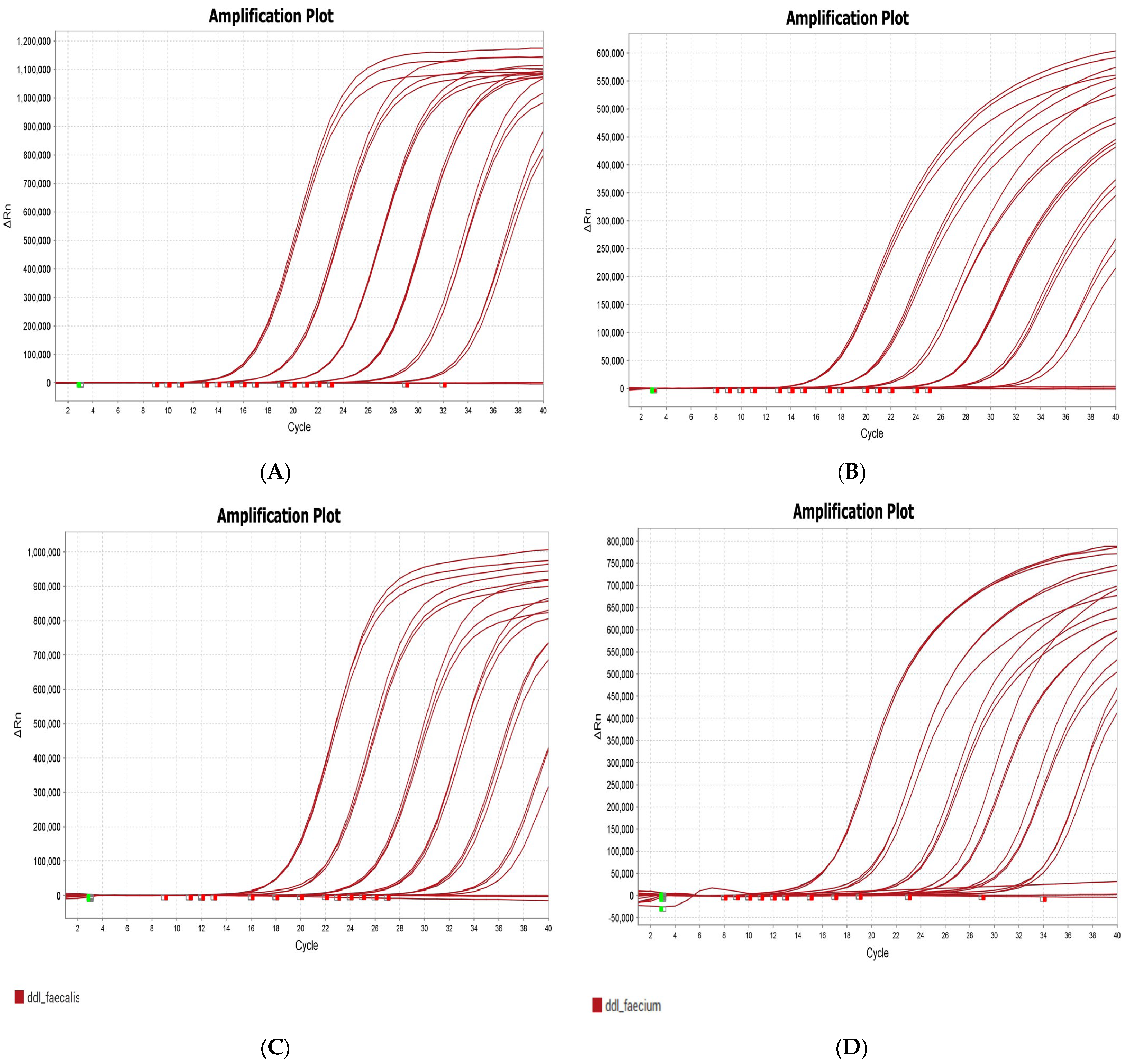
2.1. Multiplex PCR Primer Design
2.2. Optimization of Multiplex PCR for VRE
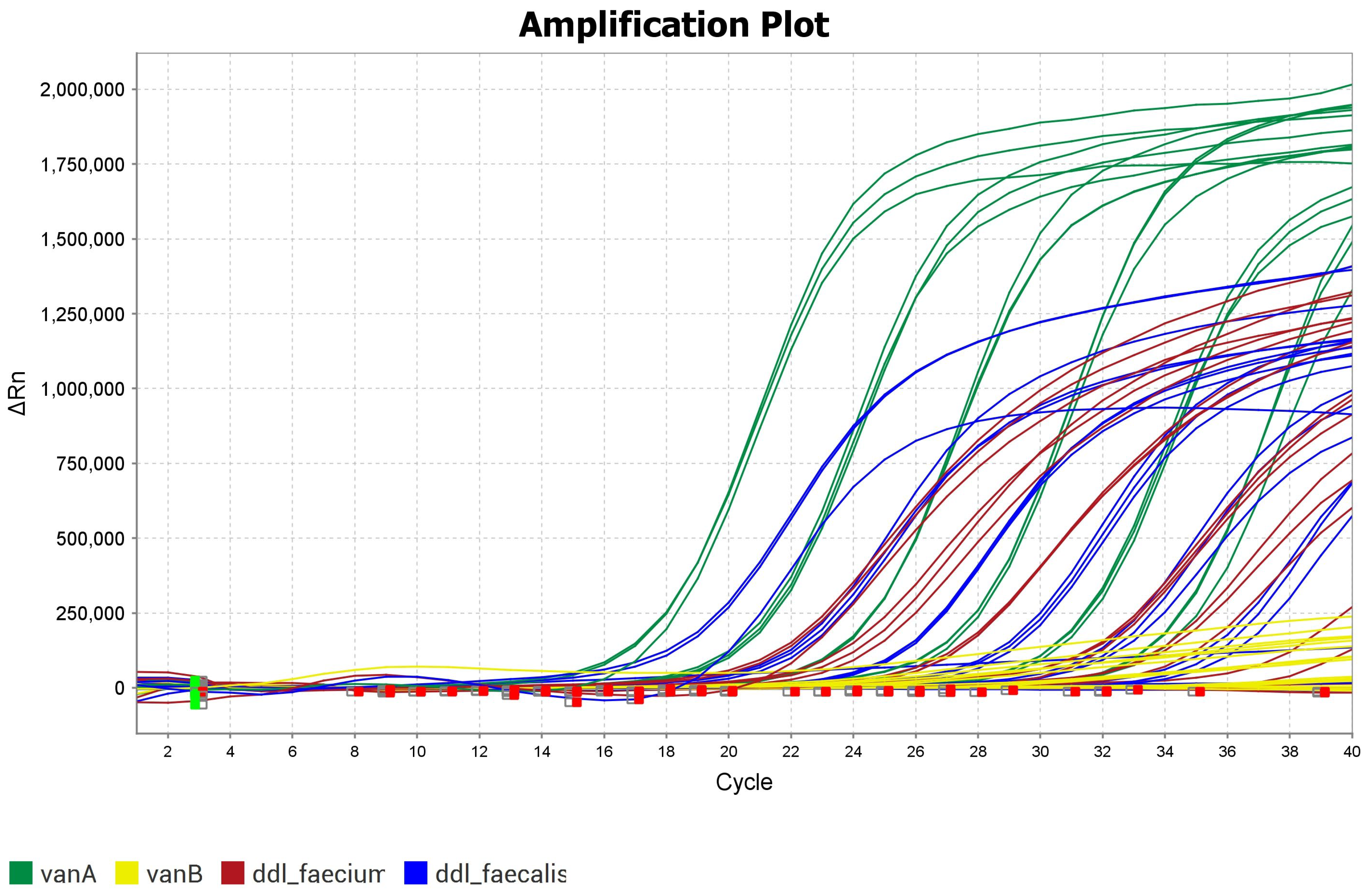
2.3. Multiplex PCR Results
2.4. Comparison Between Multiplex PCR and RPA
2.5. Heat Lysis of VRE Isolates After Cell Culture
3. Discussion
4. Materials and Methods
4.1. RPA Oligonucleotides
4.2. Samples
4.3. Fast Lysis Protocol Without Purification
4.4. DNA Dilutions
4.5. Multiplex PCR
4.6. RPA
4.7. Gel Electrophoresis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Mac, S.; Fitzpatrick, T.; Johnstone, J.; Sander, B. Vancomycin-resistant enterococci (VRE) screening and isolation in the general medicine ward: A cost-effectiveness analysis. Antimicrob. Resist. Infect. Control. 2019, 8, 168. [Google Scholar] [CrossRef] [PubMed]
- Osadare, I.E.; Monecke, S.; Abdilahi, A.; Müller, E.; Collatz, M.; Braun, S.; Reissig, A.; Schneider-Brachert, W.; Kieninger, B.; Eichner, A.; et al. Fast and Economic Microarray-Based Detection of Species-, Resistance-, and Virulence-Associated Genes in Clinical Strains of Vancomycin-Resistant Enterococci (VRE). Sensors 2024, 24, 6476. [Google Scholar] [CrossRef]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Klare, I.; Witte, W.; Wendt, C.; Werner, G. Vancomycin-resistant enterococci (VRE). Recent results and trends in development of antibiotic resistance. Bundesgesundheitsbl. Gesundheitsforschung Gesundheitsschutz 2012, 55, 1387–1400. [Google Scholar] [CrossRef]
- Ziakas, P.D.; Thapa, R.; Rice, L.B.; Mylonakis, E. Trends and significance of VRE colonization in the ICU: A meta-analysis of published studies. PLoS ONE 2013, 8, e75658. [Google Scholar] [CrossRef] [PubMed]
- Furuya, K.; Yamagishi, T.; Suzuki, K.; Sugiyama, K.; Yamamoto, M.; Koyama, M.; Yamada, A.; Sasaki, R.; Kurioka, J.; Kurai, H.; et al. Cumulative incidence of vancomycin-resistant Enterococcus faecium detection by patient characteristics or possible exposures: Prioritization of patients for active screening culture. J. Hosp. Infect. 2024, 154, 70–76. [Google Scholar] [CrossRef]
- Austin, D.J.; Bonten, M.J.M.; Weinstein, R.A.; Slaughter, S.; Anderson, R.M. Vancomycin-resistant enterococci in intensive-care hospital settings: Transmission dynamics, persistence, and the impact of infection control programs. Proc. Natl. Acad. Sci. USA 1999, 96, 6908–6913. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. In the MMWR Recommendations and Reports “Recommendations for Preventing the Spread of Vancomycin Resistance: Recommendations of the Hospital Infection Control Practices Advisory Committee (HICPAC)”. Morb. Mortal. Wkly. Rep. 1995, 44, 1–13. [Google Scholar]
