
An Insight into Rational Drug Design: The Development of In-House Azole Compounds with Antimicrobial Activity

 ,
,  ,
,  , ,
, ,  , and
, and 
Abstract
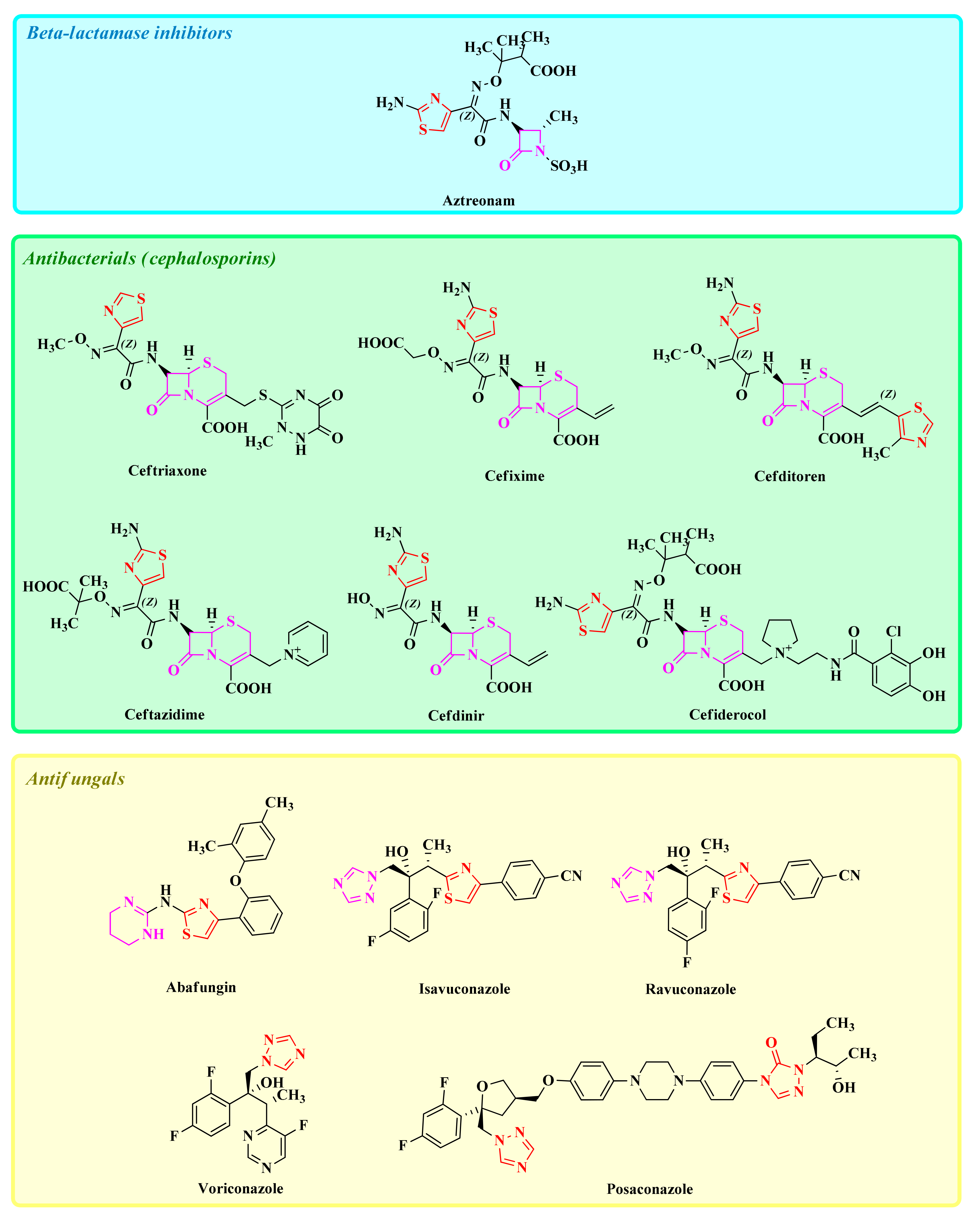
1. Introduction
2. The Development of In-House Azole Compounds with Antimicrobial Activity
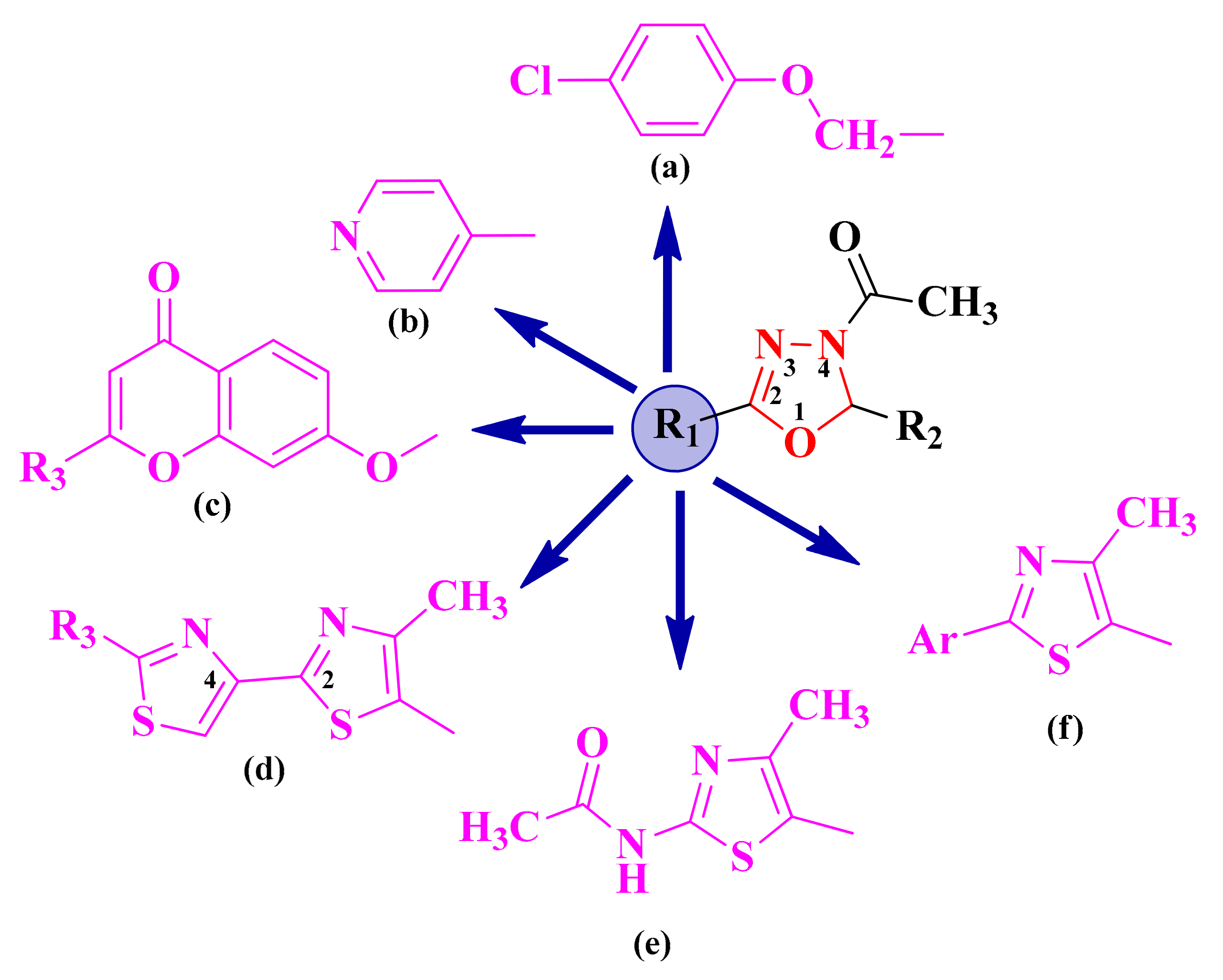
2.1. Synthesis and Antimicrobial Assay of Aryl and Hetaryl-1,3,4-Oxadiazoline Compounds
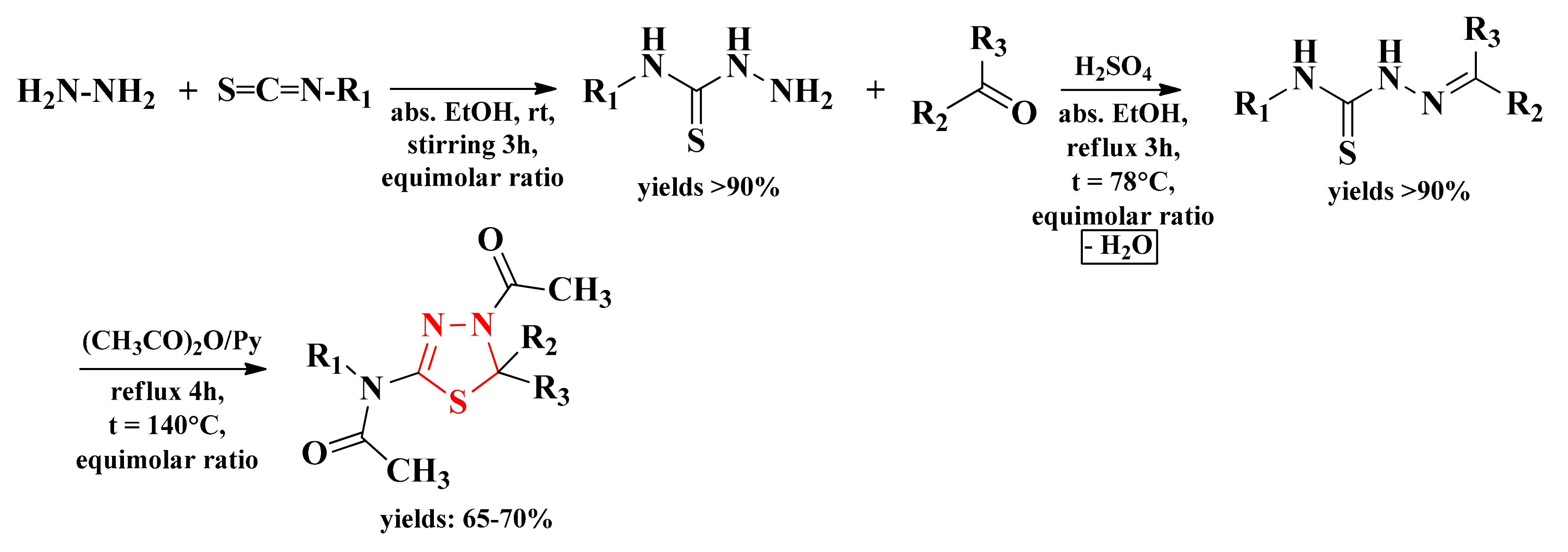
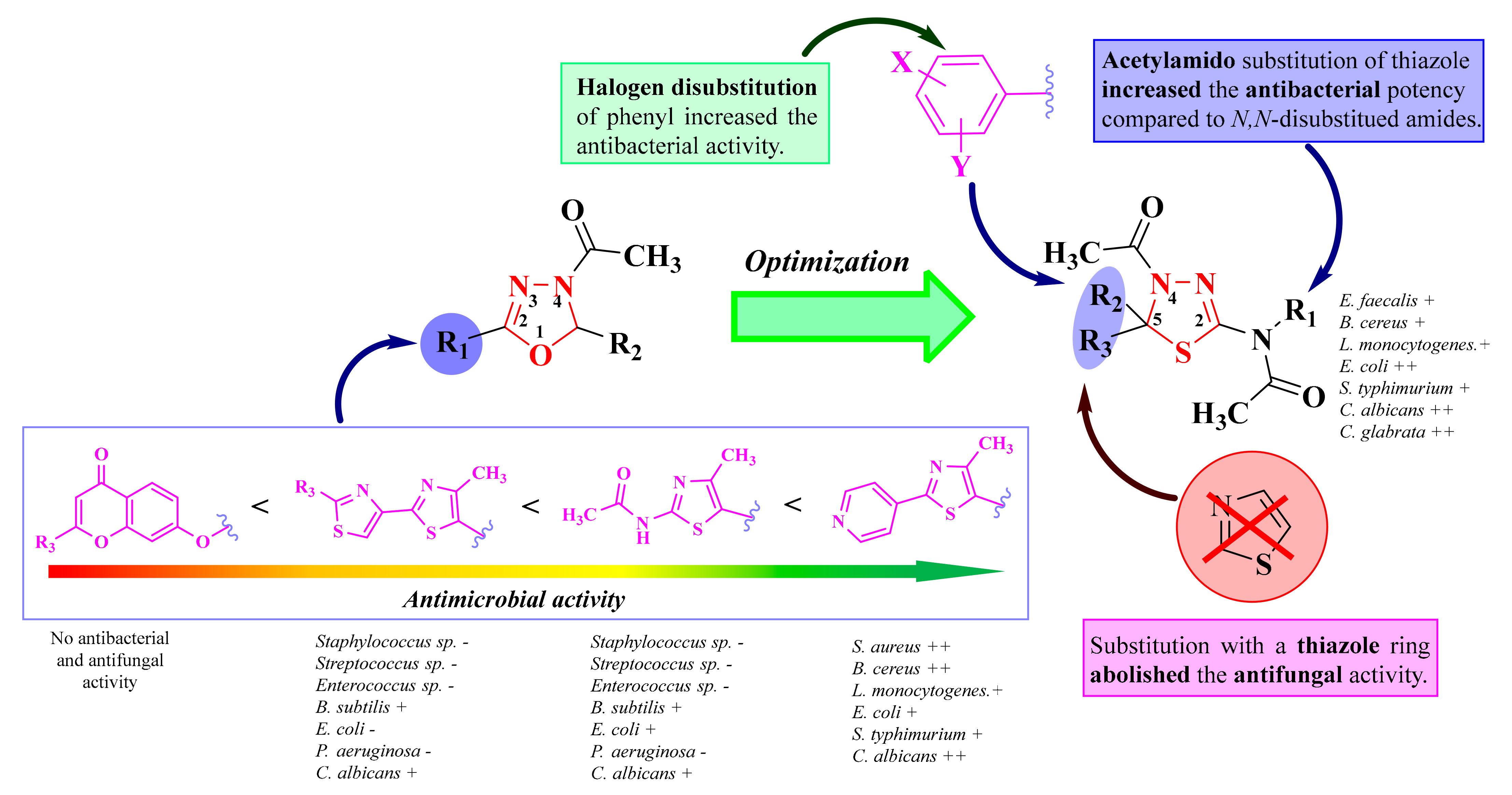
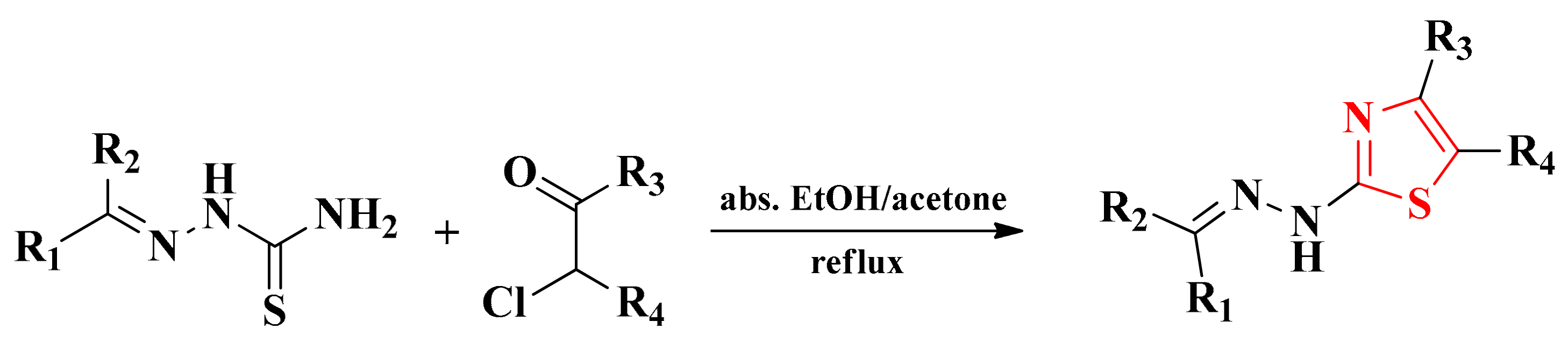
2.2. Synthesis and Antimicrobial Assay of Aryl and Hetaryl-1,3,4-Thiadiazoline Compounds
