
The Tick Saliva Peptide HIDfsin2 TLR4-Dependently Inhibits the Tick-Borne Severe Fever with Thrombocytopenia Syndrome Virus in Mouse Macrophages

 ,
, 
Abstract

1. Introduction
2. Results
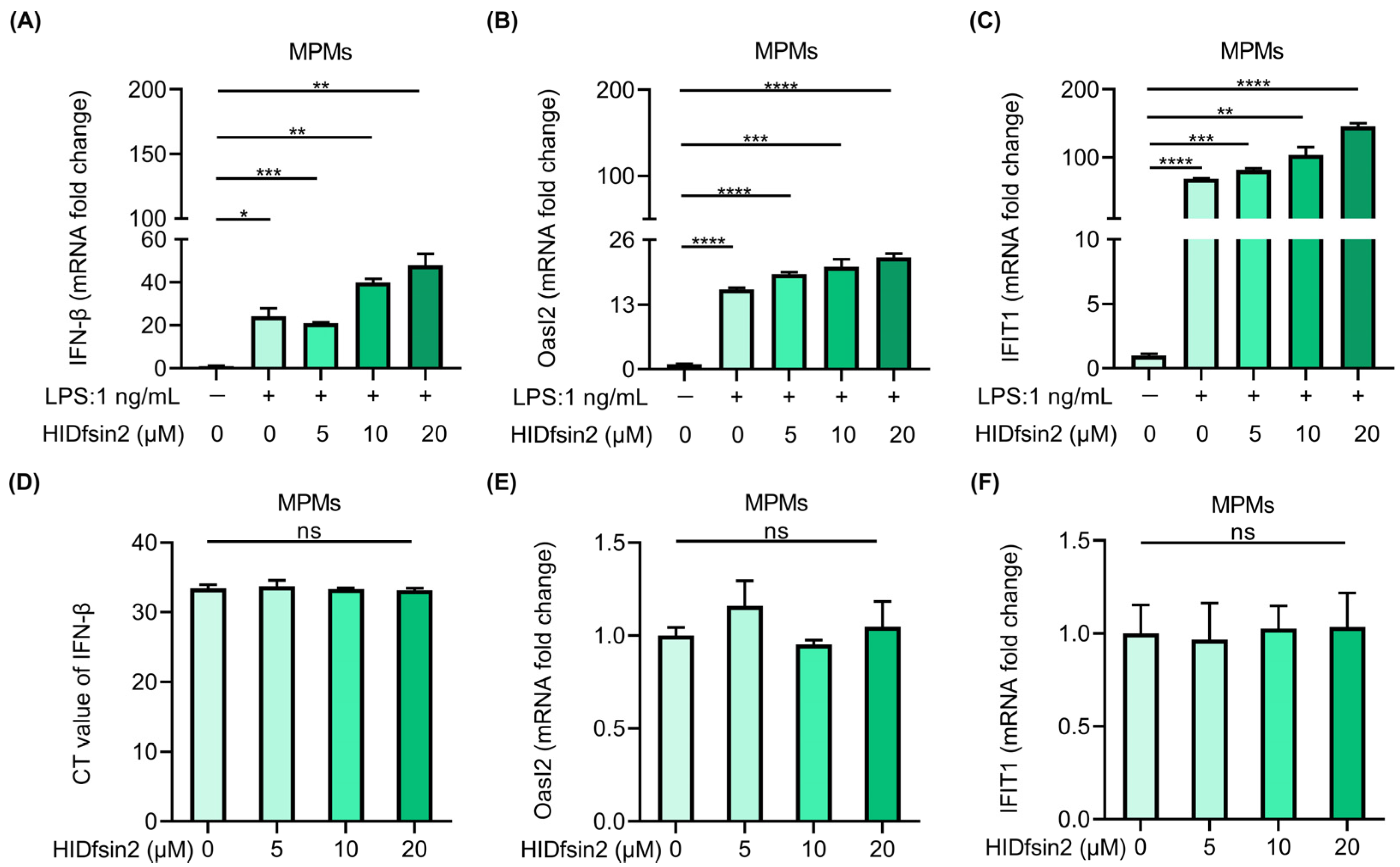
2.1. HIDfsin2 Inhibited SFTSV Replication in MPMs