- Nilsson, O. Vancomycin resistant enterococci in farm animals—Occurrence and importance. Infect. Ecol. Epidemiol. 2012, 2, 16959. [Google Scholar] [CrossRef]
- Kühn, I.; Iversen, A.; Finn, M.; Greko, C.; Burman, L.G.; Blanch, A.R.; Vilanova, X.; Manero, A.; Taylor, H.; Caplin, J.; et al. Occurrence and relatedness of vancomycin-resistant enterococci in animals, humans, and the environment in different European regions. Appl. Environ. Microbiol. 2005, 71, 5383–5390. [Google Scholar] [CrossRef] [PubMed]
- Acar, J.; Casewell, M.; Freeman, J.; Friis, C.; Goossens, H. Avoparcin and virginiamycin as animal growth promoters: A plea for science in decision-making. Clin. Microbiol. Infect. 2000, 6, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Klare, I.; Badstübner, D.; Konstabel, C.; Böhme, G.; Claus, H.; Witte, W. Decreased incidence of VanA-type vancomycin-resistant enterococci isolated from poultry meat and from fecal samples of humans in the community after discontinuation of avoparcin usage in animal husbandry. Microb. Drug Resist. 1999, 5, 45–52. [Google Scholar] [CrossRef]
- Xie, O.; Slavin, M.A.; Teh, B.W.; Bajel, A.; Douglas, A.P.; Worth, L.J. Epidemiology, treatment and outcomes of bloodstream infection due to vancomycin-resistant enterococci in cancer patients in a vanB endemic setting. BMC Infect. Dis. 2020, 20, 228. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.R.; Tedim, A.P.; Francia, M.V.; Jensen, L.B.; Novais, C.; Peixe, L.; Sánchez-Valenzuela, A.; Sundsfjord, A.; Hegstad, K.; Werner, G.; et al. Multilevel population genetic analysis of vanA and vanB Enterococcus faecium causing nosocomial outbreaks in 27 countries (1986–2012). J. Antimicrob. Chemother. 2016, 71, 3351–3366. [Google Scholar] [CrossRef]
- Freitas, A.R.; Tedim, A.P.; Francia, M.V.; Jensen, L.B.; Novais, C.; Peixe, L.; Sánchez-Valenzuela, A.; Sundsfjord, A.; Hegstad, K.; Werner, G.; et al. Prevalence and antimicrobial susceptibility pattern of Enterococcus species isolated from different clinical samples at Black Lion Specialized Teaching Hospital, Addis Ababa, Ethiopia. BMC Res. Notes 2018, 11, 793. [Google Scholar]
- García-Solache, M.; Louis, B.R. The Enterococcus: A Model of Adaptability to Its Environment. Clin. Microbiol. Rev. 2019, 32, e00058-18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Willems, R.J.L.; Friedrich, A.W.; Rossen, J.W.A.; Bathoorn, E. Enterococcus faecium: From microbiological insights to practical recommendations for infection control and diagnostics. Antimicrob. Resist. Infect. Control 2020, 9, 130. [Google Scholar] [CrossRef]