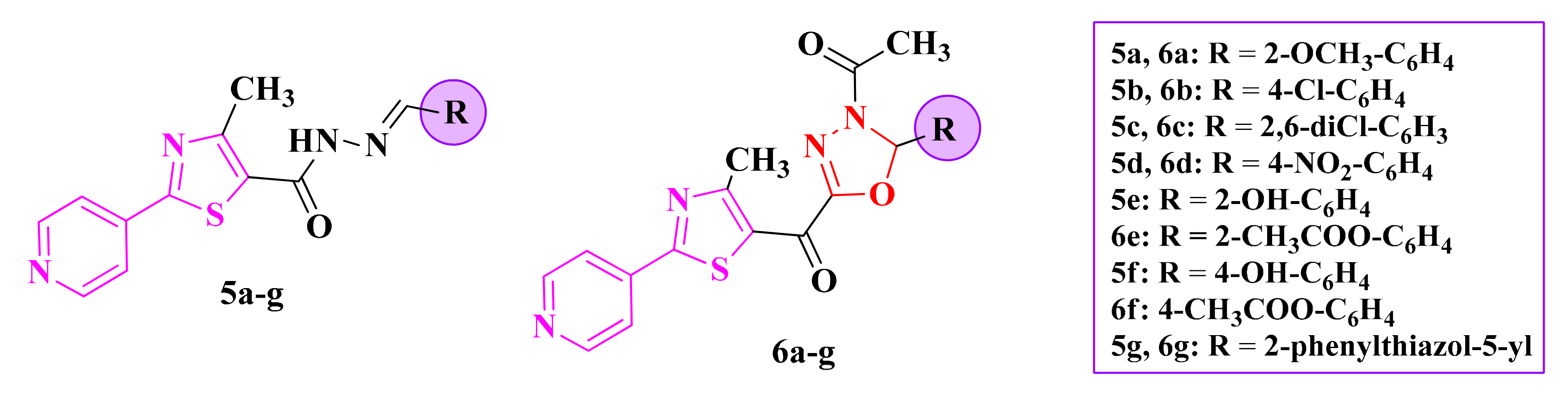
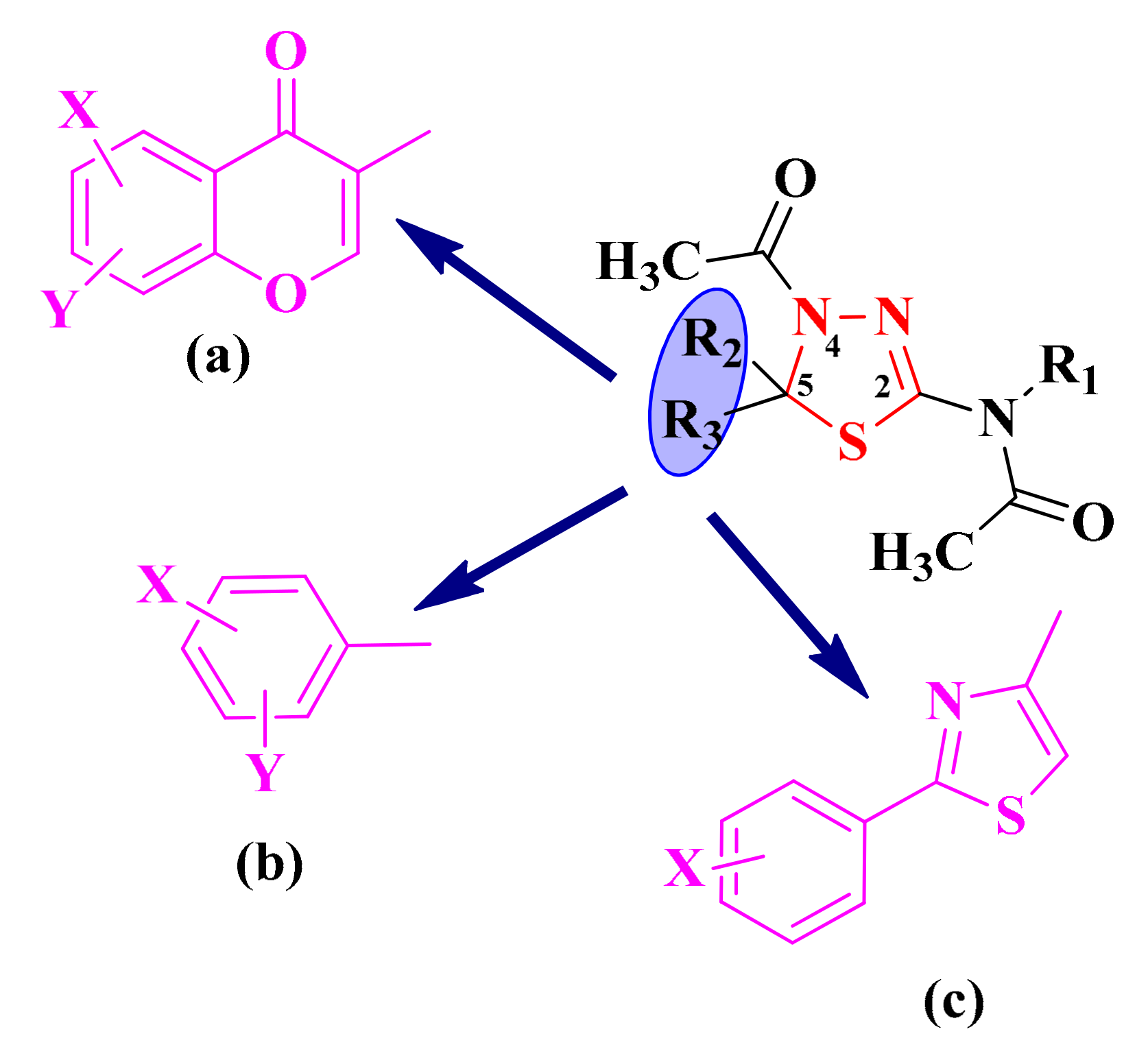
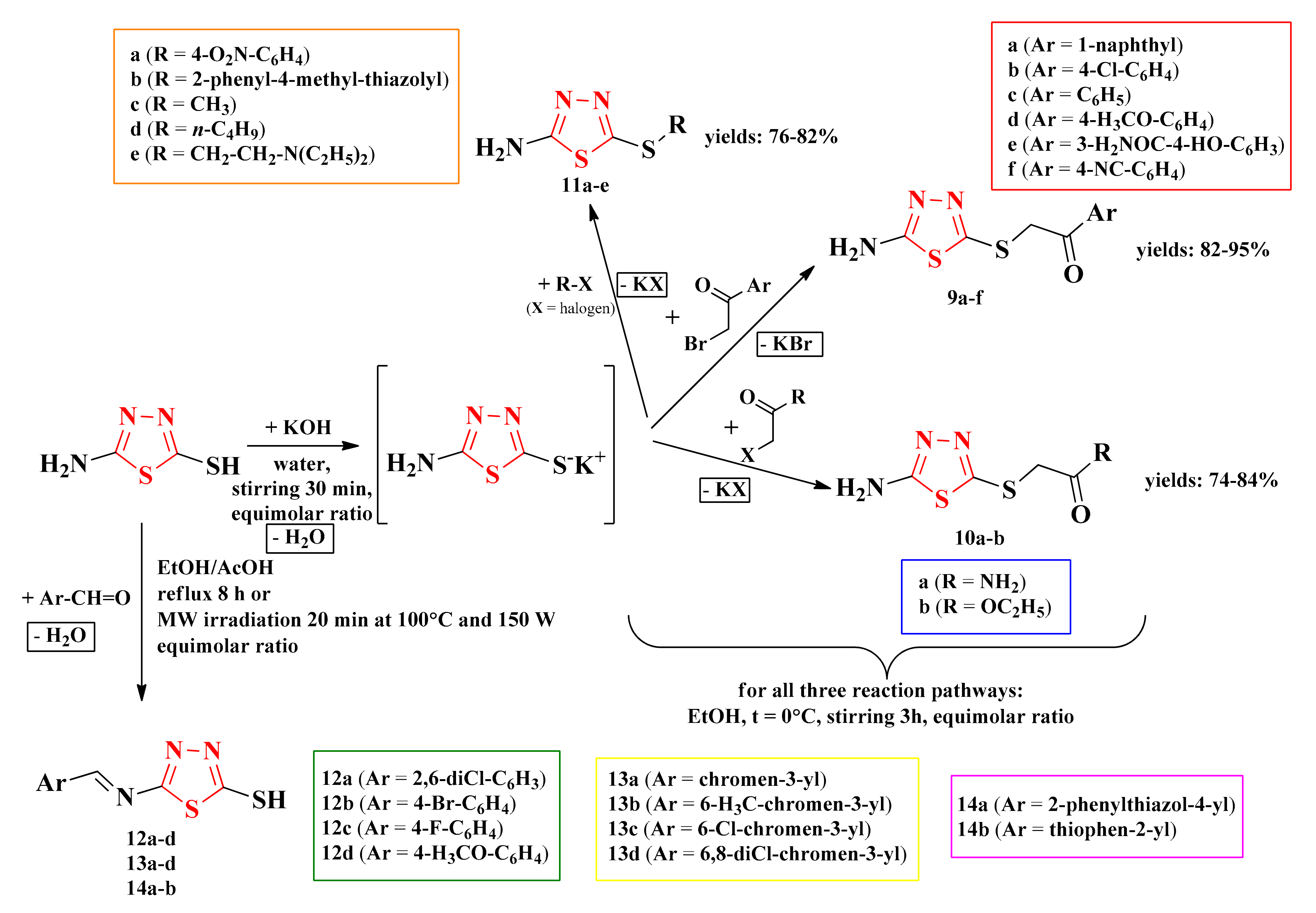
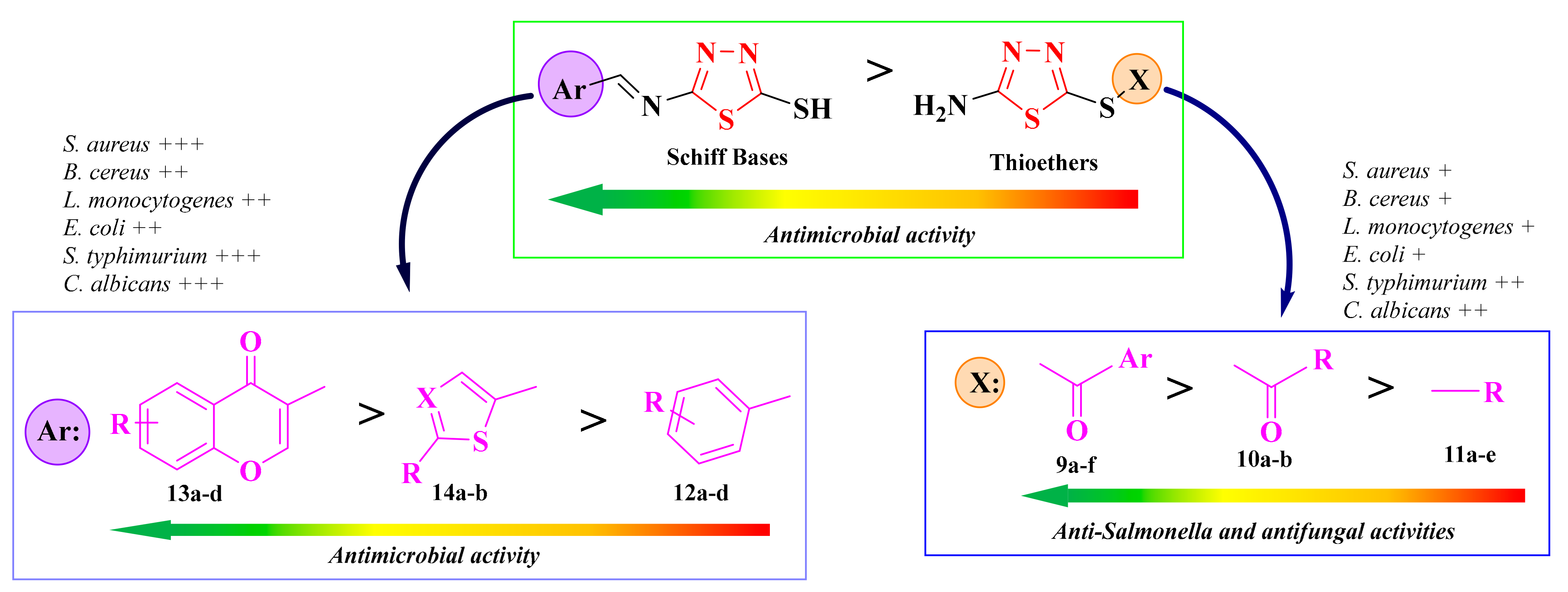
2.3. 1,3,4-Thiadiazolyl-Thioethers and Schiff Bases
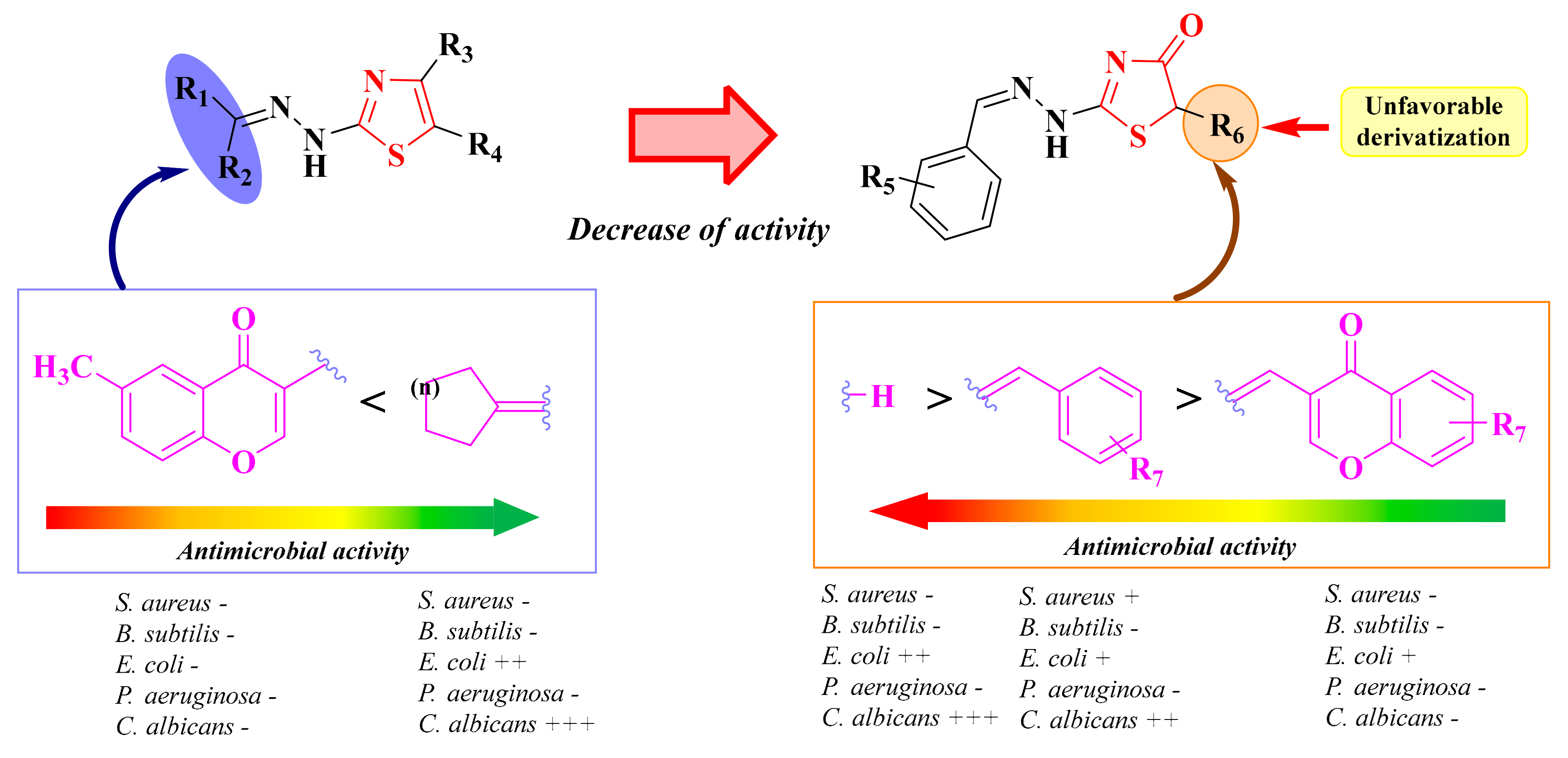
2.4. Alkylidene-Hydrazinyl-Thiazoles
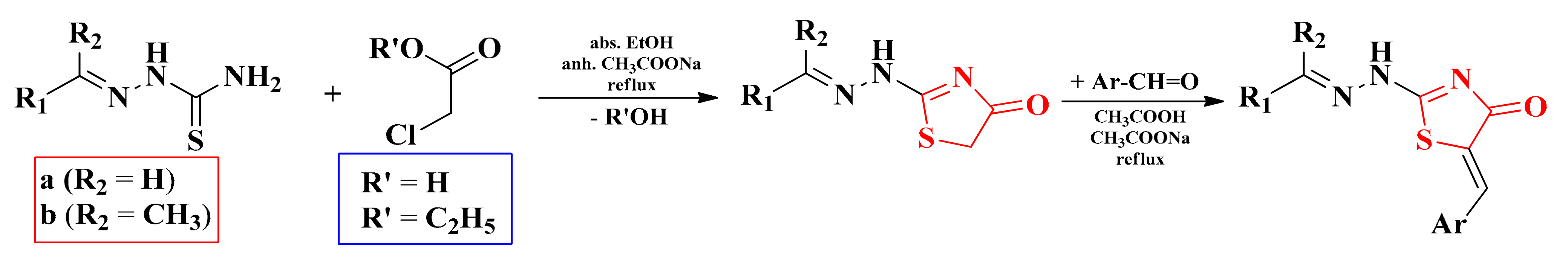
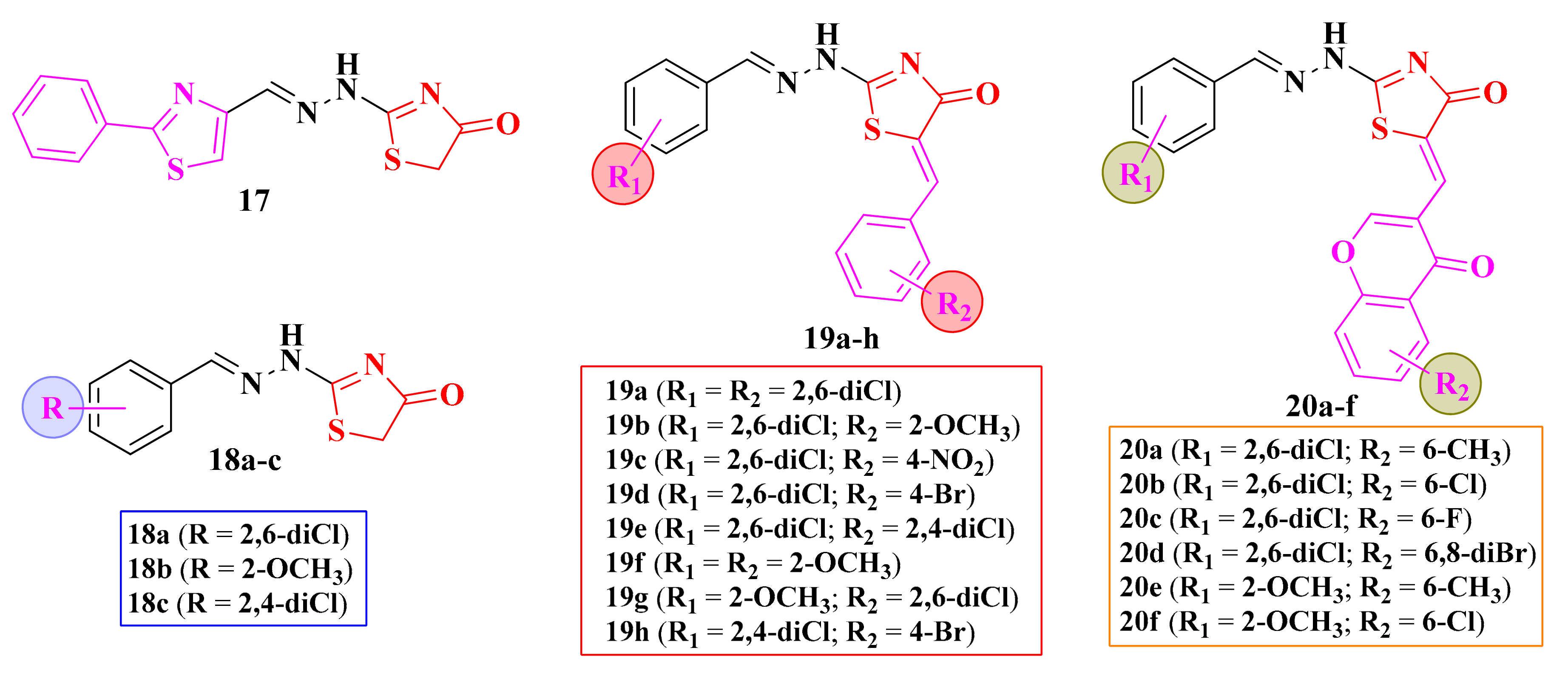
2.5. Alkylidene- and Arylidene-Hydrazinyl-Thiazolin-4-ones
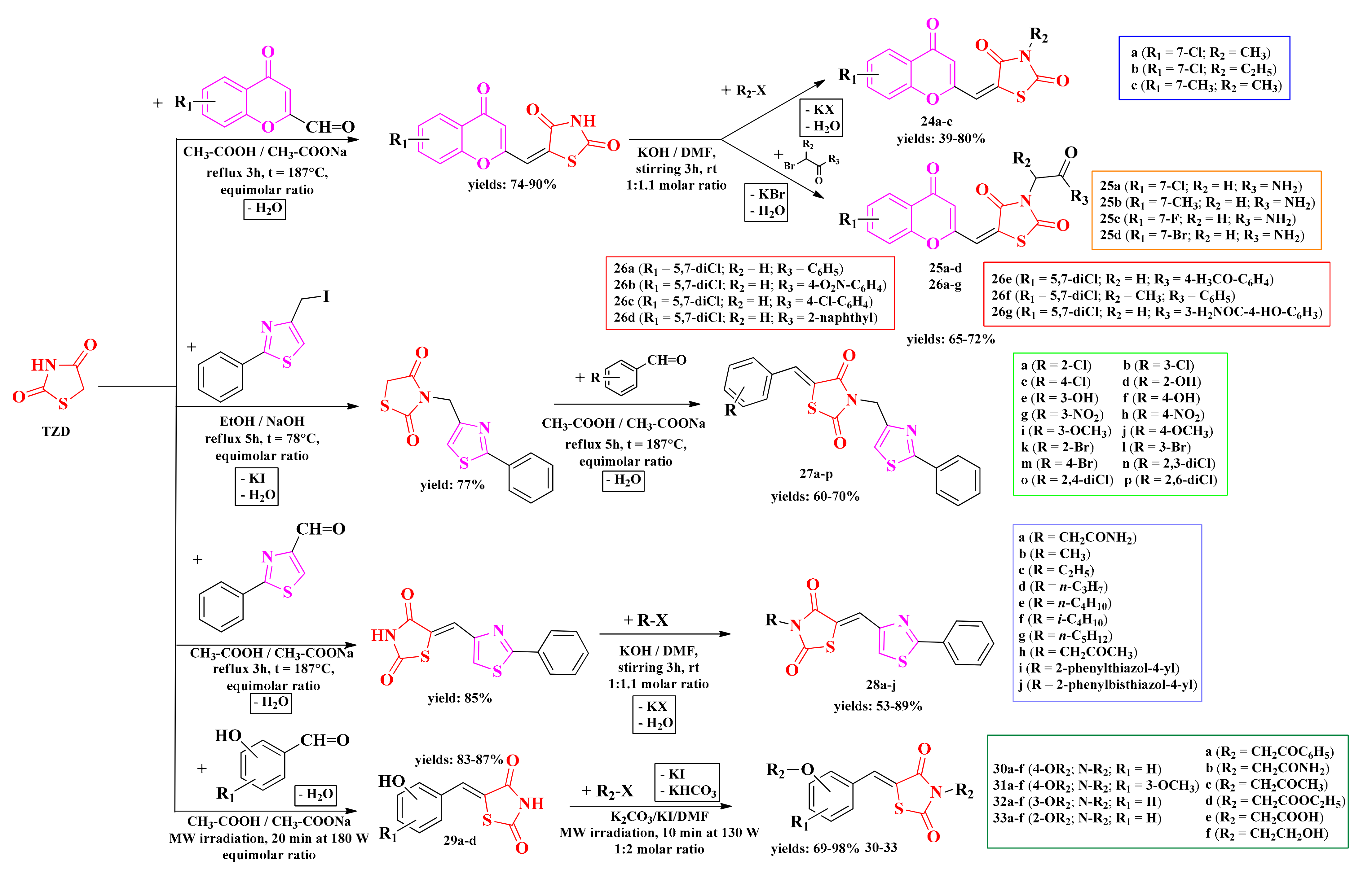
2.6. Aryl- and Hetaryl-Thiazolidine-2,4-dione Compounds
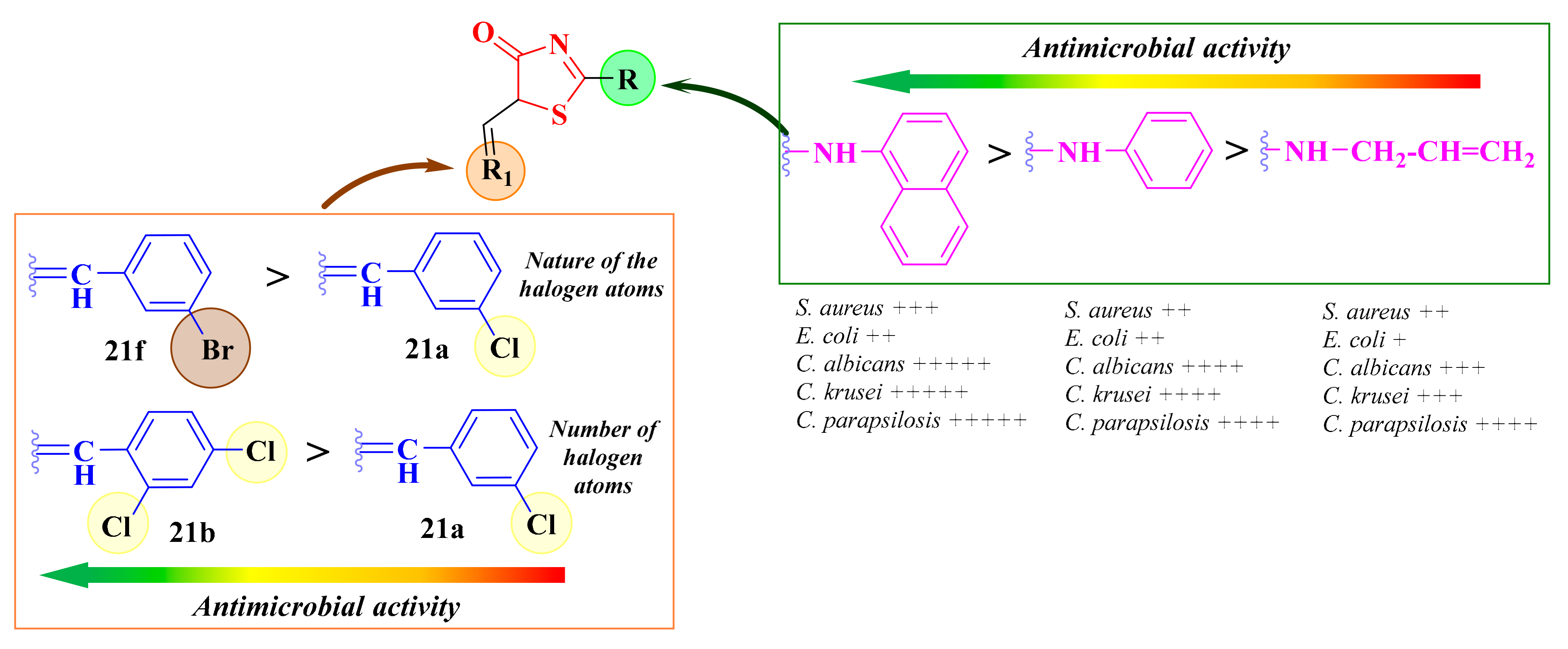
2.6.1. 3,5-Disubstituted-Thiazolidine-2,4-diones