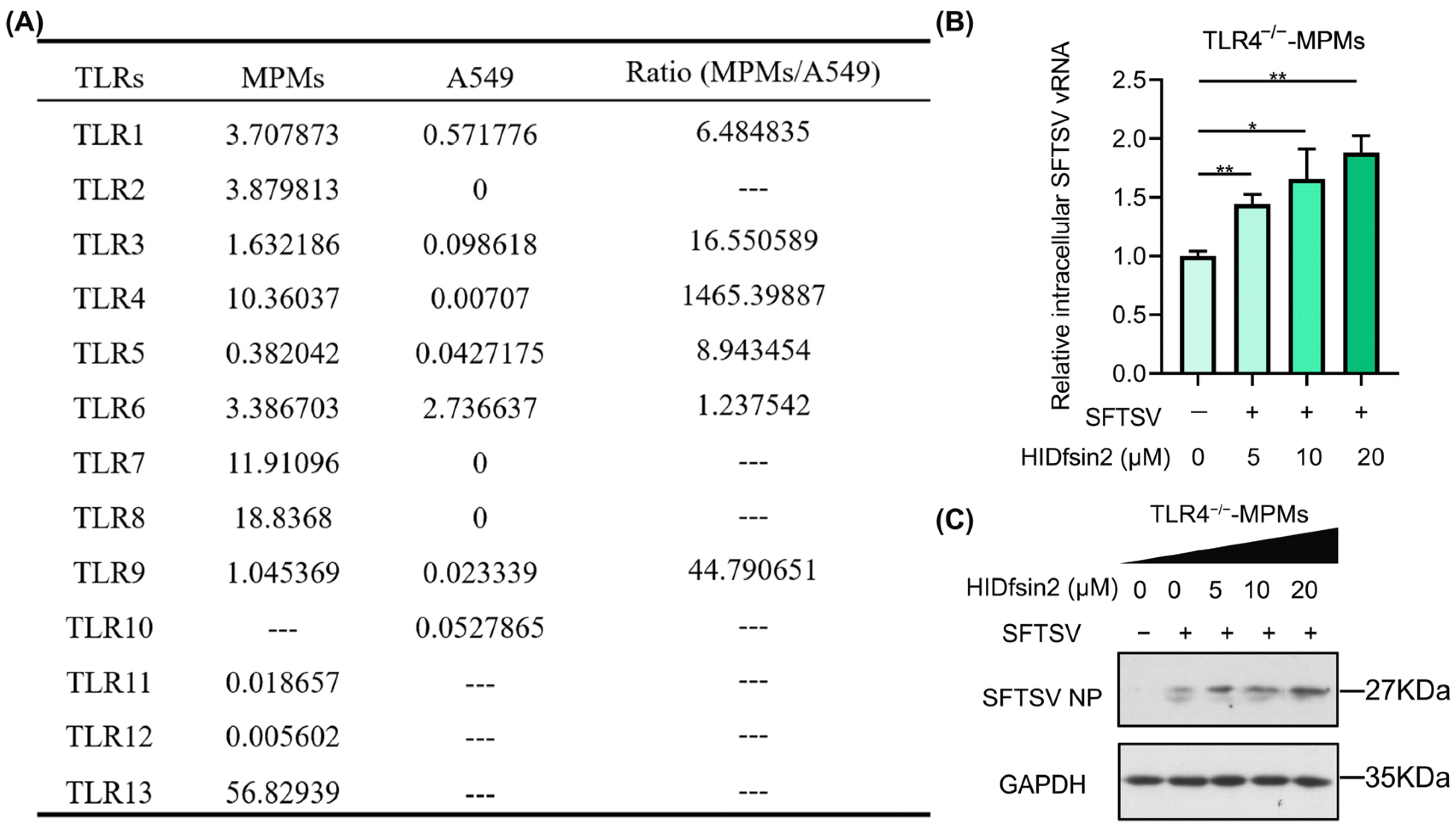
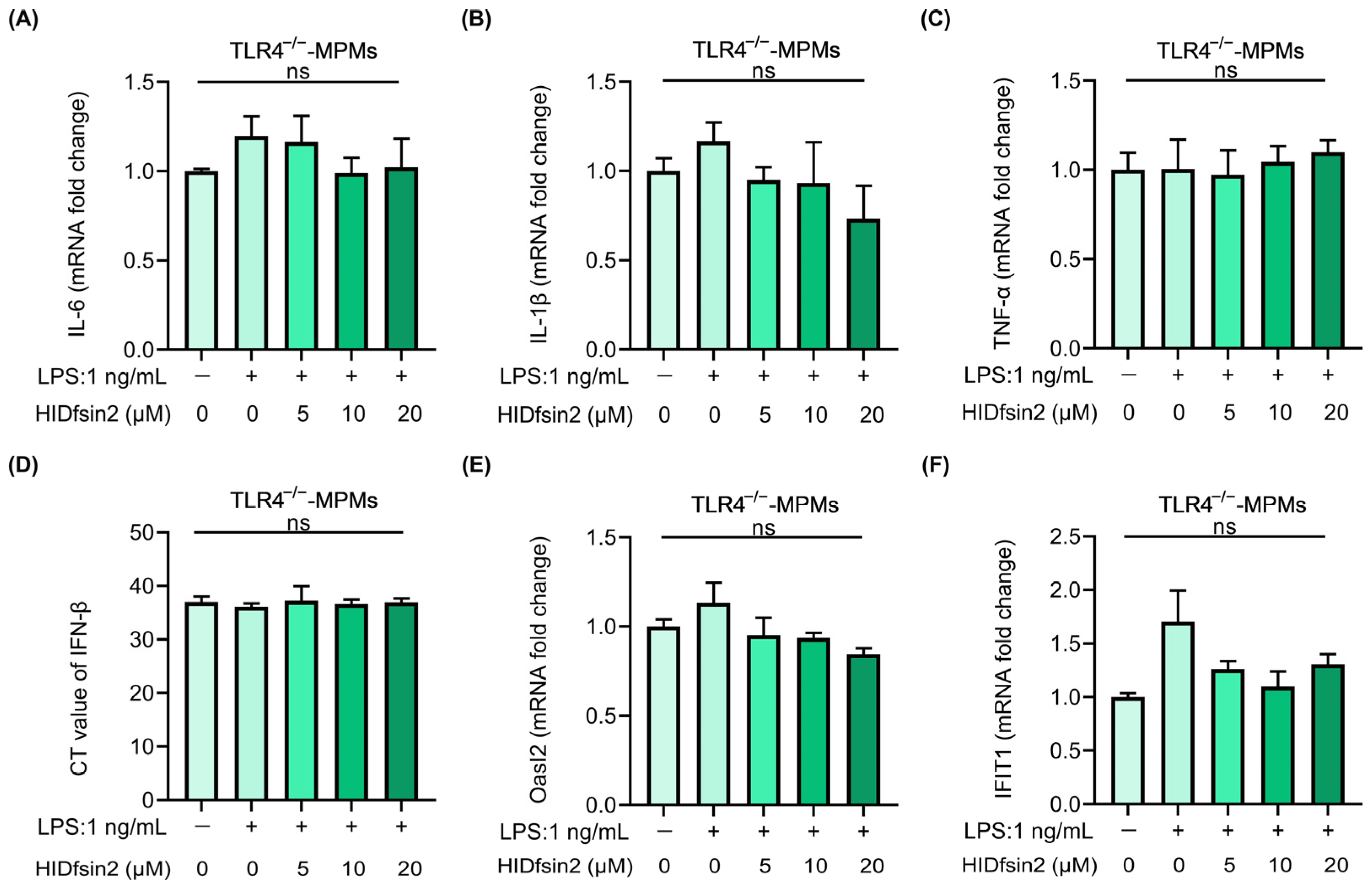
2.2. HIDfsin2 Promoted SFTSV Replication in TLR4-Knockout MPMs
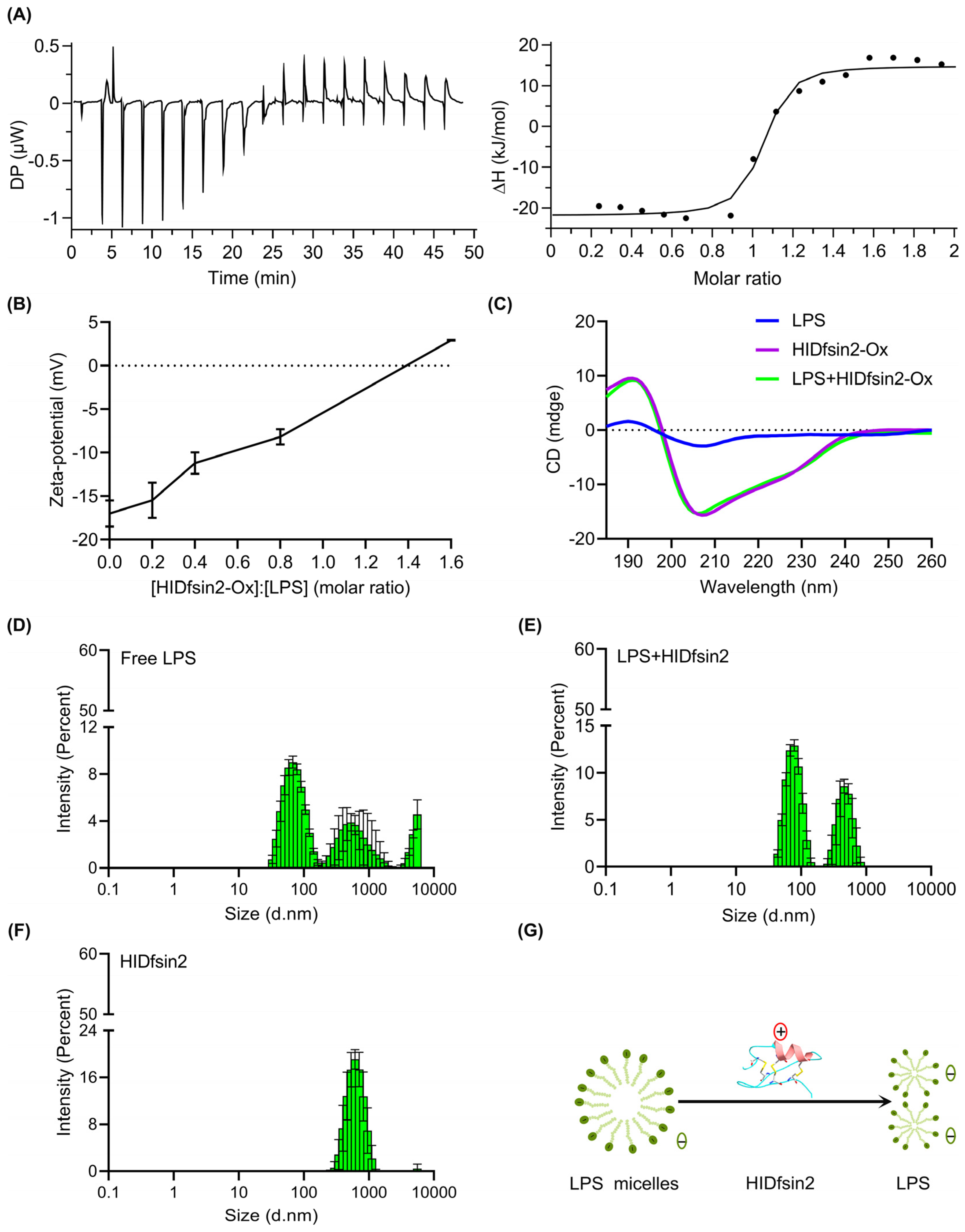
2.3. HIDfsin2 Interacted with the TLR4 Ligand LPS and Depolymerized LPS Micelles