- Dadashi, M.; Sharifian, P.; Bostanshirin, N.; Hajikhani, B.; Bostanghadiri, N.; Khosravi-Dehaghi, N.; van Belkum, A.; Darban-Sarokhalil, D. The Global Prevalence of Daptomycin, Tigecycline, and Linezolid-Resistant Enterococcus faecalis and Enterococcus faecium Strains From Human Clinical Samples: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 720647. [Google Scholar]
- Moosavian, M.; Ghadri, H.; Samli, Z. Molecular detection of vanA and vanB genes among vancomycin-resistant enterococci in ICU-hospitalized patients in Ahvaz in southwest of Iran. Infect. Drug Resist. 2018, 11, 2269–2275. [Google Scholar]
- Arthur, M.; Quintiliani, R., Jr. Regulation of VanA- and VanB-type glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 2001, 45, 375–381. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. ACS Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef] [PubMed]
- Bar-Meir, M.; Berliner, E.; Kashat, L.; Zeevi, D.A.; Assous, M.V. The utility of MALDI-TOF MS for outbreak investigation in the neonatal intensive care unit. Eur. J. Pediatr. 2020, 179, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, S.; Lorino, G.; Gherardi, G.; Battistoni, F.; De Cesaris, M.; Dicuonzo, G. Routine molecular identification of enterococci by gene-specific PCR and 16S ribosomal DNA sequencing. J. Clin. Microbiol. 2001, 39, 794–797. [Google Scholar] [CrossRef]
- Cebeci, T. Species prevalence, virulence genes, and antibiotic resistance of enterococci from food-producing animals at a slaughterhouse in Turkey. Sci. Rep. 2024, 14, 13191. [Google Scholar] [CrossRef]
- Kaprou, G.D.; Bergšpica, I.; Alexa, E.A.; Alvarez-Ordóñez, A.; Prieto, M. Rapid Methods for Antimicrobial Resistance Diagnostics. Antibiotics 2021, 10, 209. [Google Scholar] [CrossRef]
- Jenkins, D.R.; Auckland, C.; Chadwick, C.; Dodgson, A.; Enoch, D.; Goldenberg, S.; Hussain, A.; Martin, J.; Spooner, E.; Whalley, T. A practical approach to screening for carbapenemase-producing Enterobacterales- views of a group of multidisciplinary experts from English hospitals. BMC Infect. Dis. 2024, 24, 444. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jin, X.; Cheng, J.; Zhou, H.; Zhang, Y.; Dai, Y. Advances in the application of molecular diagnostic techniques for the detection of infectious disease pathogens (Review). Mol. Med. Rep. 2023, 27, 104. [Google Scholar] [CrossRef]