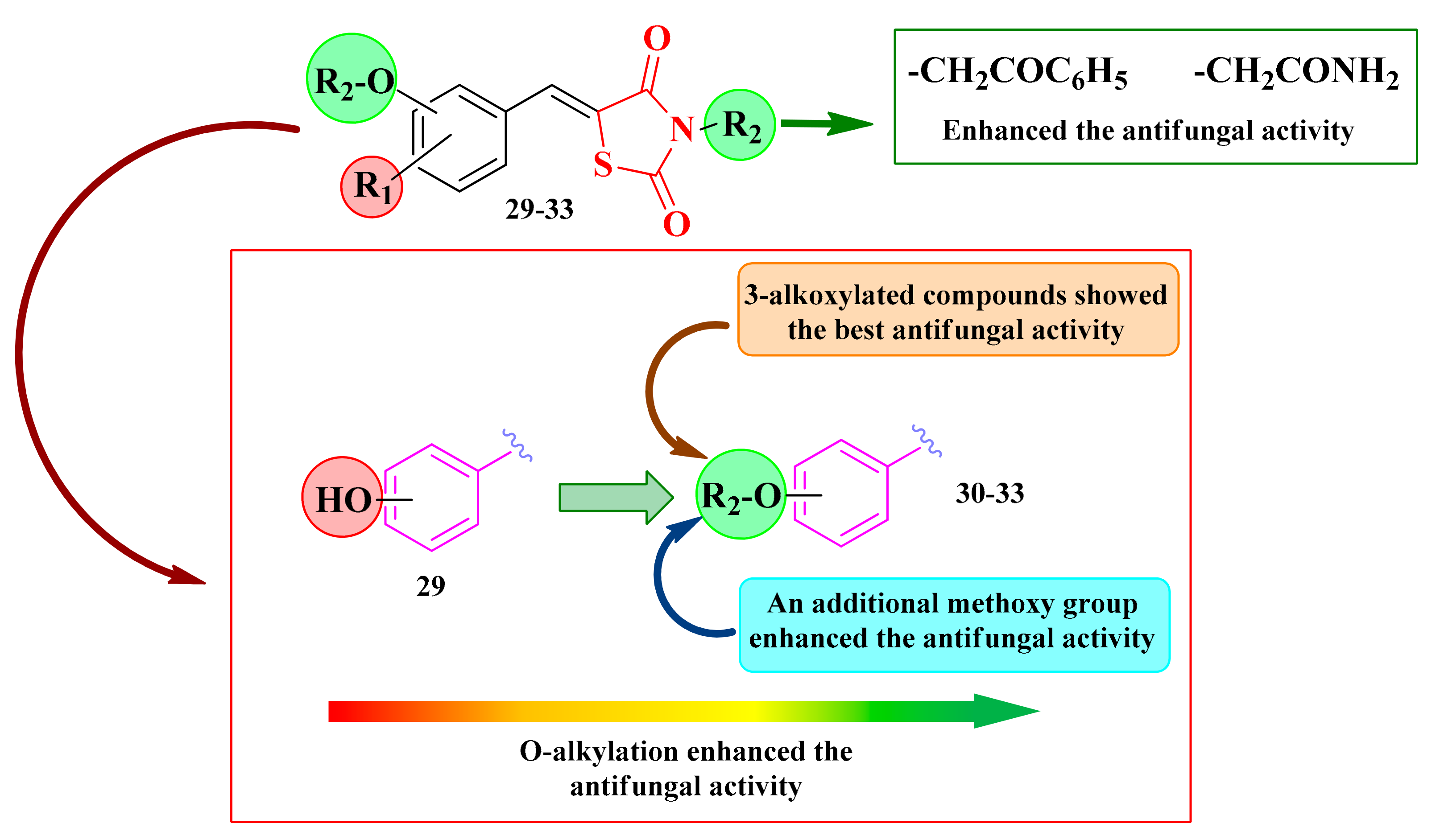
2.6.2. N-Substituted-5-Hydroxyarylidene-Thiazolidine-2,4-diones
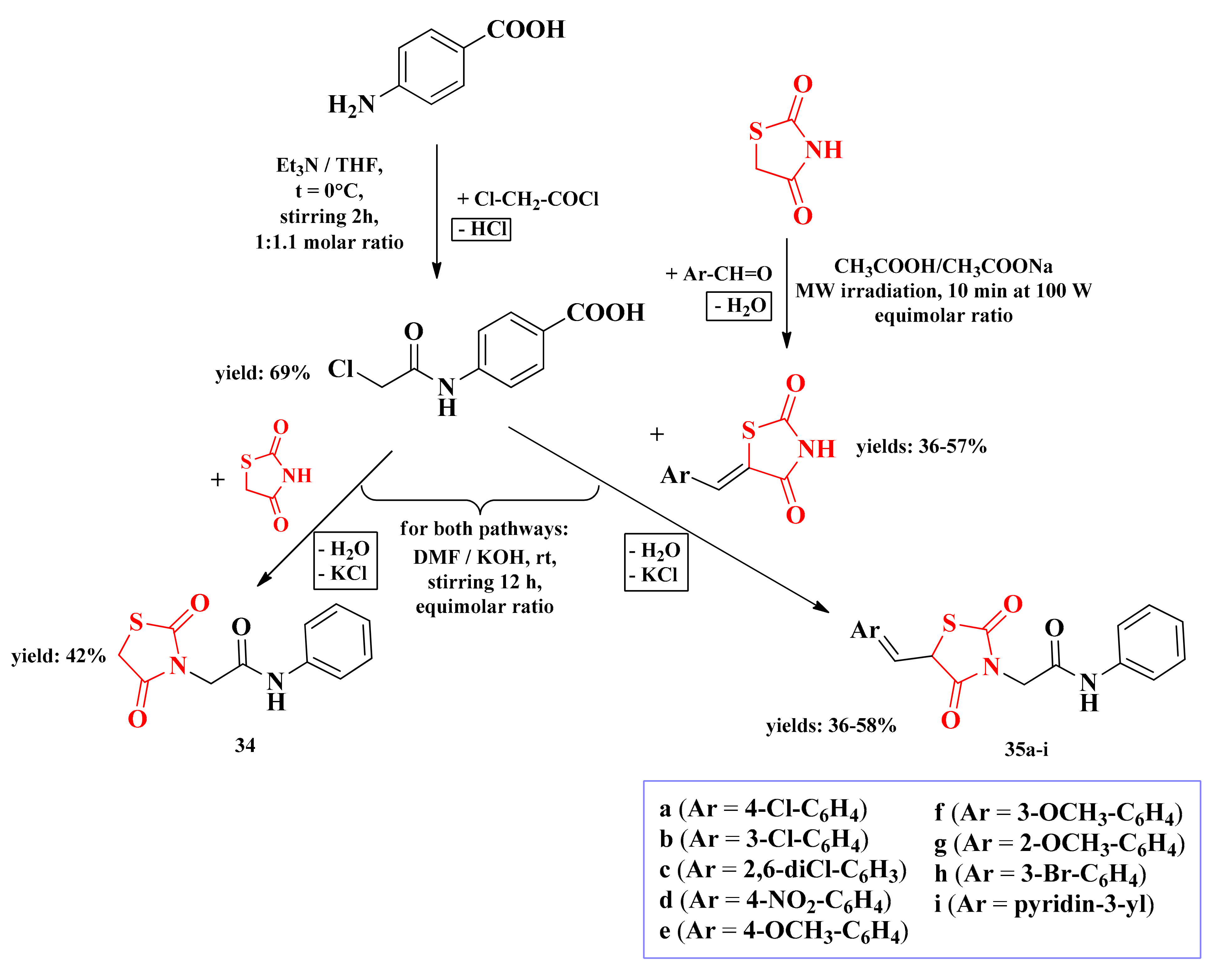
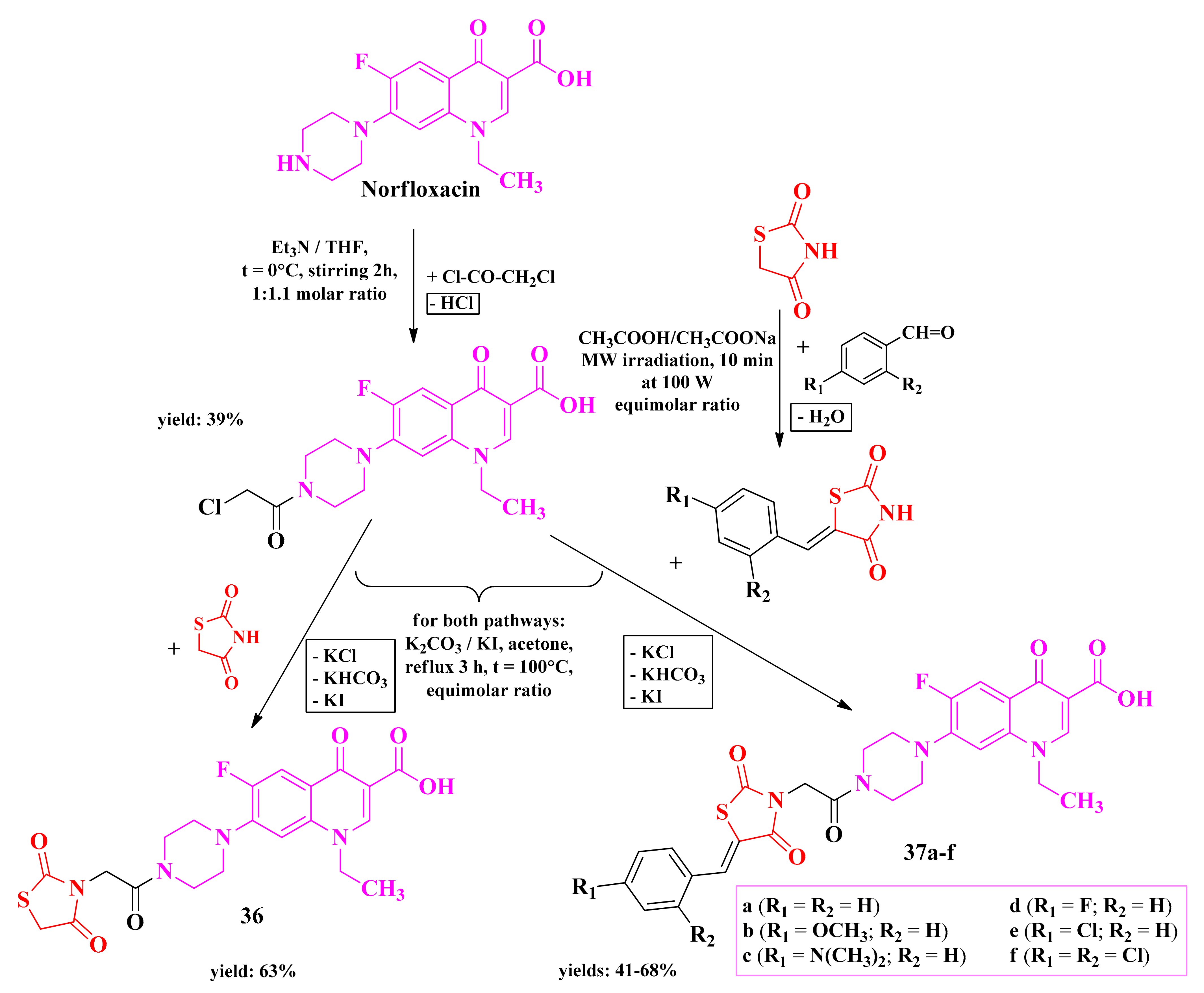
2.6.3. Piperazin-4-yl-(Acetyl-Thiazolidine-2,4-dione) Norfloxacin Analogues
2.7. Thiazolyl-1,2,4-Triazole Schiff Bases

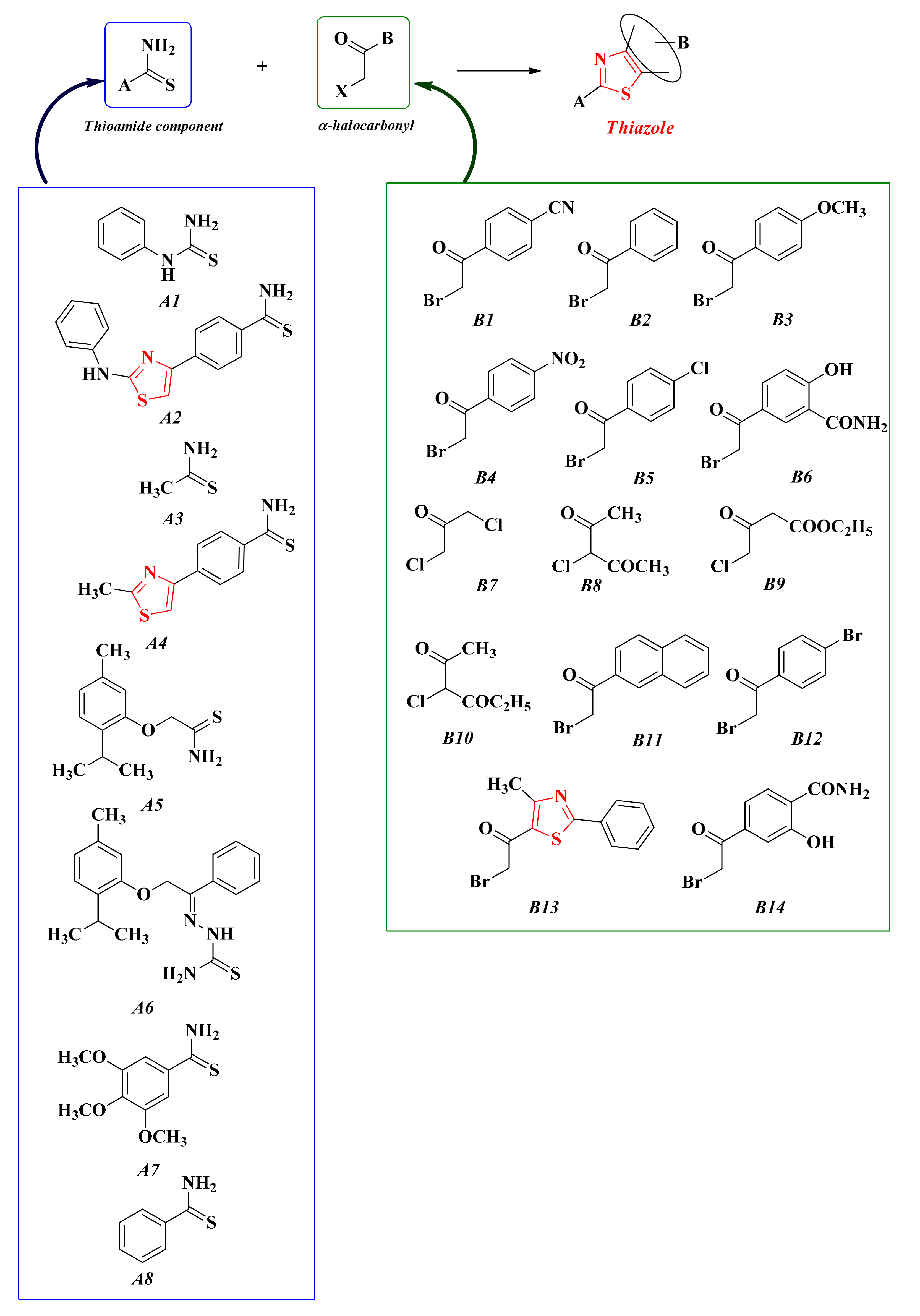
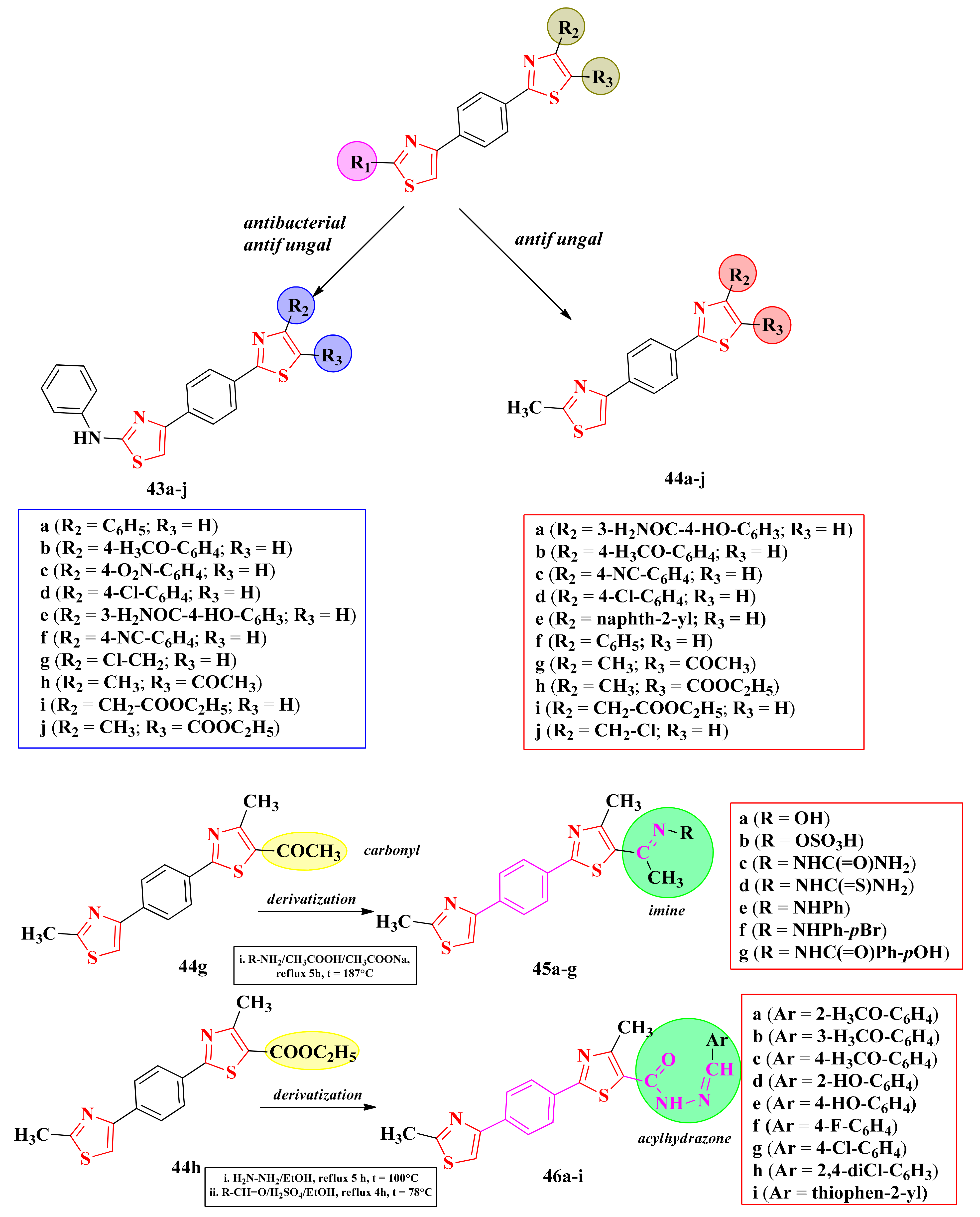
2.8. Thiazoles and Bisthiazoles
2.8.1. Thiazol-4-yl-1,4-Phenylene-2-Thiazoles
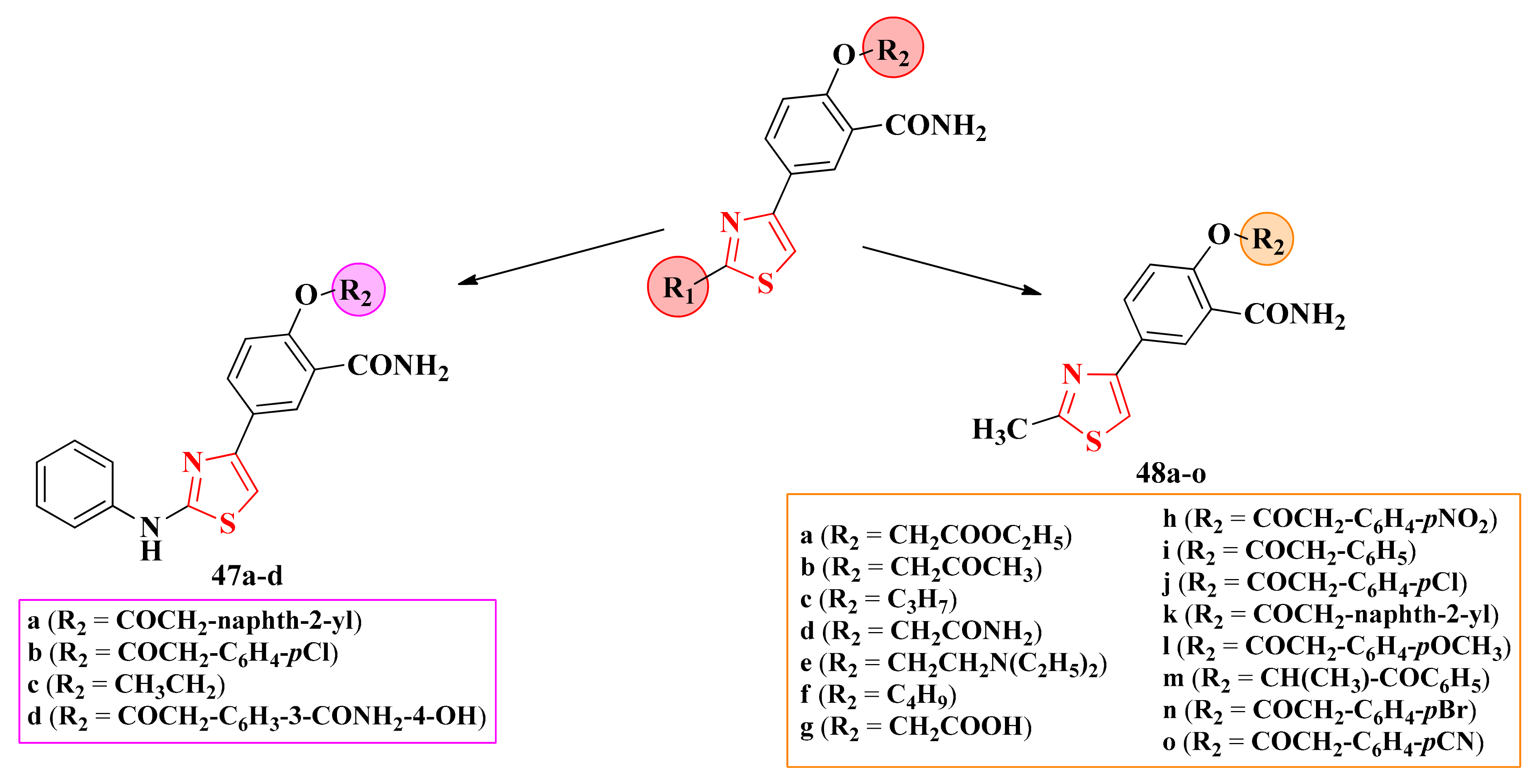
2.8.2. 4-(5-Salicylamide)-Thiazoles
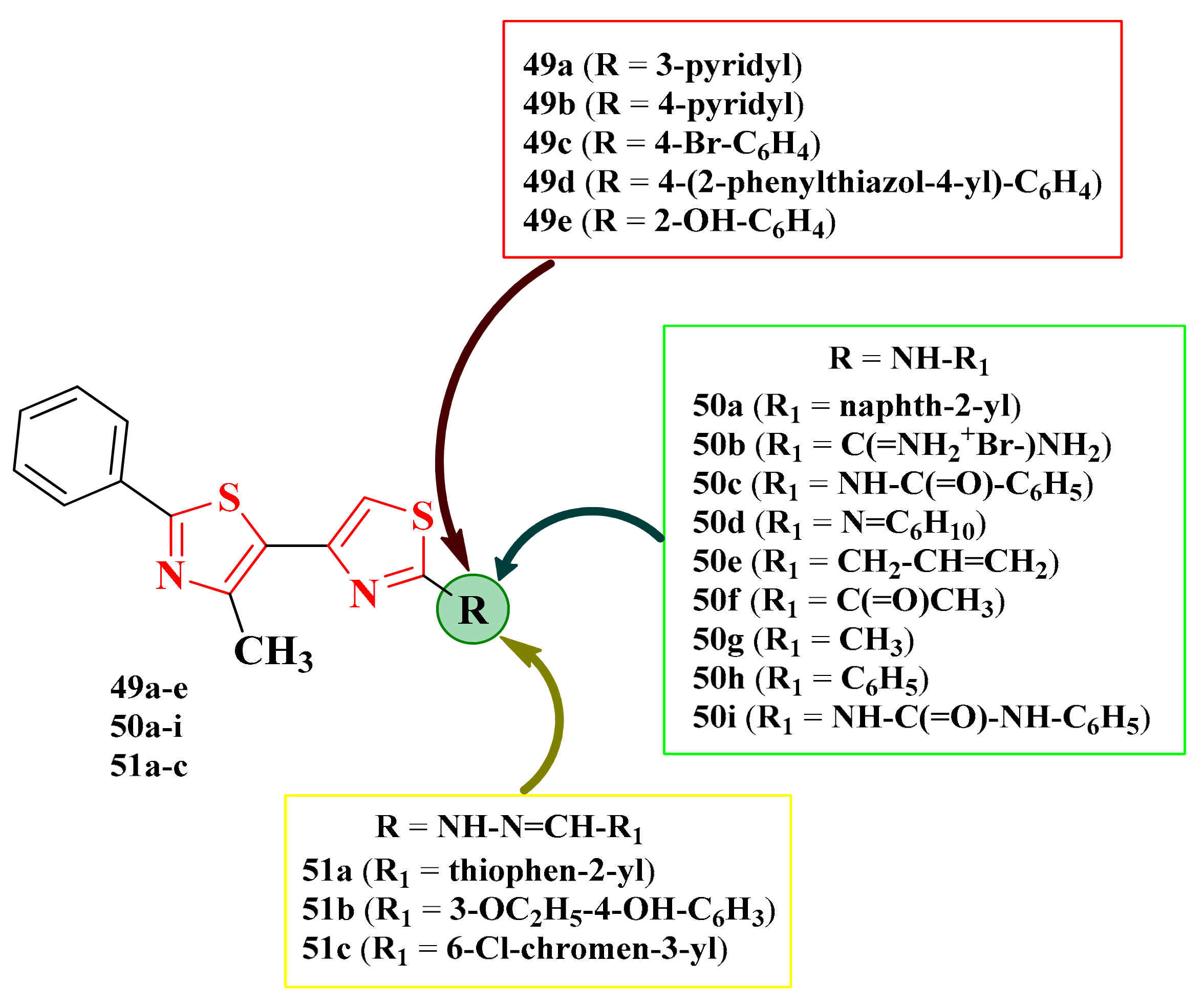
2.8.3. 4,5′-Bisthiazoles
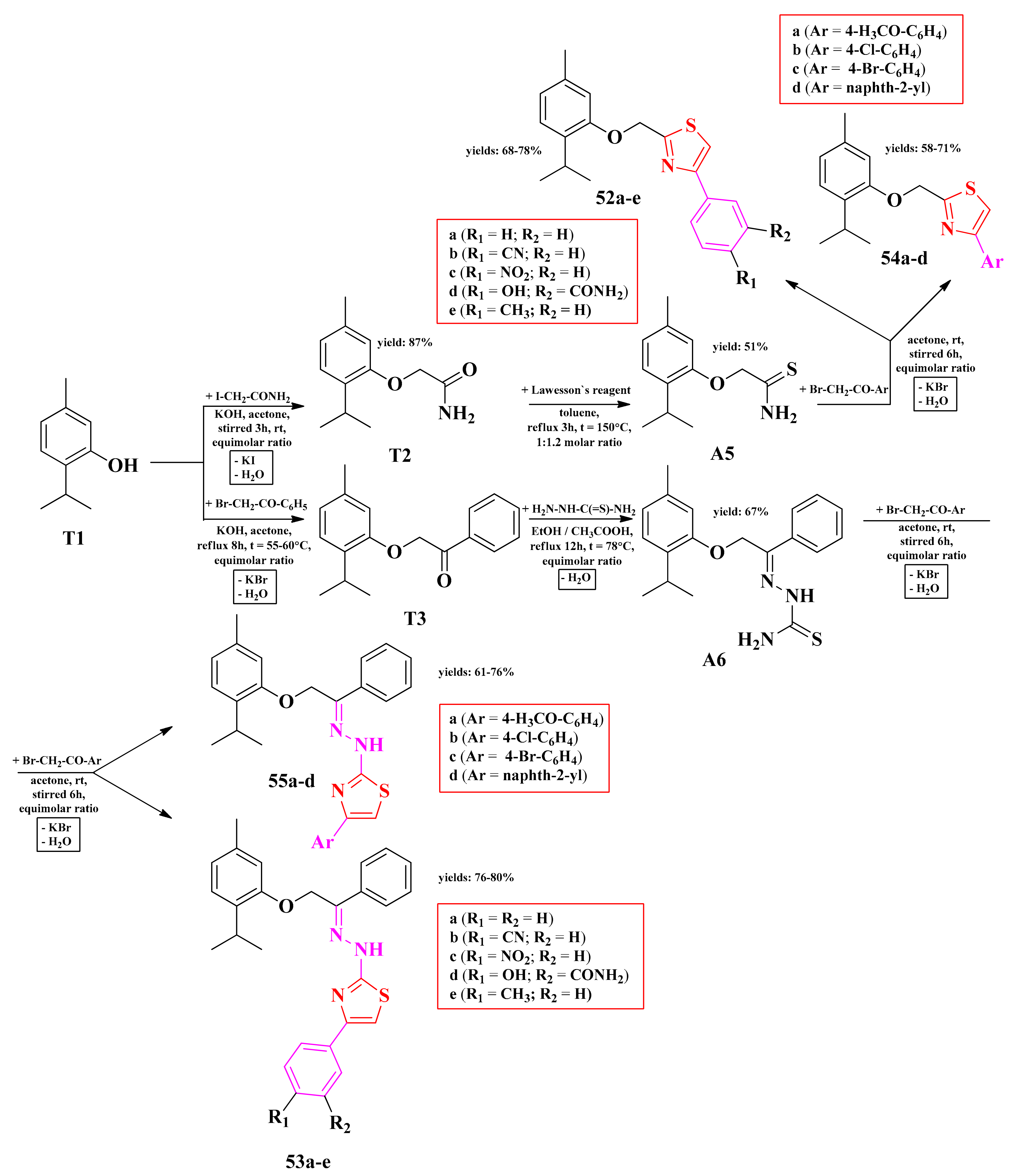
2.9. Thymolyl-Azoles
2.9.1. Thymolyl-Thiazoles
2.9.2. Thimolyl-Triazoles
3. The Development of In-House Azole Compounds with Antibiofilm Activity
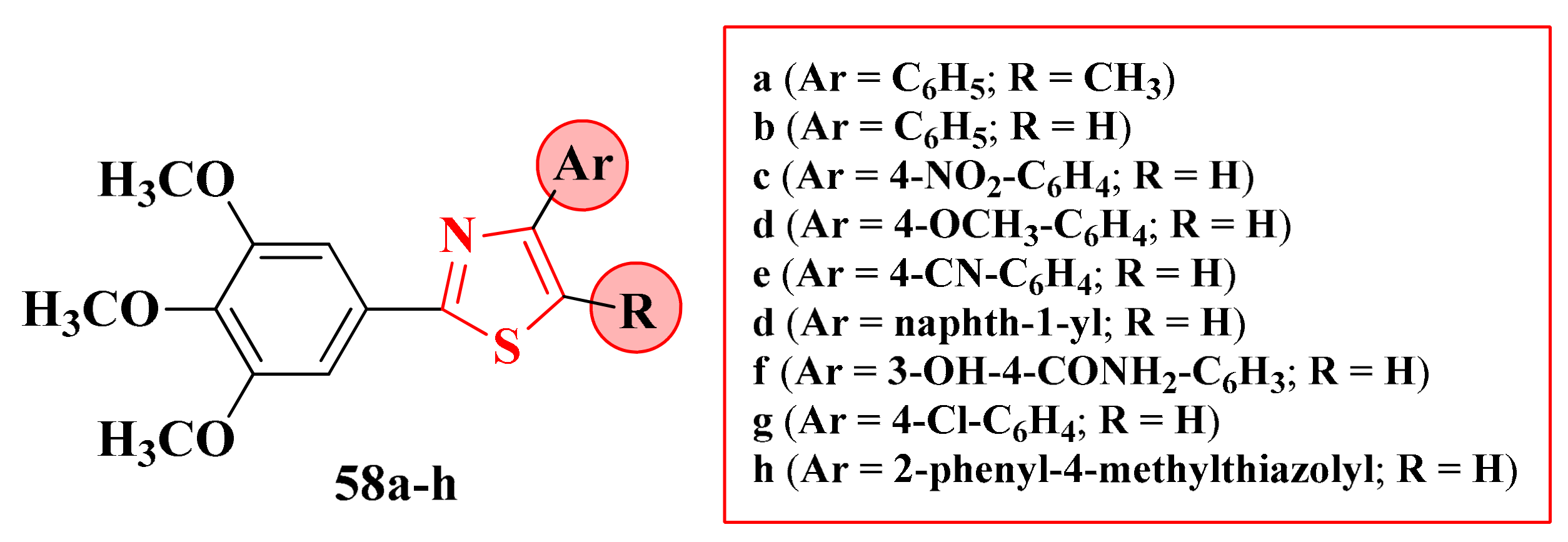
3.1. 2-(3,4,5-Trimethoxyphenyl)-4-Ar-5-R-Thiazoles
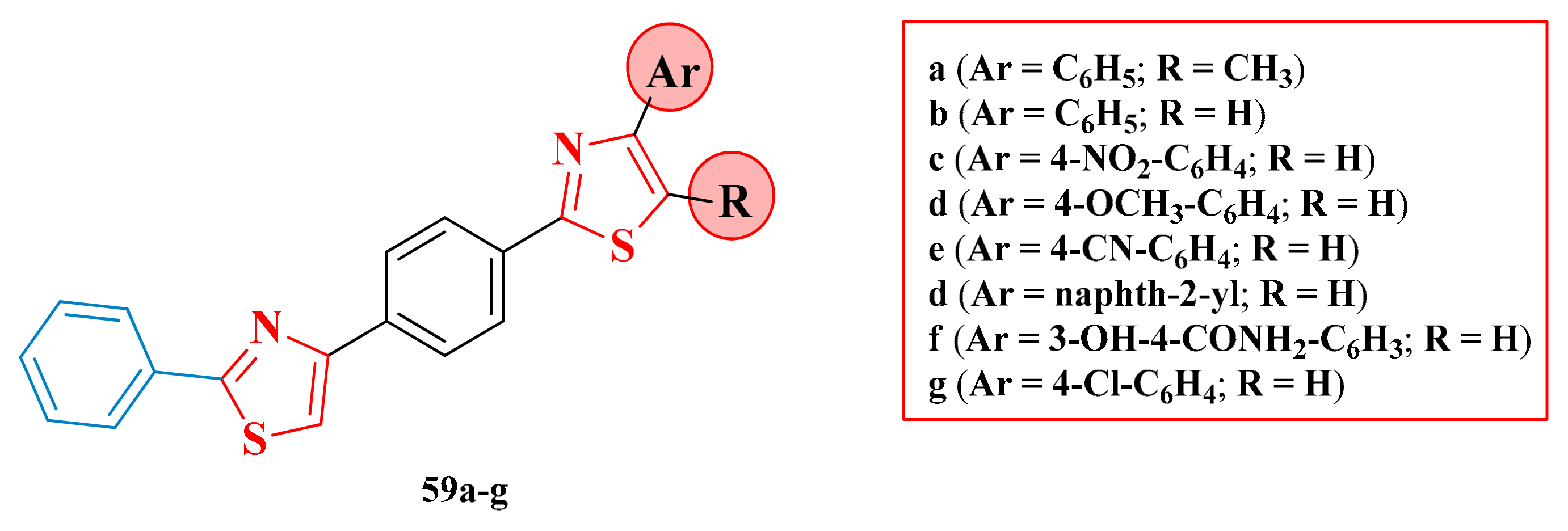
3.2. Thiazol-4-yl-1,4-Phenylene-2-Thiazoles
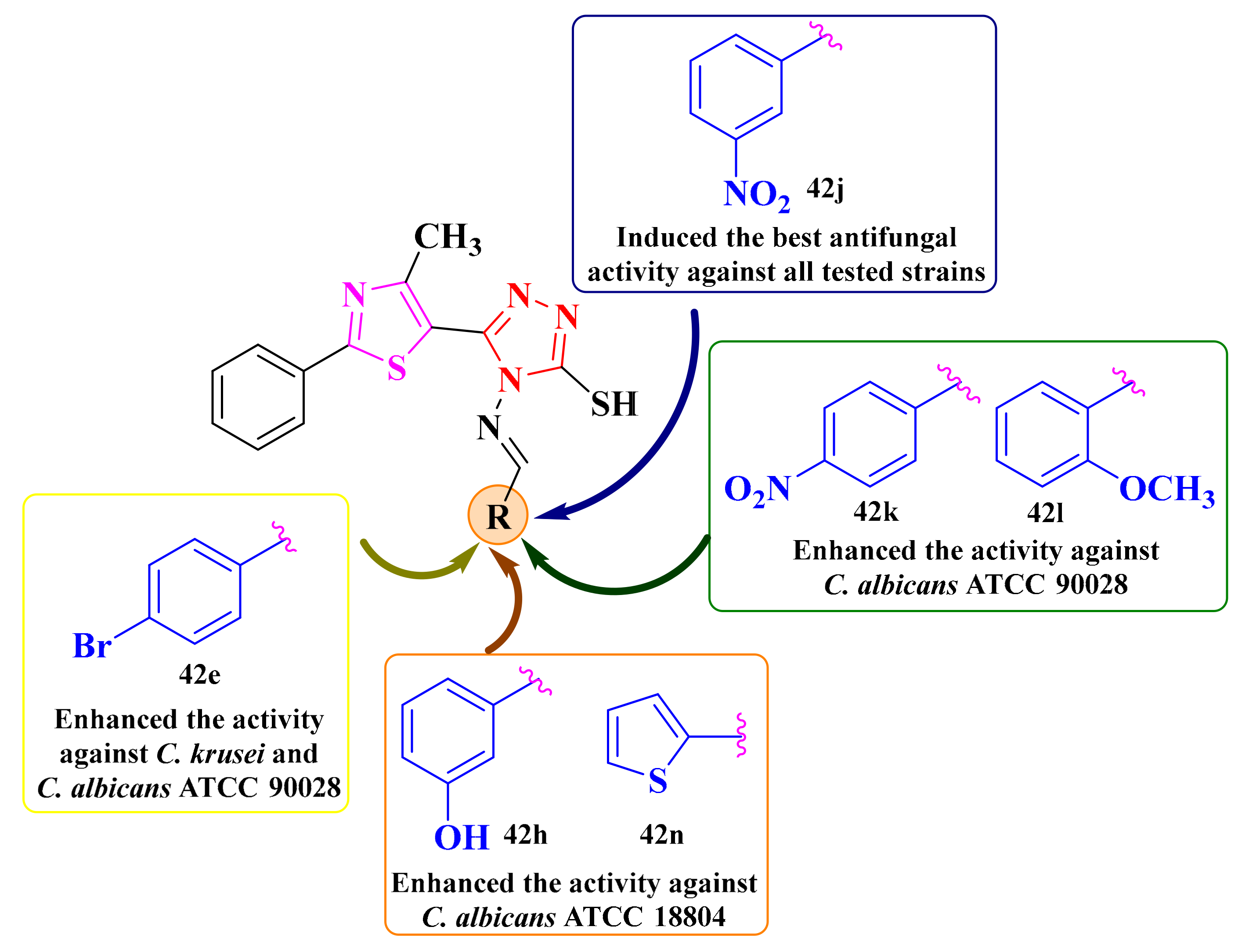
3.3. 2-(3-Pyridyl)-Thiazolyl-1,3,4-Oxadiazolines
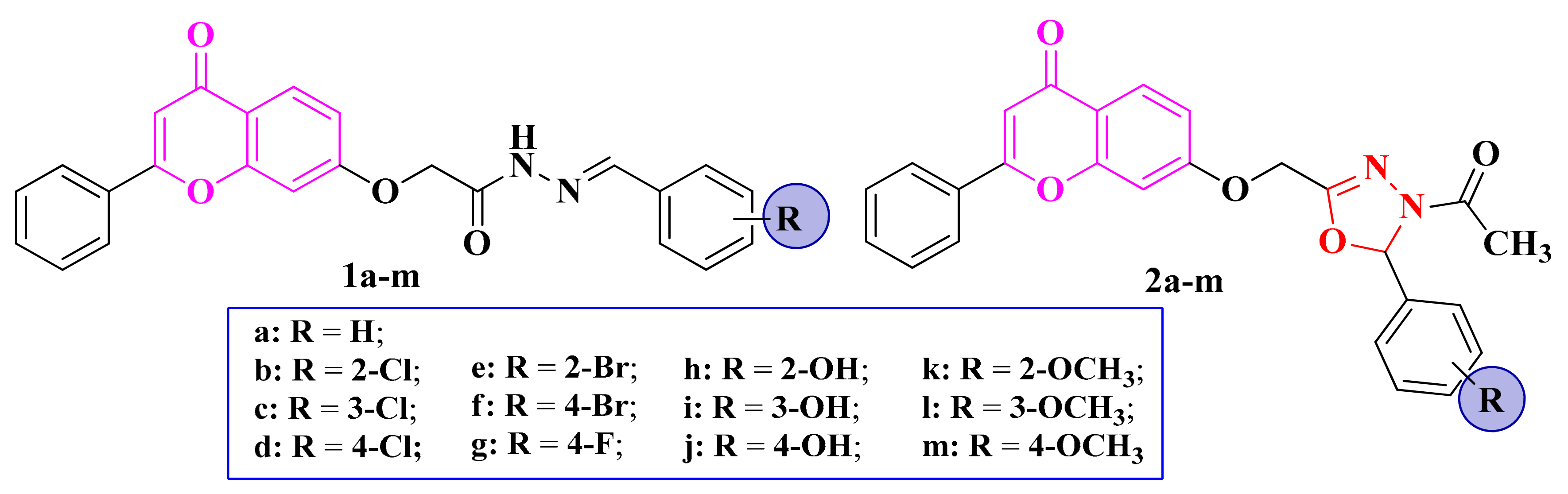
3.4. N-(Oxazolylmethyl)-Thiazolidine-2,4-diones
3.5. Piperazin-4-yl-(Acetyl-Thiazolidine-2,4-dione) Norfloxacin Analogues
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Allel, K.; Day, L.; Hamilton, A.; Lin, L.; Furuya-Kanamori, L.; Moore, C.E.; Van Boeckel, T.; Laxminarayan, R.; Yakob, L. Global Antimicrobial-Resistance Drivers: An Ecological Country-Level Study at the Human–Animal Interface. Lancet Planet. Health 2023, 7, e291–e303. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report: 2022. 2022. Available online: https://www.who.int/publications/i/item/9789240062702 (accessed on 2 August 2024).
- WHO Antimicrobial Resistance. 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 2 August 2024).