- Debnath, M.; Prasad, G.B.; Bisen, P.S. Molecular Microbiological Testing. In Molecular Diagnostics: Promises and Possibilities; Springer: Berlin/Heidelberg, Germany, 2009; pp. 227–243. [Google Scholar]
- Mullis, K.B. The unusual origin of the polymerase chain reaction. Sci. Am. 1990, 262, 56–61+64–65. [Google Scholar] [CrossRef]
- Garibyan, L.; Avashia, N. Polymerase chain reaction. J. Investig. Dermatol. 2013, 133, 1–4. [Google Scholar] [CrossRef]
- Artika, I.M.; Dewi, Y.P.; Nainggolan, I.M.; Siregar, J.E.; Antonjaya, U. Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis. Genes 2022, 13, 2387. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Liao, C.; Liang, L.; Yi, X.; Zhou, Z.; Wei, G. Recent advances in recombinase polymerase amplification: Principle, advantages, disadvantages and applications. Front. Cell. Infect. Microbiol. 2022, 12, 1019071. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Collatz, M.; Braun, S.D.; Monecke, S.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Ideal Consensus Primer Design. BioMedInformatics 2022, 2, 637–642. [Google Scholar] [CrossRef]
- Collatz, M.; Reinicke, M.; Diezel, C.; Braun, S.D.; Monecke, S.; Reissig, A.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Efficient Consensus Primer Design—Detection of Various Resistance and Virulence Factors in MRSA—A Case Study. BioMedInformatics 2024, 4, 1249–1261. [Google Scholar] [CrossRef]
- He, Y.; Ruan, G.; Hao, H.; Xue, F.; Zhu, S.; Xiao, B.; Zheng, B. Evaluation of Quadruple Real-Time PCR Method to Detect Enterococci Carrying Vancomycin-Resistant Genes vanA, vanB, vanM in Rectal Swabs. Front. Med. 2020, 7, 403. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Ali, J.; Johansen, W.; Ahmad, R. Short turnaround time of seven to nine hours from sample collection until informed decision for sepsis treatment using nanopore sequencing. Sci. Rep. 2024, 14, 6534. [Google Scholar] [CrossRef]
- Forde, B.M.; Bergh, H.; Cuddihy, T.; Hajkowicz, K.; Hurst, T.; Playford, E.G.; Henderson, B.C.; Runnegar, N.; Clark, J.; Jennison, A.V.; et al. Clinical Implementation of Routine Whole-genome Sequencing for Hospital Infection Control of Multi-drug Resistant Pathogens. Clin. Infect. Dis. 2022, 76, e1277–e1284. [Google Scholar] [CrossRef]
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar]
- Brownie, J.; Shawcross, S.; Theaker, J.; Whitcombe, D.; Ferrie, R.; Newton, C.; Little, S. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 1997, 25, 3235–3241. [Google Scholar] [CrossRef] [PubMed]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef]
- Zhang, S.; Duan, M.; Li, S.; Hou, J.; Qin, T.; Teng, Z.; Hu, J.; Zhang, H.; Xia, X. Current status of recombinase polymerase amplification technologies for the detection of pathogenic microorganisms. Diagn. Microbiol. Infect. Dis. 2024, 108, 116097. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Toepfer, S.; Steinmetzer, K.; Ruettger, M.; Walz, I.; Kanitz, L.; Lemuth, O.; Hubold, S.; Fritsch, F.; Loncarevic-Barcena, I.; et al. DNA-Binding Magnetic Nanoreactor Beads for Digital PCR Analysis. Anal. Chem. 2023, 95, 14175–14183. [Google Scholar] [CrossRef]
- Palladino, S.; Kay, I.D.; Flexman, J.P.; Boehm, I.; Costa, A.M.G.; Lambert, E.J.; Christiansen, K.J. Rapid detection of vanA and vanB genes directly from clinical specimens and enrichment broths by real-time multiplex PCR assay. J. Clin. Microbiol. 2003, 41, 2483–2486. [Google Scholar] [CrossRef]
- Xiang, S.; Zhang, H.; Cha, X.; Lin, Y.; Shang, Y. A New Duplex Recombinase Polymerase Amplification (D-RPA) Method for the Simultaneous and Rapid Detection of Shigella and Bacillus cereus in Food. Foods 2023, 12, 1889. [Google Scholar] [CrossRef] [PubMed]
- Panpru, P.; Srisrattakarn, A.; Panthasri, N.; Tippayawat, P.; Chanawong, A.; Tavichakorntrakool, R.; Daduang, J.; Wonglakorn, L.; Lulitanond, A. Rapid detection of Enterococcus and vancomycin resistance using recombinase polymerase amplification. PeerJ 2021, 9, e12561. [Google Scholar] [CrossRef]
- Raja, B.; Goux, H.; Marapadaga, A.; Rajagopalan, S.; Kourentzi, K.; Willson, R. Development of a panel of recombinase polymerase amplification assays for detection of common bacterial urinary tract infection pathogens. J. Appl. Microbiol. 2017, 123, 544–555. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer–Probe Set | Sequence (5′-3′) |
|---|---|---|
| ddl_faecalis | primer_left_0_sequence_fwd (5′-3′) | AAGTAGCCATTTTAGGAAAT |
| primer_right_0_sequence_revcomp (5′-3′) | GCATCATAATCATAGAAAGC | |
| probe_internal_0_sequence | CCGTACGACTTTACCTGGTGAAGTGG | |
| ddl_faecium | primer_left_0_sequence_fwd (5′-3′) | ACATTGAATATGCCTTATGT |
| primer_right_0_sequence_revcomp (5′-3′) | TTGGTCATGATTTTATCCAT | |
| probe_internal_0_sequence | CAGGCGTATTGACCAGTGCATGTG | |
| vanA | primer_left_0_sequence_fwd (5′-3′) | CATGTTGATGTAGCATTTTC |
| primer_right_0_sequence_revcomp (5′-3′) | AATTCAAACAGACCTTGTAT | |
| probe_internal_0_sequence | GGCAAGTCAGGTGAAGATGGATCC | |
| vanB | primer_left_0_sequence_fwd (5′-3′) | TTGCTCGGAGGAACATGAT |
| primer_right_0_sequence_revcomp (5′-3′) | TCTTGCATAGCTTCCATA | |