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Prabhakaran, V.-S.; Kim, K. The Multi-Faceted Potential of Plant-Derived Metabolites as Antimicrobial Agents against Multidrug-Resistant Pathogens. Microb. Pathog. 2018, 116, 209–214. [Google Scholar] [CrossRef]
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A.A. Antimicrobial Resistance: A Growing Serious Threat for Global Public Health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the Sustainable Discovery and Development of New Antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Emran, T.B.; Dhama, K.; Ripon, M.K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic Resistance in Microbes: History, Mechanisms, Therapeutic Strategies and Future Prospects. J. Infect. Public Health 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Chifiriuc, M.C.; Filip, R.; Constantin, M.; Pircalabioru, G.G.; Bleotu, C.; Burlibasa, L.; Ionica, E.; Corcionivoschi, N.; Mihaescu, G. Common Themes in Antimicrobial and Anticancer Drug Resistance. Front. Microbiol. 2022, 13, 960693. [Google Scholar] [CrossRef] [PubMed]
- Lazăr, V.; Gheorghe, I.; Curutiu, C.; Savin, I.; Marinescu, F.; Cristea, V.C.; Dobre, D.; Popa, G.L.; Chifiriuc, M.C.; Popa, M.I. Antibiotic Resistance Profiles in Cultivable Microbiota Isolated from Some Romanian Natural Fishery Lakes Included in Natura 2000 Network. BMC Vet. Res. 2021, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Pitt, S.J.; Gunn, A. The One Health Concept. Br. J. Biomed. Sci. 2024, 81, 12366. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Foxman, B.; Mody, L.; Snitkin, E.S. Network of Microbial and Antibiotic Interactions Drive Colonization and Infection with Multidrug-Resistant Organisms. Proc. Natl. Acad. Sci. USA 2017, 114, 10467–10472. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, D.; Schultze, N.; Borchardt, J.; Böttcher, I.; Schaufler, K.; Guenther, S. Synergistic Antimicrobial Activities of Epigallocatechin Gallate, Myricetin, Daidzein, Gallic Acid, Epicatechin, 3-Hydroxy-6-Methoxyflavone and Genistein Combined with Antibiotics against ESKAPE Pathogens. J. Appl. Microbiol. 2022, 132, 949–963. [Google Scholar] [CrossRef]
- Jackson, N.; Czaplewski, L.; Piddock, L.J. V Discovery and Development of New Antibacterial Drugs: Learning from Experience? J. Antimicrob. Chemother. 2018, 73, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Silberg, A.; Simiti, I.; Mantsch, H. Contributions to the Study of Thiazoles. I. On the Preparation and Properties of 2-Aryl-4-Halogenmethyl-Thiazoles. Chem. Ber. 1961, 94, 2887–2894. [Google Scholar] [CrossRef]
- Simiti, I.; Farkas, M. Contribution to the Study of Some Heterocycles IX. Reactions of Thyazole Aldehydes with Diazomethane. Bull. Soc. Chim. Fr. 1968, 9, 3862–3866. [Google Scholar]
- Simiti, I.; Chindriş, E. Heterocycles 39. Representation of the 2-Aryl-4-Formyloxazole the Sommelet Reaction. Arch. Pharm. 1975, 308, 688–692. [Google Scholar] [CrossRef]
- Simiti, I.; Hintz, G. Heterocyclic Compounds 35, Nitration and Bromination of 2-Benzyl-4-Chloromethylthiazole and Use of the Sommelet-Reaction in This Series of Substances. Pharmazie 1974, 29, 443–445. [Google Scholar]
- Zaharia, V.; Palage, M.; Simiti, I. The Application of Sommelet and Kröhnke Reactions in the 2-Aryl-5- Halomethyl-6R-Thiazolo[3,2-b][1,2,4] Triazole Series. Farmacia 2000, 48, 57–64. [Google Scholar]
- Simiti, I.; Farkas, M. Contributions to the Study of Some Heterocycles XXXI. The Application of the Sommelet Reaction to 2-Aryl-4-Chloromethyl-5-Bromothiazoles. Arch. Pharm. 1974, 307, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Simiti, I.; Proinov, L. The Delepine Reaction with 2- Arylamino-5-Chloromethyl-1,3,4-Thiadiazoles. Heterocyclic Compounds. XIII. Arch. Pharm. 1968, 303, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Simiti, I.; Hintz, G. Studies on Heterocyclic Compounds. 19. Synthesis and Properties of 2-Benzyl-4-Formylthiazole. Pharmazie 1972, 27, 146–147. [Google Scholar] [PubMed]
- Simiti, I.; Demian, H. Contribution to the Study of Some XLI Heterocycles. Bromination of 2-Anilino-4- Chloromethylthiazols and Obtaining 2-Anilino-4-Formyl5-Brom-Thiazols. Ann. Chim. 1975, 10, 317–321. [Google Scholar]
- Simiti, I.; Farkas, M.; Silberg, A. Contributions to the Study of Some Heterocycles, II. On the Preparation of Some 2-Anilino-4-Chloromethyl- and 2-Anilino-4- Formyl-Thiazoles. Chem. Berichte 1962, 95, 2672–2679. [Google Scholar] [CrossRef]
- Simiti, I.; Proinov, L. The Synthesis of Some 2-Anilino-5- Chloromethyl- and 2-Anilino-5-Formil-1,3,4-Thiodiazoles. VI. Rev. Roum. Chim. 1966, 11, 429–440. [Google Scholar]
- Simiti, I.; Muresan, A. Application of the Sommelet Reaction in the Series of 2-Aryl-4methyl-5-Chlormethyl-Thiazole. Rev. Roum. Chim. 1976, 7, 1073–1081. [Google Scholar]
- Simiti, I.; Oniga, O. Heterocycles 71. Sommelet and Kröhnke Reactions in the Series of 2,4’-Bisthiazoles. Monatshefte Fur Chem. 1996, 127, 733–737. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, Z.; Zha, G.; Long, S.; Sridhara, M.B.; Kumar, K.S.S.; Rakesh, K.P. Triazole Derivatives as Potential Antifungal Agents: A Structure-Activity Relationship (SAR) Studies. Process Biochem. 2023, 135, 102–118. [Google Scholar] [CrossRef]
- Tian, G.; Song, Q.; Liu, Z.; Guo, J.; Cao, S.; Long, S. Recent Advances in 1,2,3- and 1,2,4-Triazole Hybrids as Antimicrobials and Their SAR: A Critical Review. Eur. J. Med. Chem. 2023, 259, 115603. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.; Pradhan, T.; Chawla, G. An Updated Review on Diverse Range of Biological Activities of 1,2,4-Triazole Derivatives: Insight into Structure Activity Relationship. J. Mol. Struct. 2023, 1274, 134487. [Google Scholar] [CrossRef]
- Salma, U.; Ahmad, S.; Zafer Alam, M.; Khan, S.A. A Review: Synthetic Approaches and Biological Applications of Triazole Derivatives. J. Mol. Struct. 2024, 1301, 137240. [Google Scholar] [CrossRef]
- Singh, A.; Malhotra, D.; Singh, K.; Chadha, R.; Bedi, P.M.S. Thiazole Derivatives in Medicinal Chemistry: Recent Advancements in Synthetic Strategies, Structure Activity Relationship and Pharmacological Outcomes. J. Mol. Struct. 2022, 1266, 133479. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K. Fluorinated Isoxazolines and Isoxazoles: A Synthetic Perspective. J. Fluor. Chem. 2015, 180, 55–97. [Google Scholar] [CrossRef]
- Kane, A.; Carter, D.A. Augmenting Azoles with Drug Synergy to Expand the Antifungal Toolbox. Pharmaceuticals 2022, 15, 482. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the Development of Antifungal Azoles: A Review on Structures, SAR, and Mechanism of Action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef] [PubMed]
- Devasia, J.; Nizam, A.; Vasantha, V.L. Azole-Based Antibacterial Agents: A Review on Multistep Synthesis Strategies and Biology. Polycycl. Aromat. Compd. 2022, 42, 5474–5495. [Google Scholar] [CrossRef]
- King, A.; Bethune, L.; Phillips, I. The Comparative In Vitro Activity of FK-037 (Cefoselis), a New Broad-Spectrum Cephalosporin. Clin. Microbiol. Infect. 1995, 1, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Geria, A.N.; Scheinfeld, N.S. Pramiconazole, a Triazole Compound for the Treatment of Fungal Infections. IDrugs 2008, 11, 661–670. [Google Scholar] [PubMed]
- Mishra, I.; Mishra, R.; Mujwar, S.; Chandra, P.; Sachan, N. A Retrospect on Antimicrobial Potential of Thiazole Scaffold. J. Heterocycl. Chem. 2020, 57, 2304–2329. [Google Scholar] [CrossRef]
- Rodríguez, D.; González-Bello, C. Siderophores: Chemical Tools for Precise Antibiotic Delivery. Bioorg. Med. Chem. Lett. 2023, 87, 129282. [Google Scholar] [CrossRef] [PubMed]
- Ampomah-Wireko, M.; Chen, S.; Li, R.; Gao, C.; Wang, M.; Qu, Y.; Kong, H.; Nininahazwe, L.; Zhang, E. Recent Advances in the Exploration of Oxazolidinone Scaffolds from Compound Development to Antibacterial Agents and Other Bioactivities. Eur. J. Med. Chem. 2024, 269, 116326. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, D.-B.; Sui, L.-L.; Luan, T. Natural Products-Isoxazole Hybrids: A Review of Developments in Medicinal Chemistry. Arab. J. Chem. 2024, 17, 105794. [Google Scholar] [CrossRef]
- Zhu, J.; Mo, J.; Lin, H.; Chen, Y.; Sun, H. The Recent Progress of Isoxazole in Medicinal Chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- McNaught, A.G.; Wilkinson, A. The IUPAC Compendium of Chemical Terminology; Gold, V., Ed.; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 2019; ISBN 0-9678550-9-8. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 9780471720911. [Google Scholar]
- Gilchrist, T.L. Heterocyclic Chemistry, 3rd ed.; Addison Wesley: Essex, UK, 1997; ISBN 0-582-27843-0. [Google Scholar]
- Laniel, D.; Weck, G.; Gaiffe, G.; Garbarino, G.; Loubeyre, P. High-Pressure Synthesized Lithium Pentazolate Compound Metastable under Ambient Conditions. J. Phys. Chem. Lett. 2018, 9, 1600–1604. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Synthesis, and Applications, 3rd ed.; Wiley-VCH Verlag & Co: Weinheim, Germany, 2003; ISBN 9783527307203. [Google Scholar]
- Maertens, J.A. History of the Development of Azole Derivatives. Clin. Microbiol. Infect. 2004, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, S.; Tahlan, S.; Lim, S.M.; Ramasamy, K.; Mani, V.; Shah, S.A.A.; Narasimhan, B. Benzoxazole Derivatives: Design, Synthesis and Biological Evaluation. Chem. Cent. J. 2018, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, C.; Schneider, K.; Bruntner, C.; Irran, E.; Nicholson, G.; Bull, A.T.; Jones, A.L.; Brown, R.; Stach, J.E.M.; Goodfellow, M.; et al. Caboxamycin, a New Antibiotic of the Benzoxazole Family Produced by the Deep-Sea Strain Streptomyces Sp. NTK 937. J. Antibiot. 2009, 62, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Gan, L.-L.; Wang, H.; Zhou, C.-H. New Progress in Azole Compounds as Antimicrobial Agents. Mini-Rev. Med. Chem. 2016, 17, 122–166. [Google Scholar] [CrossRef]
- Peng, X.-M.; Cai, G.-X.; Zhou, C.-H. Recent Developments in Azole Compounds as Antibacterial and Antifungal Agents. Curr. Top. Med. Chem. 2013, 13, 1963–2010. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G. Current Bioactive Azole-Containing Natural Products. Curr. Bioact. Compd. 2010, 6, 284–308. [Google Scholar] [CrossRef]
- Kaur, R.; Ranjan Dwivedi, A.; Kumar, B.; Kumar, V. Recent Developments on 1,2,4-Triazole Nucleus in Anticancer Compounds: A Review. Anticancer. Agents Med. Chem. 2016, 16, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Q.; Liu, L.-X.; Li, Y.; Sun, C.-J.; Chen, W.; Li, L.; Zhang, H.-B.; Yang, X.-D. Design, Synthesis and Biological Evaluation of Novel Hybrid Compounds of Imidazole Scaffold-Based 2-Benzylbenzofuran as Potent Anticancer Agents. Eur. J. Med. Chem. 2013, 62, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Khaybullin, R.N.; Liang, X.; Cisneros, K.; Qi, X. Synthesis and Anticancer Evaluation of Complex Unsaturated Isosteviol-Derived Triazole Conjugates. Future Med. Chem. 2015, 7, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Kumar, V.; Gupta, G.K. Trifluoromethylpyrazoles as Anti-Inflammatory and Antibacterial Agents: A Review. J. Fluor. Chem. 2015, 178, 306–326. [Google Scholar] [CrossRef]
- Brauer, V.S.; Rezende, C.P.; Pessoni, A.M.; De Paula, R.G.; Rangappa, K.S.; Nayaka, S.C.; Gupta, V.K.; Almeida, F. Antifungal Agents in Agriculture: Friends and Foes of Public Health. Biomolecules 2019, 9, 521. [Google Scholar] [CrossRef]
- Niu, Z.X.; Wang, Y.T.; Zhang, S.N.; Li, Y.; Chen, X.B.; Wang, S.Q.; Liu, H.M. Application and Synthesis of Thiazole Ring in Clinically Approved Drugs. Eur. J. Med. Chem. 2023, 250, 115172. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, H.; Chen, F.; He, Q.; Zhang, X.; Lan, L.; Yang, C. Design, Synthesis and Biological Evaluation of Thiazolyl-Halogenated Pyrroles or Pyrazoles as Novel Antibacterial and Antibiofilm Agents. Eur. J. Med. Chem. 2024, 268, 116221. [Google Scholar] [CrossRef] [PubMed]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xie, Z.; Ruan, W.; Tang, Q.; Qiao, D.; Zhu, W. Thiazole-Based Analogues as Potential Antibacterial Agents against Methicillin-Resistant Staphylococcus Aureus (MRSA) and Their SAR Elucidation. Eur. J. Med. Chem. 2023, 259, 115689. [Google Scholar] [CrossRef] [PubMed]
- Nahar, D.; Mohite, P.; Lonkar, A.; Chidrawar, V.R.; Dodiya, R.; Uddin, M.J.; Singh, S.; Prajapati, B.G. An Insight into New Strategies and Targets to Combat Antifungal Resistance: A Comprehensive Review. Eur. J. Med. Chem. Rep. 2024, 10, 100120. [Google Scholar] [CrossRef]
- Alhadi, A.A.; Othman, R.; Yehye, W.A.; Rahman, N.A. Formation of 1,3,4-Oxadiazolines and 1,3,4-Oxadiazepines through Acetylation of Salicylic Hydrazones. Tetrahedron Lett. 2015, 56, 573–576. [Google Scholar] [CrossRef]
- Yang, J.; Cao, H.; Liu, H.; Li, B.; Ma, Y. Synthesis and Bioactivity of Novel Bisheterocyclic Compounds Containing Pyrazole and Oxadiazoline. J. Chin. Chem. Soc. 2011, 58, 369–375. [Google Scholar] [CrossRef]
- Li, H.; Qin, J.; Dhondi, P.; Zhou, W.; Vicarel, M.; Bara, T.; Cole, D.; Josien, H.; Pissarnitski, D.; Zhu, Z.; et al. The Discovery of Fused Oxadiazepines as Gamma Secretase Modulators for Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2013, 23, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Fuloria, N.K.; Singh, V.; Shaharyar, M.; Ali, M. Synthesis and Antimicrobial Evaluation of Some New Oxadiazoles Derived from Phenylpropionohydrazides. Molecules 2009, 14, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Manojkumar, P.; Ravi, T.; Subbuchettiar, G. Synthesis of Coumarin Heterocyclic Derivatives with Antioxidant Activity and in Vitro Cytotoxic Activity against Tumour Cells. Acta Pharm. 2009, 59, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Kashaw, S.K.; Jatav, V.; Mishra, P. Synthesis and Antimicrobial Activity of Some New 3–[5-(4-Substituted) Phenyl-1,3,4-Oxadiazole-2yl]-2- Styrylquinazoline-4(3H)-Ones. Med. Chem. Res. 2008, 17, 205–211. [Google Scholar] [CrossRef]
- Yusuf, M.; Jain, P. Synthesis and Biological Significances of 1,3,4-Thiadiazolines and Related Heterocyclic Compounds. Arab. J. Chem. 2014, 7, 525–552. [Google Scholar] [CrossRef]
- Loncle, C.; Brunel, J.M.; Vidal, N.; Dherbomez, M.; Letourneux, Y. Synthesis and Antifungal Activity of Cholesterol-Hydrazone Derivatives. Eur. J. Med. Chem. 2004, 39, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou-Garoufalias, S.; Pouli, N.; Marakos, P.; Chytyroglou-Ladas, A. Synthesis Antimicrobial and Antifungal Activity of Some New 3-Substituted Derivatives of 4-(2,4-Dichlorophenyl)-5-Adamantyl-1H-1,2,4-Triazole. Farmaco 2002, 57, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Tiperciuc, B.; Oniga, O.; Palage, M.; Ghiran, D.; Chirtoc, I. Heterocicli 81. Obtinerea Si Caracterizarea Unor 3-N-Acetil-2R-5-[2’-Fenil-4’-Metil-Tiazol-5’-Il]-D2-Oxadiazoline. Clujul Med. 1999, 72, 99–103. [Google Scholar]
- Tiperciuc, B.; Pârvu, A.E.; Palage, M.; Oniga, O.; Ghiran, D. Heterocycles 82. The Synthesis and the Study of the Antiinflammatory Activity of Some 3-N-Acetyl-2-R-5-[2’-Aryl-4’-Methyl-Thiazole-5’-Yl]- D2 -1,3,4 Oxadiazoline. Farmacia 1999, 47, 77–84. [Google Scholar]
- Vicini, P.; Zani, F.; Cozzini, P.; Doytchinova, I. Hydrazones of 1,2-Benzisothiazole Hydrazides: Synthesis, Antimicrobial Activity and QSAR Investigations. Eur. J. Med. Chem. 2002, 37, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, A.R.; de Miranda, A.L.P.; da Silva, K.C.M.; Parrini, S.C.; Barreiro, E.J. Synthesis and Evaluation of Analgesic, Antiinflammatory and Antiplatelet Properties of New 2-Pyridylarylhydrazone Derivatives. Eur. J. Med. Chem. 1998, 33, 189–199. [Google Scholar] [CrossRef]
- Patole, J.; Sandbhor, U.; Padhye, S.; Deobagkar, D.N.; Anson, C.E.; Powell, A. Structural Chemistry and In Vitro Antitubercular Activity of Acetylpyridine Benzoyl Hydrazone and Its Copper Complex against Mycobacterium Smegmatis. Bioorg. Med. Chem. Lett. 2003, 13, 51–55. [Google Scholar] [CrossRef]