| probe_internal_0_sequence | GAAAAATTCGATCCGCACTACATCGG |
| Target | VRE Strains | Dye | Lift Δ Rn (Mean) D5 | Calibration Curve Efficiency |
|---|---|---|---|---|
| vanA | 95735_UK040 | Cy5 | 1,150,000 | 98 |
| vanB | 95738_UK043 | ATTO 550 | 600,000 | 95 |
| ddl_faecalis | 95737_UK045 | FAM | 827,212 | 94 |
| ddl_faecium | 95738_UK043 | ROX | 547,649 | 90 |
| Multiplex RT PCR | RPA | |||||||
|---|---|---|---|---|---|---|---|---|
| Strains ID | ddl_faecalis | ddl_faecium | vanA | vanB | rpoA_faecalis | ddl_faecium | vanA | vanB |
| 97629 | N | P | P | N | N | P | P | N |
| 97903 | N | P | P | N | - | P | P | N |
| 97718 | N | P | P | N | N | P | P | N |
| 97618 | N | P | N | P | N | P | - | P |
| 97721 | N | P | N | P | - | P | N | P |
| 97728 | N | P | N | P | - | P | N | P |
| 97731 | N | P | N | P | - | P | N | P |
| 97778 | N | P | N | P | N | P | N | P |
| 97875 | N | P | P | N | - | P | P | - |
| 97878 | N | P | P | N | - | P | P | - |
| 97914 | N | P | P | N | - | P | P | - |
| 97631 | P | N | P | N | P | - | P | - |
| 97635 | P | N | P | N | P | - | P | - |
| 97636 | P | N | P | N | P | N | P | - |
| 97633 | P | N | P | N | P | N | P | - |
| 97643 | P | N | P | N | P | N | P | - |
| 97644 | P | N | P | N | P | N | P | - |
| 97880 | N | P | P | N | - | P | P | - |
| 97889 | N | P | P | N | - | P | P | - |
| 97925 | N | P | N | P | - | P | - | P |
| Parameters | Multiplex RT PCR | RPA | WGS | VRE DNA Microarray |
|---|---|---|---|---|
| Working time | Between 2 and 3 h [37]. | About 20 min [38]. | Between 7 and 9 h, starting from VRE cultures [39]. In real-life clinical settings, time between sample collection and result can range from 24 h to 33 days [40]. | About 4–5 h, starting from VRE cultures [3]. |
| Scope of application | Screening method/confirmation of susceptibility results. | Screening method/confirmation of susceptibility results. | Characterization of isolates. | Characterization of isolates. |
| Number of target genes detected | Four to six [37]. | One. | Thousands, all that are present [41]. | Multiple genes to more than 300 [3]. |
| Disadvantages | Requires use of expensive equipment. Requires trained personnel. Possibility of false amplification products, because of formation of primer dimers, resulting in poor annealing and extension rates [42]. Prone to contamination. Only specified targets can be detected. | Only few commercially available kits. Not suitable for large-scale experiments. High possibility of non-specific amplification [38]. Prone to contamination. Only specified target can be detected. | Requires trained personnel to obtain, analyze, and correctly interpret results [40]. Restricted to specialized laboratories. Large dataset. Not suitable for quick screening.Relatively expensive. Requires expensive hardware and/or expensive reagents. | Requires minimal training. Only targets present on microarray can be detected. Compared to PCR methods, “proofreading” can be achieved by using multiple primers/probes per target or target operon. |
| Advantages | Quick sample preparation, reagents are relatively affordable, there are many commercially available kits for PCR. DNA quantity of 1–5 ng is sufficient [43]. Suitable for screening. | Does not require expensive cycling equipment. Short run time [44]. Suitable for screening. | Can detect new genes or genes that are not pre-defined in panel. | Affordable. More than 300 targets genes can be detected. Suitable for use in outbreaks, for detection and tracking of transmission chains. |
| Target | Primer–Probe Set | Sequence (5′-3′) | Product Length |
|---|---|---|---|
| ddl | ddl-F | ACCCAAGTGGACAGACAGAGGAAGGCTTTA | 156 bp |
| ddl-R | TTCCATCTTCCCCGTTTGGCCCATGTAAAACT | ||
| ddl-F_revcomp | TAAAGCCTTCCTCTGTCTGTCCACTTGGGT | ||
| ddl-R_revcomp | AGTTTTACATGGGCCAAACGGGGAAGATGGAA | ||
| vanA | vanA-F | TTGCGCGGAATGGGAAAACGACAATTGCTATT | 194 bp |
| vanA-R | CAAAAGGGATACCGGACAATTCAAACAGACC | ||
| vanA-F_revcomp | AATAGCAATTGTCGTTTTCCCATTCCGCGCAA | ||
| vanA-R_revcomp | GGTCTGTTTGAATTGTCCGGTATCCCTTTTG | ||
| vanB | vanB-F | GAGGATGATTTGATTGTCGGCGAAGTGGAT | 165 bp |
| vanB-R | TTTGCCGTTTCTTGCACCCGATTTCGTTCCTC | ||
| vanB-F_revcomp | ATCCACTTCGCCGACAATCAAATCATCCTC | ||
| vanB-R_revcomp | GAGGAACGAAATCGGGTGCAAGAAACGGCAAA | ||
| rpoA | rpoA-F | GGACCCGCTACCGTGACTGCCGGCGATATTATCG | 197 bp |
| rpoA-R | GAATCAACTGGAAGTACACCGATTGGCATATC | ||
| rpoA-F_revcomp | CGATAATATCGCCGGCAGTCACGGTAGCGGGTCC | ||
| rpoA-R_revcomp | GATATGCCAATCGGTGTACTTCCAGTTGATTC |
| Substances | Unit | Final Concentration | Volume/Well (µL) |
|---|---|---|---|
| qPCR Buffer | mM | 1 | 2.00 |
| MgCl2 | mM | 3 | 1.20 |
| dNTP/dUTP (BTR) | mM | 0.2 | 0.40 |
| ddl-faecalis_fwd_left_0 | µM | 0.2 | 0.04 |
| ddl-faecium_fwd_left_0 | µM | 0.2 | 0.04 |
| vanA_fwd_left_0 | µM | 0.2 | 0.04 |
| vanB_fwd_left_0 | µM | 0.2 | 0.04 |
| ddl-faecalis_revcomp_rigth_0 | µM | 0.2 | 0.04 |
| ddl-faecium_revcomp_rigth_0 | µM | 0.2 | 0.04 |
| vanA_revcomp_rigth_0 | µM | 0.2 | 0.04 |
| vanB_revcomp_rigth_0 | µM | 0.2 | 0.04 |