- Islam, M.A.-A.-A.-A.; Mumit, M.A.; Pal, T.K.; Alam, M.A. Structural Evaluation and Antibacterial Sensitivity of Iron (III) Complexes of 4-n-Hexyloxybenzoylhydrazine with Some Aliphatic and Aromatic. J. Sci. Res. 2012, 4, 635–647. [Google Scholar] [CrossRef]
- Tiperciuc, B.; Ghiran, D.; Verité, P. Studii Asupra Unor 2-Aril-5-Para-Clor Fenoximetilen D2–1,3,4–Oxadiazoline. Clujul Med. 1997, 70, 85–90. [Google Scholar]
- Rada, F.; Tiperciuc, B.; Chirtoc, I.; Ionescu, M.; Oniga, O.; Ghiran, D. The Synthesis and the Study of the Antimicrobial Activity of Some Cromananon-Oxymethyl-D2-1,3,4-Oxadiazoline. Rev. Med. Şi Farm. Tg. Mureş 2004, 50, 191–195. [Google Scholar]
- Oniga, S.; Oniga, O.; Tiperciuc, B.; Palage, M.; Muresan, A.; Ghiran, D. Heterocycles 83. Synthesis and Antimicrobial Activity of Some New 5-Dithiazolyl-2-R—1,3,4-D4-Oxadiazoline Derivatives. Farmacia 2000, 48, 65–73. [Google Scholar]
- Paruch, K.; Popiołek, Ł.; Wujec, M. Antimicrobial and Antiprotozoal Activity of 3-Acetyl-2,5-Disubstituted-1,3,4-Oxadiazolines: A Review. Med. Chem. Res. 2020, 29, 1–16. [Google Scholar] [CrossRef]
- Oniga, S.; Oniga, O.; Chirtoc, I.; Ionescu, M.; Tiperciuc, B.; Ghiran, D. Obtinerea Si Activitatea Antimicrobiana a Unor 3-N-Acetil-5-(2’-(Acetylamino)-4’-Amino-4’-Metil-5’-Tiazolil-2-Aril—D4-1,3,4-Oxadiazoline. Farmacia 2005, 53, 28–35. [Google Scholar]
- Tiperciuc, B.; Suciu, R.; Oniga, I. La Synthese Des 4-Acetyl-2-(4’-Pyridil)-5-R-2,3-Dihidro-1,3,4-Oxadiazolines. In Proceedings of the XV-th Edition of Balkan Medical Days, Iași, Romania, 28–30 April 1999; pp. 107–115. [Google Scholar]
- Oniga, S.; Duma, M.; Oniga, O.; Tiperciuc, B.; Pirnau, A.; Araniciu, C.; Palage, M. Synthesis of Some New 4-Methyl-2-(4-Pyridyl)-Thiazoles-5-Yl-Azoles as Potential Antimicrobial Agents. Farmacia 2015, 62, 171–178. [Google Scholar]
- Degola, F.; Morcia, C.; Bisceglie, F.; Mussi, F.; Tumino, G.; Ghizzoni, R.; Pelosi, G.; Terzi, V.; Buschini, A.; Restivo, F.M.; et al. In Vitro Evaluation of the Activity of Thiosemicarbazone Derivatives against Mycotoxigenic Fungi Affecting Cereals. Int. J. Food Microbiol. 2015, 200, 104–111. [Google Scholar] [CrossRef]
- Ebrahimi, H.P.; Hadi, J.S.; Alsalim, T.A.; Ghali, T.S.; Bolandnazar, Z. A Novel Series of Thiosemicarbazone Drugs: From Synthesis to Structure. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 137, 1067–1077. [Google Scholar] [CrossRef]
- Sarmiento, G.P.; Rouge, P.D.; Fabian, L.; Vega, D.; Ortuño, R.M.; Moltrasio, G.Y.; Moglioni, A.G. Efficient Synthesis of Chiral Δ2-1,3,4-Thiadiazolines from α-Pinene and Verbenone. Tetrahedron Asymmetry 2011, 22, 1924–1929. [Google Scholar] [CrossRef]
- Ionuţ, I.; Nastasǎ, C.; Ndongo, J.T.; Bruyère, C.; Leclercqz, H.; Tiperciuc, B.; Lefranc, F.; Pîrnǎu, A.; Kiss, R.; Oniga, O. Synthesis and in Vitro Anticancer Activity of New Thiadiazolines and Thiazolinones Containing a Chromenyl Scaffold. Dig. J. Nanomater. Biostructures 2013, 8, 1509–1523. [Google Scholar]
- Ionuţ, I.; Nastasă, C.; Tiperciuc, B.; Oniga, S.; Pîrnău, A.; Vlase, L.; Colosi, I.; Duma, M.; Oniga, O. Synthesis and Antimicrobial Evaluation of Some N1-Arylidenethiosemicarbazone and 1,3,4-Thiadiazoline Derivatives. Clujul Med. 2013, 86, 27–33. [Google Scholar]
- Rad, O.; Rosza, T.; Duma, M.; Vlase, L.; Pîrnău, A.; Tiperciuc, B.; Ionuţ, I.; Oniga, O. Synthesis and Antimicrobial Activity of Some 5-Amino-2-Mercapto-1,3,4-Thiadiazole Derivatives Thioeters and Schiff Bases. Stud. UBB Chem. 2016, LXI, 17–31. [Google Scholar]
- Karegoudar, P.; Karthikeyan, M.S.; Prasad, D.J.; Mahalinga, M.; Holla, B.S.; Kumari, N.S. Synthesis of Some Novel 2,4-Disubstituted Thiazoles as Possible Antimicrobial Agents. Eur. J. Med. Chem. 2008, 43, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bǎdiceanu, C.D.; Larion, C. Antimicrobial Activity of Some New Thioureides from 2-Thiopheneacetic Acid. Farmacia 2009, 57, 473–478. [Google Scholar]
- Vicini, P.; Geronikaki, A.; Incerti, M.; Busonera, B.; Poni, G.; Cabras, C.A.; La Colla, P. Synthesis and Biological Evaluation of Benzo[d]Isothiazole, Benzothiazole and Thiazole Schiff Bases. Bioorg. Med. Chem. 2003, 11, 4785–4789. [Google Scholar] [CrossRef]
- Bharti, S.K.; Nath, G.; Tilak, R.; Singh, S.K. Synthesis, Anti-Bacterial and Anti-Fungal Activities of Some Novel Schiff Bases Containing 2,4-Disubstituted Thiazole Ring. Eur. J. Med. Chem. 2010, 45, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Cukurovali, A.; Yilmaz, İ.; Gur, S.; Kazaz, C. Synthesis, Antibacterial and Antifungal Activity of Some New Thiazolylhydrazone Derivatives Containing 3-Substituted Cyclobutane Ring. Eur. J. Med. Chem. 2006, 41, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Rollas, S.; Küçükgüzel, S. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Vipan Kumar, C. Hydrazone: A Promising Pharmacophore in Medicinal Chemistry. J. Pharmacogn. Phytochem. 2018, 7, 40–43. [Google Scholar]
- Oniga, S.; Tiperciuc, B.; Palage, M.; Pîrnău, A.; Verité, P.; Crişan, O.; Oniga, O. Synthesis of Some Novel Thiazoles, Bistriazoles, Thiazolin-4-Ones and 1,3,4-Thiadiazoline Compounds Starting from 3-Acetyl-2-Phenyl-4-Methyl-Thiazolyl-Thiosemicarbazone. Farmacia 2013, 61, 929–930. [Google Scholar]
- Nastasă, C.; Tiperciuc, B.; Oniga, S.; Pîrnău, A.; Ionescu, M.; Tărlungeanu, D.; Palage, M.; Verité, P.; Oniga, O. Synthesis and Antimicrobial Activity of Some Novel 2-Aryliden-Hydrazone Thiazoles. Farmacia 2013, 61, 1027–1036. [Google Scholar]
- Oniga, O.; Moldovan, C.; Oniga, S.; Tiperciuc, B.; Pîrnău, A.; Verité, P.; Crişan, O.; Ionuţ, I. Synthesis of Some 2-(Acetophenonhydrazin)-Thiazoles and 2-(4-Thiazolylmethinhydrazin)-Thiazoles as Potential Antibacterial and Antifungal Agents. Farmacia 2010, 58, 825–833. [Google Scholar]
- Jin, S.; Liang, X.; Yang, X.; Chen, F.; Wang, D. Synthesis, Structural Characterization and Solubility of 2-(1-Substituted-1,11-Undecylidene)-5-Arylimino-Δ3-1,3,4-Thiadiazolines. Chin. Chem. Lett. 2009, 20, 1267–1270. [Google Scholar] [CrossRef]
- Shih, M.-H.; Wu, C.-L. Efficient Syntheses of Thiadiazoline and Thiadiazole Derivatives by the Cyclization of 3-Aryl-4-Formylsydnone Thiosemicarbazones with Acetic Anhydride and Ferric Chloride. Tetrahedron 2005, 61, 10917–10925. [Google Scholar] [CrossRef]
- Oniga, O.; Ndongo, J.T.; Moldovan, C.; Tiperciuc, B.; Oniga, S.; Pîrnău, A.; Vlase, L.; Verité, P. Synthesis and Antimicrobial Activity of Some New 2-Hydrazone-Thiazoline-4-Ones. Farmacia 2012, 60, 785–797. [Google Scholar]
- Raza, S.; Srivastava, S.P.; Srivastava, D.S.; Srivastava, A.K.; Haq, W.; Katti, S.B. Thiazolidin-4-One and Thiazinan-4-One Derivatives Analogous to Rosiglitazone as Potential Antihyperglycemic and Antidyslipidemic Agents. Eur. J. Med. Chem. 2013, 63, 611–620. [Google Scholar] [CrossRef]
- Epple, R.; Cow, C.; Xie, Y.; Azimioara, M.; Russo, R.; Wang, X.; Wityak, J.; Karanewsky, D.S.; Tuntland, T.; Nguyêñ-Trân, V.T.B.; et al. Novel Bisaryl Substituted Thiazoles and Oxazoles as Highly Potent and Selective Peroxisome Proliferator-Activated Receptor δ Agonists. J. Med. Chem. 2010, 53, 77–105. [Google Scholar] [CrossRef] [PubMed]
- Dow, R.L.; Bechle, B.M.; Chou, T.T.; Clark, D.A.; Hulin, B.; Stevenson, R.W. Benzyloxazolidine-2,4-Diones as Potent Hypoglycemic Agents. J. Med. Chem. 1991, 34, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, H.; Onogi, S.; Tanaka, M.; Shibata, T.; Iwao, M.; Wakitani, K.; Uchida, I. Isoxazolidine-3,5-Dione and Noncyclic 1,3-Dicarbonyl Compounds as Hypoglycemic Agents. J. Med. Chem. 1998, 41, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Byrjalsen, I.; Nielsen, R.H.; Madsen, A.N.; Larsen, L.K.; Christiansen, C.; Beck-Nielsen, H.; Karsdal, M.A. A Comparison of Glycemic Control, Water Retention, and Musculoskeletal Effects of Balaglitazone and Pioglitazone in Diet-Induced Obese Rats. Eur. J. Pharmacol. 2009, 616, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Vodnar, D.C.; Marc, G.; Benedec, D.; Tiperciuc, B.; Tamaian, R.; Oniga, O. Antioxidant Activity and Antibacterial Evaluation of New Thiazolin-4-One Derivatives as Potential Tryptophanyl-TRNA Synthetase Inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yu, K.; Xu, B.; Chen, L.; Chen, X.; Mao, J.; Danchin, A.; Shen, X.; Qu, D.; Jiang, H. Potent and Selective Inhibitors of Staphylococcus Epidermidis Tryptophanyl-TRNA Synthetase. J. Antimicrob. Chemother. 2007, 60, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.-L.; Alkhalaf, L.M.; Ryan, K.S. In Vitro Reconstitution of Indolmycin Biosynthesis Reveals the Molecular Basis of Oxazolinone Assembly. Proc. Natl. Acad. Sci. USA 2015, 112, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, T.; Nakano, Y.; Toyoda, Y.; Miyagawa, K.-I.; Tada, M.; Kaisho, T.; Nakao, M. In Vitro and in Vivo Antibacterial Activities of TAK-083, an Agent for Treatment of Helicobacter Pylori Infection. Antimicrob. Agents Chemother. 2001, 45, 2455–2459. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Vodnar, D.C.; Tamaian, R.; Pîrnău, A.; Vlase, L.; Ionuț, I.; Oniga, O.; Tiperciuc, B. Design, Synthesis and Antifungal Activity Evaluation of New Thiazolin-4-Ones as Potential Lanosterol 14α-Demethylase Inhibitors. Int. J. Mol. Sci. 2017, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-J.; Xiong, N.; Chen, B.-C.; Luo, F.; Huang, M.; Ding, Z.-S.; Qian, C.-D. The Antibacterial Properties of 4, 8, 4′, 8′-Tetramethoxy (1,1′-Biphenanthrene) -2,7,2′,7′-Tetrol from Fibrous Roots of Bletilla Striata. Indian J. Microbiol. 2021, 61, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Becher, R.; Wirsel, S.G.R. Fungal Cytochrome P450 Sterol 14??-Demethylase (CYP51) and Azole Resistance in Plant and Human Pathogens. Appl. Microbiol. Biotechnol. 2012, 95, 825–840. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Naim, M.J.; Alam, M.J.; Ahmad, S.; Nawaz, F.; Shrivastava, N.; Sahu, M.; Alam, O. Therapeutic Journey of 2,4-Thiazolidinediones as a Versatile Scaffold: An Insight into Structure Activity Relationship. Eur. J. Med. Chem. 2017, 129, 218–250. [Google Scholar] [CrossRef] [PubMed]
- Day, C. Thiazolidinediones: A New Class of Antidiabetic Drugs. Diabet. Med. 1999, 16, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Tuncbilek, M.; Altanlar, N. Synthesis of New 3-(Substituted Phenacyl)-5-[3′-(4H-4-Oxo-1-Benzopyran-2-Yl)-Benzylidene]-2,4-Thiazolidinediones and Their Antimicrobial Activity. Arch. Pharm. 2006, 339, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Bahare, R.S.; Ganguly, S.; Choowongkomon, K.; Seetaha, S. Synthesis, HIV-1 RT Inhibitory, Antibacterial, Antifungal and Binding Mode Studies of Some Novel N-Substituted 5-Benzylidine-2,4-Thiazolidinediones. DARU J. Pharm. Sci. 2015, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Ottanà, R.; Curinga, C.; Vigorita, M.G.; Rakowitz, D.; Steindl, T.; Langer, T. Structure–Activity Relationships and Molecular Modelling of 5-Arylidene-2,4-Thiazolidinediones Active as Aldose Reductase Inhibitors. Bioorg. Med. Chem. 2005, 13, 2809–2823. [Google Scholar] [CrossRef] [PubMed]
- Chinthala, Y.; Kumar Domatti, A.; Sarfaraz, A.; Singh, S.P.; Kumar Arigari, N.; Gupta, N.; Satya, S.K.V.N.; Kotesh Kumar, J.; Khan, F.; Tiwari, A.K.; et al. Synthesis, Biological Evaluation and Molecular Modeling Studies of Some Novel Thiazolidinediones with Triazole Ring. Eur. J. Med. Chem. 2013, 70, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Tilekar, K.; Mehendale-Munj, S.; Mohan, R.; Ramaa, C.S. Synthesis and Primary Cytotoxicity Evaluation of New 5-Benzylidene-2,4-Thiazolidinedione Derivatives. Eur. J. Med. Chem. 2010, 45, 4539–4544. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, C.; Madhusudhan, G.; Sahadev, K.; Maheedhara Reddy, C.; Sarma, M.R.; Reddy, G.O.; Chakrabarti, R.; Seshagiri Rao, C.; Kumar, T.D.; Rajagopalan, R. Synthesis and Biological Activity of Novel Thiazolidinediones. Bioorg. Med. Chem. Lett. 1998, 8, 2725–2730. [Google Scholar] [CrossRef] [PubMed]
- Brackman, G.; Al Quntar, A.A.A.; Enk, C.D.; Karalic, I.; Nelis, H.J.; Van Calenbergh, S.; Srebnik, M.; Coenye, T. Synthesis and Evaluation of Thiazolidinedione and Dioxazaborocane Analogues as Inhibitors of AI-2 Quorum Sensing in Vibrio Harveyi. Bioorg. Med. Chem. 2013, 21, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Pinheiro, D.; Santos, E.N.D., Jr.; Consolini, G.; Aguiar, M.J.; de Oliveira Silva, R.R.; de Oliveira Vieira, R.; Palma, M.S.A. Optimized Synthesis and Characterization of Thiazolidine-2, 4-Dione for Pharmaceutical Application. MOJ Bioorganic Org. Chem. 2017, 1, 122–126. [Google Scholar] [CrossRef]
- Nastasă, C.; Duma, M.; Marie, C.; Scherman, D.; Tiperciuc, B.; Oniga, O. New N-Substituted 5-Chromenyl-Thiazolidinediones as Antimicrobial and Antiproliferative Agents. Dig. J. Nanomater. Biostructures 2013, 8, 1079–1087. [Google Scholar]
- Nastasă, C.; Tiperciuc, B.; Vlase, L.; Pîrnău, A.; Oniga, O. Synthesis of New 5-(Chromene-3-Yl)-Methylene-2,4-Thiazolidinediones. Stud. UBB Chem. 2015, 60, 239–246. [Google Scholar]
- Nastasă, C.; Duma, M.; Pîrnău, A.; Vlase, L.; Tiperciuc, B.; Oniga, O. Development of New 5-(Chromen-3-Yl)-Methylene-2,4-Thiazolidinediones as Antimicrobial Agents. Clujul Med. 2016, 89, 122–127. [Google Scholar] [PubMed]
- Nastasă, C.; Tiperciuc, B.; Pârvu, A.; Duma, M.; Ionuţ, I.; Oniga, O. Synthesis of New N-Substituted 5-Arylidene-2,4-thiazolidinediones as Anti-Inflammatory and Antimicrobial Agents. Arch. Pharm. 2013, 346, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Tiperciuc, B.; Duma, M.; Vlase, L.; Crişan, O.; Pîrnău, A.; Oniga, O. Synthesis and Antimicrobial Activity of Some New N-Substituted-5-Arylidene-Thiazolidine-2,4-Diones. J. Heterocycl. Chem. 2014, 51, 411–417. [Google Scholar] [CrossRef]
- Marc, G.; Oniga, S.; Pîrnău, A.; Duma, M.; Vlase, L.; Oniga, O. Rational Synthesis of Some New Para-Aminobenzoic Acid Hybrids with Thiazolidin-2,4-Diones with Antimicrobial Properties ADMET and Molecular Docking Evaluation. Rev. Chim. 2019, 70, 769–775. [Google Scholar] [CrossRef]
- Fischer, M.; Thöny, B.; Leimkühler, S. The Biosynthesis of Folate and Pterins and Their Enzymology. In Comprehensive Natural Products II; Elsevier: Amsterdam, The Netherlands, 2010; pp. 599–648. [Google Scholar]
- Shah, J.J.; Khedkar, V.; Coutinho, E.C.; Mohanraj, K. Design, Synthesis and Evaluation of Benzotriazole Derivatives as Novel Antifungal Agents. Bioorg. Med. Chem. Lett. 2015, 25, 3730–3737. [Google Scholar] [CrossRef]
- Khedr, M. Stepwise Design, Synthesis, and in Vitro Antifungal Screening of (Z)-Substituted-Propenoic Acid Derivatives with Potent Broad-Spectrum Antifungal Activity. Drug Des. Devel. Ther. 2015, 9, 4501–4513. [Google Scholar] [CrossRef] [PubMed]
- Marc, G.; Ionuț, I.; Pirnau, A.; Vlase, L.; Vodnar, D.C.; Duma, M.; Tiperciuc, B.; Oniga, O. Microwave Assisted Synthesis of 3,5-Disubstituted Thiazolidine-2,4-Diones with Antifungal Activity. Design, Synthesis, Virtual and in Vitro Antifungal Screening. Farmacia 2017, 65, 414–422. [Google Scholar]
- Marc, G.; Stana, A.; Pîrnău, A.; Vlase, L.; Vodnar, D.C.; Duma, M.; Tiperciuc, B.; Oniga, O. 3,5-Disubstituted Thiazolidine-2,4-Diones: Design, Microwave-Assisted Synthesis, Antifungal Activity, and ADMET Screening. SLAS Discov. Adv. Life Sci. RD 2018, 23, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Marc, G.; Araniciu, C.; Oniga, S.D.; Vlase, L.; Pîrnău, A.; Nadăș, G.C.; Novac, C.Ș.; Matei, I.A.; Chifiriuc, M.C.; Măruțescu, L.; et al. Design, Synthesis and Biological Evaluation of New Piperazin-4-Yl-(Acetyl-Thiazolidine-2,4-Dione) Norfloxacin Analogues as Antimicrobial Agents. Molecules 2019, 24, 3959. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, J.; Cao, Y.; Chai, X.; Zou, Y.; Wu, Q.; Zhang, D.; Jiang, Y.; Sun, Q. Synthesis, in Vitro Evaluation and Molecular Docking Studies of New Triazole Derivatives as Antifungal Agents. Bioorg. Med. Chem. Lett. 2011, 21, 4471–4475. [Google Scholar] [CrossRef]