| ddl-faecalis_probe_0 | µM | 0.2 | 0.04 |
| ddl-faecium_probe_0 | µM | 0.2 | 0.04 |
| vanA_probe_0 | µM | 0.2 | 0.04 |
| vanB_probe_0 | µM | 0.2 | 0.04 |
| Polymerase (BTR) | U/µL | 0.2 | 0.80 |
| BSA (NEB) | mg/ml | 1 | 1.00 |
| Uracil-DNA Glycosylase (BTR) | U/µL | 0.01 | 0.20 |
| Template (DNA) | - | - | 8.00 |
| water, nuclease-free | - | - | 5.92 |
| Total volume | - | - | 20.00 |
| Components | Concentration | ||
|---|---|---|---|
| Stock | Reaction | Reaction | |
| 1 bead | |||
| Water (H2O) | 17.80 µL | ||
| RPA reconstitution buffer 10 | 2× | 1.17 | 25.00 µL |
| Total volume | 42.8 µL |
| Components | Concentration | ||
|---|---|---|---|
| Stock | Reaction | ||
| Reconstituted LyoBead RPA | 1.17× | 1× | 42.8 µL |
| Volume of pre-mix | 43.5 µL | ||
| Template (DNA) | 2.0 µL | ||
| fw_Primer | 20 µM | 0.2 µM | 0.5 µL |
| rev_Primer | 20 µM | 0.2 µM | 0.5 µL |
| EvaGreen (dye) | 50× | 1× | 1.0 µL |
| RPA reaction initiator | 20× | 1× | 2.5 µL |
| Total volume per reaction | 50.0 µL | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osadare, I.E.; Abdilahi, A.; Reinicke, M.; Diezel, C.; Collatz, M.; Reissig, A.; Monecke, S.; Ehricht, R. Multiplex Real-Time Polymerase Chain Reaction and Recombinase Polymerase Amplification: Methods for Quick and Cost-Effective Detection of Vancomycin-Resistant Enterococci (VRE). Antibiotics 2025, 14, 295. https://doi.org/10.3390/antibiotics14030295
Osadare IE, Abdilahi A, Reinicke M, Diezel C, Collatz M, Reissig A, Monecke S, Ehricht R. Multiplex Real-Time Polymerase Chain Reaction and Recombinase Polymerase Amplification: Methods for Quick and Cost-Effective Detection of Vancomycin-Resistant Enterococci (VRE). Antibiotics. 2025; 14(3):295. https://doi.org/10.3390/antibiotics14030295
Chicago/Turabian StyleOsadare, Ibukun Elizabeth, Abdinasir Abdilahi, Martin Reinicke, Celia Diezel, Maximilian Collatz, Annett Reissig, Stefan Monecke, and Ralf Ehricht. 2025. "Multiplex Real-Time Polymerase Chain Reaction and Recombinase Polymerase Amplification: Methods for Quick and Cost-Effective Detection of Vancomycin-Resistant Enterococci (VRE)" Antibiotics 14, no. 3: 295. https://doi.org/10.3390/antibiotics14030295
APA StyleOsadare, I. E., Abdilahi, A., Reinicke, M., Diezel, C., Collatz, M., Reissig, A., Monecke, S., & Ehricht, R. (2025). Multiplex Real-Time Polymerase Chain Reaction and Recombinase Polymerase Amplification: Methods for Quick and Cost-Effective Detection of Vancomycin-Resistant Enterococci (VRE). Antibiotics, 14(3), 295. https://doi.org/10.3390/antibiotics14030295