- Barbuceanu, S.-F.; Ilies, D.; Saramet, G.; Uivarosi, V.; Draghici, C.; Radulescu, V. Synthesis and Antioxidant Activity Evaluation of New Compounds from Hydrazinecarbothioamide and 1,2,4-Triazole Class Containing Diarylsulfone and 2,4-Difluorophenyl Moieties. Int. J. Mol. Sci. 2014, 15, 10908–10925. [Google Scholar] [CrossRef] [PubMed]
- Chazin, E.; Sanches, P.; Lindgren, E.; Vellasco Júnior, W.; Pinto, L.; Burbano, R.; Yoneda, J.; Leal, K.; Gomes, C.; Wardell, J.; et al. Synthesis and Biological Evaluation of Novel 6-Hydroxy-Benzo[d][1,3]Oxathiol-2-One Schiff Bases as Potential Anticancer Agents. Molecules 2015, 20, 1968–1983. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Chohan, Z.H. Design, Spectral Characterization and Biological Studies of Transition Metal(II) Complexes with Triazole Schiff Bases. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 104, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Aouad, M. Synthesis, Characterization and Antimicrobial Evaluation of Some New Schiff, Mannich and Acetylenic Mannich Bases Incorporating a 1,2,4-Triazole Nucleus. Molecules 2014, 19, 18897–18910. [Google Scholar] [CrossRef] [PubMed]
- Aswathanarayanappa, C.; Bheemappa, E.; Bodke, Y.D.; Krishnegowda, P.S.; Venkata, S.P.; Ningegowda, R. Synthesis and Evaluation of Antioxidant Properties of Novel 1,2,4-T-Riazole-Based Schiff Base Heterocycles. Arch. Pharm. 2013, 346, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Stana, A.; Enache, A.; Vodnar, D.; Nastasă, C.; Benedec, D.; Ionuț, I.; Login, C.; Marc, G.; Oniga, O.; Tiperciuc, B. New Thiazolyl-Triazole Schiff Bases: Synthesis and Evaluation of the Anti-Candida Potential. Molecules 2016, 21, 1595. [Google Scholar] [CrossRef] [PubMed]
- Nastasă, C.; Vodnar, D.; Ionuţ, I.; Stana, A.; Benedec, D.; Tamaian, R.; Oniga, O.; Tiperciuc, B. Antibacterial Evaluation and Virtual Screening of New Thiazolyl-Triazole Schiff Bases as Potential DNA-Gyrase Inhibitors. Int. J. Mol. Sci. 2018, 19, 222. [Google Scholar] [CrossRef] [PubMed]
- Login, C.C.; Bâldea, I.; Tiperciuc, B.; Benedec, D.; Vodnar, D.C.; Decea, N.; Suciu, Ş. A Novel Thiazolyl Schiff Base: Antibacterial and Antifungal Effects and In Vitro Oxidative Stress Modulation on Human Endothelial Cells. Oxid. Med. Cell. Longev. 2019, 2019, 1607903. [Google Scholar] [CrossRef] [PubMed]
- Kajal, A.; Bala, S.; Kamboj, S.; Sharma, N.; Saini, V. Schiff Bases: A Versatile Pharmacophore. J. Catal. 2013, 2013, 893512. [Google Scholar] [CrossRef]
- Cui, S.-F.; Addla, D.; Zhou, C.-H. Novel 3-Aminothiazolquinolones: Design, Synthesis, Bioactive Evaluation, SARs, and Preliminary Antibacterial Mechanism. J. Med. Chem. 2016, 59, 4488–4510. [Google Scholar] [CrossRef] [PubMed]
- Brvar, M.; Perdih, A.; Renko, M.; Anderluh, G.; Turk, D.; Solmajer, T. Structure-Based Discovery of Substituted 4,5′-Bithiazoles as Novel DNA Gyrase Inhibitors. J. Med. Chem. 2012, 55, 6413–6426. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting Bacterial DNA Gyrase as a Drug Target: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Bikobo, D.S.N.; Vodnar, D.C.; Stana, A.; Tiperciuc, B.; Nastasă, C.; Douchet, M.; Oniga, O. Synthesis of 2-Phenylamino-Thiazole Derivatives as Antimicrobial Agents. J. Saudi Chem. Soc. 2017, 21, 861–868. [Google Scholar] [CrossRef]
- Borcea, A.M.; Marc, G.; Pîrnău, A.; Vlase, L.; Ionuţ, I.; Tiperciuc, B.; Oniga, O. Synthesis and Molecular Docking Study of Some New 1,4-Phenylene-Bisthiazoles as Fungal Lanosterol 14α- Demethylase Inhibitors. Farmacia 2017, 65, 683–689. [Google Scholar]
- Borcea, A.-M.; Marc, G.; Vodnar, D.C.; Vlase, L.; Oniga, O. Synthesis of Novel Thiazolyl-Phenyl-Thiazole Derivatives as Promising Anti-Candida Agents. Pak. J. Pharm. Sci. 2018, 31, 2085–2090. [Google Scholar] [PubMed]
- Borcea, A.-M.; Marc, G.; Ionuț, I.; Vodnar, D.C.; Vlase, L.; Gligor, F.; Pricopie, A.; Pîrnău, A.; Tiperciuc, B.; Oniga, O. A Novel Series of Acylhydrazones as Potential Anti-Candida Agents: Design, Synthesis, Biological Evaluation and In Silico Studies. Molecules 2019, 24, 184. [Google Scholar] [CrossRef] [PubMed]
- Pop, B.; Ionuț, I.; Marc, G.; Vodnar, D.C.; Pîrnău, A.; Vlase, L.; Oniga, O. Development of New 2-Methyl-4-Salicylamide Thiazole Derivatives: Synthesis, Antimicrobial Activity Evaluation, Lipophilicity and Molecular Docking Study. Farmacia 2021, 69, 724–731. [Google Scholar] [CrossRef]
- Rozsa, T.; Duma, M.; Vlase, L.; Ionuţ, I.; Pîrnău, A.; Tiperciuc, B.; Oniga, O. Synthesis and Antimicrobial Evaluation of Some New 4,5′-Bisthiazoles. J. Heterocycl. Chem. 2015, 52, 999–1006. [Google Scholar] [CrossRef]
- Pricopie, A.-I.; Ionuț, I.; Marc, G.; Arseniu, A.-M.; Vlase, L.; Grozav, A.; Găină, L.I.; Vodnar, D.C.; Pîrnău, A.; Tiperciuc, B.; et al. Design and Synthesis of Novel 1,3-Thiazole and 2-Hydrazinyl-1,3-Thiazole Derivatives as Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of Their Binding Interaction with Bovine Serum Albumin. Molecules 2019, 24, 3435. [Google Scholar] [CrossRef] [PubMed]
- Pricopie, A.-I.; Focșan, M.; Ionuț, I.; Marc, G.; Vlase, L.; Găină, L.-I.; Vodnar, D.C.; Simon, E.; Barta, G.; Pîrnău, A.; et al. Novel 2,4-Disubstituted-1,3-Thiazole Derivatives: Synthesis, Anti-Candida Activity Evaluation and Interaction with Bovine Serum Albumine. Molecules 2020, 25, 1079. [Google Scholar] [CrossRef] [PubMed]
- Pricopie, A.I.; Vlase, L.; Pîrnău, A.; Vodnar, D.C.; Marc, G.; Nastasă, C.; Borcea, A.M.; Ionuţ, I.; Tiperciuc, B.; Oniga, O. Design and Synthesis of Some Novel 1,2,4-Triazole-3-Yl-Mercapto Derivatives as Potential Anti-Candida Agents. Farmacia 2018, 66, 948–958. [Google Scholar] [CrossRef]
- Kamal, R.; Kumar, V.; Bhardwaj, V.; Kumar, V.; Aneja, K.R. Synthesis, Characterization and in Vitro Antimicrobial Evaluation of Some Novel Hydrazone Derivatives Bearing Pyrimidinyl and Pyrazolyl Moieties as a Promising Heterocycles. Med. Chem. Res. 2015, 24, 2551–2560. [Google Scholar] [CrossRef]
- Kaplancıklı, Z.A.; Altıntop, M.D.; Sever, B.; Cantürk, Z.; Özdemir, A. Synthesis and In Vitro Evaluation of New Thiosemicarbazone Derivatives as Potential Antimicrobial Agents. J. Chem. 2016, 2016, 1692540. [Google Scholar] [CrossRef]
- Chimenti, F.; Bizzarri, B.; Bolasco, A.; Secci, D.; Chimenti, P.; Granese, A.; Carradori, S.; D’Ascenzio, M.; Lilli, D.; Rivanera, D. Synthesis and Biological Evaluation of Novel 2,4-Disubstituted-1,3- Thiazoles as Anti-Candida Spp. Agents. Eur. J. Med. Chem. 2011, 46, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.T.; Bertout, S.; Quinonéro, O.; Akalin, G.; Turan-Zitouni, G.; Fulcrand, P.; Demirci, F.; Martinez, J.; Masurier, N. Synthesis and Anti-Candida Activity of Novel 2-Hydrazino-1,3-Thiazole Derivatives. Bioorganic Med. Chem. Lett. 2013, 23, 1803–1807. [Google Scholar] [CrossRef] [PubMed]
- Nastasă, C.; Tiperciuc, B.; Duma, M.; Benedec, D.; Oniga, O. New Hydrazones Bearing Thiazole Scaffold: Synthesis, Characterization, Antimicrobial, and Antioxidant Investigation. Molecules 2015, 20, 17325–17338. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Kollar, P.; Ferreira, A.L.; Palma, D.; Duarte, A.; Lopes, M.M.; Bartos, M.; Pauk, K.; Imramovsky, A.; Jampilek, J. Antimicrobial Effect of Salicylamide Derivatives against Intestinal Sulfate-Reducing Bacteria. J. Appl. Biomed. 2016, 14, 125–130. [Google Scholar] [CrossRef]
- Pospisilova, S.; Michnova, H.; Kauerova, T.; Pauk, K.; Kollar, P.; Vinsova, J.; Imramovsky, A.; Cizek, A.; Jampilek, J. In Vitro Activity of Salicylamide Derivatives against Vancomycin-Resistant Enterococci. Bioorganic Med. Chem. Lett. 2018, 28, 2184–2188. [Google Scholar] [CrossRef] [PubMed]
- Galal, A.M.F.; Fayad, W.; Mettwally, W.S.A.; Gomaa, S.K.; Ahmed, E.R.; El-Refai, H.A.; Hanna, A.G. Cytotoxicity of Multicellular Cancer Spheroids, Antibacterial, and Antifungal of Selected Sulfonamide Derivatives Coupled with a Salicylamide and/or Anisamide Scaffold. Med. Chem. Res. 2019, 28, 1425–1440. [Google Scholar] [CrossRef]
- Borcea, A.-M.; Ionuț, I.; Crișan, O.; Oniga, O. An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 2021, 26, 624. [Google Scholar] [CrossRef]
- Gidaro, M.C.; Alcaro, S.; Secci, D.; Rivanera, D.; Mollica, A.; Agamennone, M.; Giampietro, L.; Carradori, S. Identification of New Anti- Candida Compounds by Ligand-Based Pharmacophore Virtual Screening. J. Enzym. Inhib. Med. Chem. 2016, 31, 1703–1706. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Orhan, I.E.; Daglia, M.; Barbieri, R.; Di Lorenzo, A.; Nabavi, S.F.; Gortzi, O.; Izadi, M.; Nabavi, S.M. Antibacterial and Antifungal Activities of Thymol: A Brief Review of the Literature. Food Chem. 2016, 210, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Escobar, A.; Pérez, M.; Romanelli, G.; Blustein, G. Thymol Bioactivity: A Review Focusing on Practical Applications. Arab. J. Chem. 2020, 13, 9243–9269. [Google Scholar] [CrossRef]
- Lino, C.I.; Gonçalves de Souza, I.; Borelli, B.M.; Silvério Matos, T.T.; Santos Teixeira, I.N.; Ramos, J.P.; Maria de Souza Fagundes, E.; de Oliveira Fernandes, P.; Maltarollo, V.G.; Johann, S.; et al. Synthesis, Molecular Modeling Studies and Evaluation of Antifungal Activity of a Novel Series of Thiazole Derivatives. Eur. J. Med. Chem. 2018, 151, 248–260. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Bolasco, A.; Rivanera, D.; Mari, E.; Zicari, A.; Lotti, L.V.; Bizzarri, B. Synthesis and Cytotoxicity of Novel (Thiazol-2-Yl)Hydrazine Derivatives as Promising Anti-Candida Agents. Eur. J. Med. Chem. 2013, 65, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Strushkevich, N.; Usanov, S.A.; Park, H.-W. Structural Basis of Human CYP51 Inhibition by Antifungal Azoles. J. Mol. Biol. 2010, 397, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N. Antifungal Resistance: Current Trends and Future Strategies to Combat. Infect. Drug Resist. 2017, 10, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Shahar Yar, M.; C. Sharma, P. New Insights into Chemistry and Anti-Infective Potential of Triazole Scaffold. Curr. Med. Chem. 2011, 18, 3265–3297. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Huang, W.; Liu, M. Evaluation of the Combination Mode of Azoles Antifungal Inhibitors with CACYP51 and the Influence of Site-Directed Mutation. J. Mol. Graph. Model. 2017, 73, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Zhang, B.; Yang, H.-K.; Li, Q.; Diao, P.-C.; You, W.-W.; Zhao, P.-L. Design, Synthesis, and Biological Evaluation of Novel Alkylsulfanyl-1,2,4-Triazoles as Cis-Restricted Combretastatin A-4 Analogues. Eur. J. Med. Chem. 2017, 125, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Dufour, D.; Leung, V.; Lévesque, C.M. Bacterial Biofilm: Structure, Function, and Antimicrobial Resistance. Endod. Top. 2010, 22, 2–16. [Google Scholar] [CrossRef]
- Todd, O.A.; Peters, B.M. Candida Albicans and Staphylococcus Aureus Pathogenicity and Polymicrobial Interactions: Lessons beyond Koch’s Postulates. J. Fungi 2019, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.B.; Gulati, M.; Johnson, A.D.; Nobile, C.J. Development and Regulation of Single- and Multi-Species Candida Albicans Biofilms. Nat. Rev. Microbiol. 2018, 16, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Oniga, S.; Oniga, O.; Palage, M.; Chifiriuc, M.C.; Maruţescu, L. Antimicrobial and Anti-Pathogenic Activity Evaluation of Some 2-(Trimethoxyphenyl)-4-AR1-5-R2-Thiazoles. Farmacia 2015, 63, 40–45. [Google Scholar]
- Oniga, S.; Araniciu, C.; Palage, M.; Popa, M.; Chifiriuc, M.-C.; Marc, G.; Pirnau, A.; Stoica, C.; Lagoudis, I.; Dragoumis, T.; et al. New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules 2017, 22, 1827. [Google Scholar] [CrossRef]
- Araniciu, C.; Oniga, O.; Marc, G.; Palage, M.D.; Măruțescu, L.; Chifiriuc, M.C.; Stoica, C.I.; Ionuț, I.; Oniga, S.D. Anti-Biofilm Activity Evaluation and Molecular Docking Study of Some 2(3-Pyridyl)-Thiazolyl-1,3,4-Oxadiazolines. Farmacia 2018, 66, 627–634. [Google Scholar] [CrossRef]
- Marc, G.; Araniciu, C.; Oniga, S.; Vlase, L.; Pîrnău, A.; Duma, M.; Măruțescu, L.; Chifiriuc, M.; Oniga, O. New N-(Oxazolylmethyl)-Thiazolidinedione Active against Candida Albicans Biofilm: Potential Als Proteins Inhibitors. Molecules 2018, 23, 2522. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Pârvu, A.E.; Tiperciuc, B.; Palage, M.; Oniga, S.; Verité, P.; Oniga, O. Synthesis and Evaluation of the Anti-Inflammatory Activity of Some 2-(Trimethoxyphenyl)-4-R1-5-R2-Thiazoles. Dig. J. Nanomater. Biostructures 2013, 8, 699–709. [Google Scholar]
- Araniciu, C.; Maruţescu, L.; Oniga, S.; Oniga, O.; Chifiriuc, M.C.; Palage, M. Evaluation of the Antimicrobial and Anti-Biofilm Activity of Some 4,2 and 5,2 Bisthiazoles Derivatives. Dig. J. Nanomater. Biostructures 2014, 9, 123–131. [Google Scholar]
- Badea, M.; Crasanda, A.-M.; Chifiriuc, M.C.; Marutescu, L.; Lazar, V.; Marinescu, D.; Olar, R. Synthesis, Spectral and Thermal Study on New Fe(III) Complexes with N,N-Dimethylbiguanide as Antibacterial Agents. J. Therm. Anal. Calorim. 2013, 111, 1743–1751. [Google Scholar] [CrossRef]
- Limban, C.; Marutescu, L.; Chifiriuc, M.C. Synthesis, Spectroscopic Properties and Antipathogenic Activity of New Thiourea Derivatives. Molecules 2011, 16, 7593–7607. [Google Scholar] [CrossRef] [PubMed]
- Limban, C.; Chifiriuc, M.C. Antibacterial Activity of New Dibenzoxepinone Oximes with Fluorine and Trifluoromethyl Group Substituents. Int. J. Mol. Sci. 2011, 12, 6432–6444. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus Aureus Sortase Mutants Defective in the Display of Surface Proteins and in the Pathogenesis of Animal Infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef] [PubMed]
- Gulati, M.; Nobile, C.J. Candida Albicans Biofilms: Development, Regulation, and Molecular Mechanisms. Microbes Infect. 2016, 18, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Desai, J. Candida Albicans Hyphae: From Growth Initiation to Invasion. J. Fungi 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Filler, S.G. Candida Albicans Als3, a Multifunctional Adhesin and Invasin. Eukaryot. Cell 2011, 10, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Karale, N.N. Antibiofilm Activity of Thiazole Schiff Bases. Int. J. Chem. Sci. 2016, 14, 2535–2545. [Google Scholar]
- Romo, J.A.; Pierce, C.G.; Chaturvedi, A.K.; Lazzell, A.L.; McHardy, S.F.; Saville, S.P.; Lopez-Ribot, J.L. Development of Anti-Virulence Approaches for Candidiasis via a Novel Series of Small-Molecule Inhibitors of Candida Albicans Filamentation. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bredereck, H.; Gompper, R. Oxazol-Synthesen Aus -Halogen-Ketonen (Formamid-Reaktionen, IV. Mitteil.). Chem. Ber. 1954, 87, 700–707. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungureanu, D.; Oniga, O.; Moldovan, C.; Ionuț, I.; Marc, G.; Stana, A.; Pele, R.; Duma, M.; Tiperciuc, B. An Insight into Rational Drug Design: The Development of In-House Azole Compounds with Antimicrobial Activity. Antibiotics 2024, 13, 763. https://doi.org/10.3390/antibiotics13080763
Ungureanu D, Oniga O, Moldovan C, Ionuț I, Marc G, Stana A, Pele R, Duma M, Tiperciuc B. An Insight into Rational Drug Design: The Development of In-House Azole Compounds with Antimicrobial Activity. Antibiotics. 2024; 13(8):763. https://doi.org/10.3390/antibiotics13080763
Chicago/Turabian StyleUngureanu, Daniel, Ovidiu Oniga, Cristina Moldovan, Ioana Ionuț, Gabriel Marc, Anca Stana, Raluca Pele, Mihaela Duma, and Brîndușa Tiperciuc. 2024. "An Insight into Rational Drug Design: The Development of In-House Azole Compounds with Antimicrobial Activity" Antibiotics 13, no. 8: 763. https://doi.org/10.3390/antibiotics13080763
APA StyleUngureanu, D., Oniga, O., Moldovan, C., Ionuț, I., Marc, G., Stana, A., Pele, R., Duma, M., & Tiperciuc, B. (2024). An Insight into Rational Drug Design: The Development of In-House Azole Compounds with Antimicrobial Activity. Antibiotics, 13(8), 763. https://doi.org/10.3390/antibiotics13080763