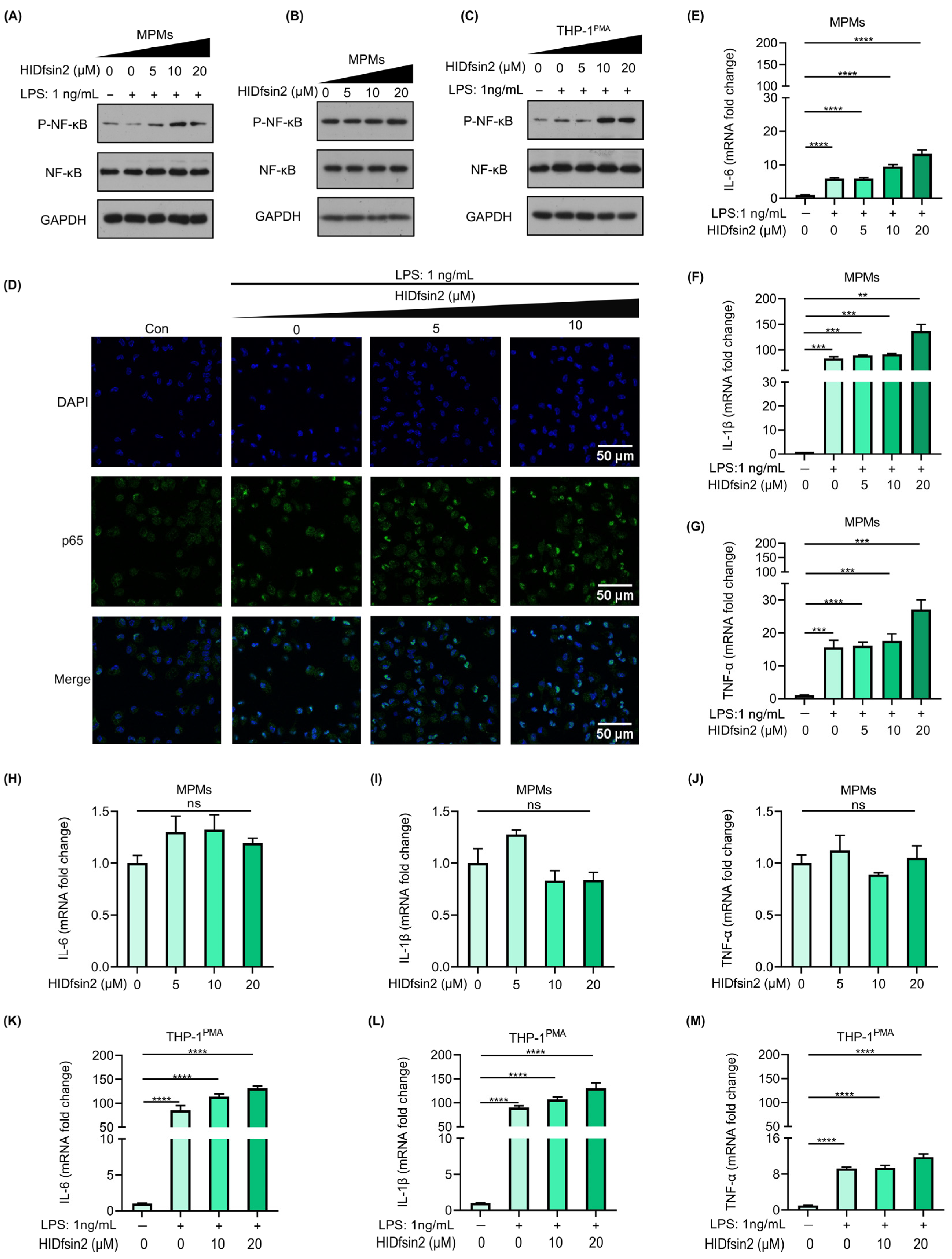
2.4. HIDfsin2 Promoted NF-κB Activation and Inflammatory Cytokine Expression
2.5. HIDfsin2 Enhanced the Type I Interferon Pathway
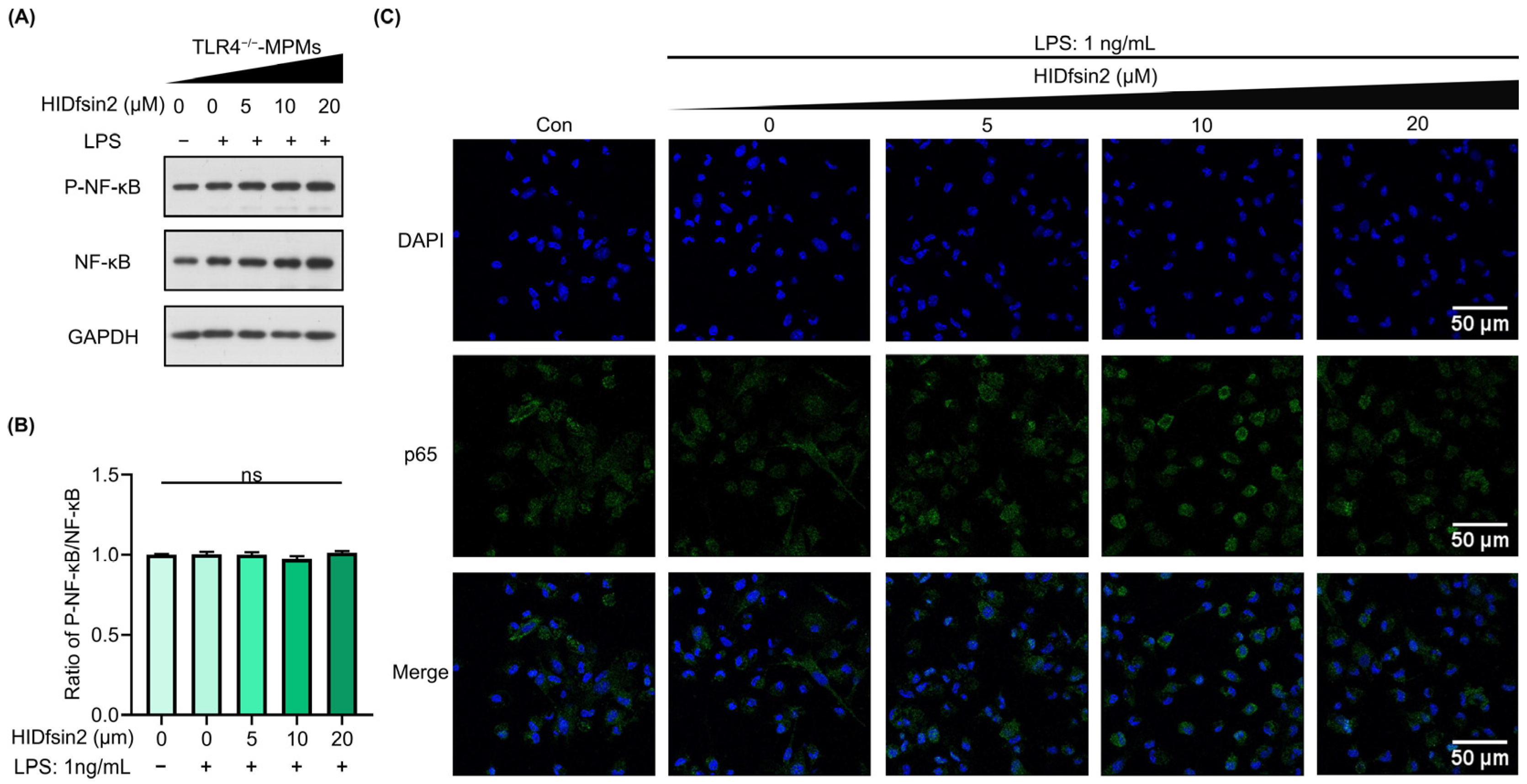
2.6. TLR4 Mediated the Enhancement of HIDfsin2 on NF-κB and Type I Interferon Activation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cells and Viruses
4.3. Reagents and Antibodies
4.4. MPM Extraction
4.5. Cell Viability
4.6. Isothermal Titration Calorimetry (ITC)
4.7. Dynamic Light Scattering (DLS)
4.8. Zeta Potential
4.9. Circular Dichroism (CD) Spectroscopy
4.10. Cellular Immunofluorescence
4.11. Quantitative Real-Time PCR (qRT-PCR)
4.12. Western Blotting
4.13. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, G.-P.; Wang, Y.-X.; Fan, Z.-W.; Ji, Y.; Liu, M.-J.; Zhang, W.-H.; Li, X.-L.; Zhou, S.-X.; Li, H.; Liang, S.; et al. Mapping ticks and tick-borne pathogens in China. Nat. Commun. 2021, 12, 1075. [Google Scholar] [CrossRef]
- Ali, A.; Zeb, I.; Alouffi, A.; Zahid, H.; Almutairi, M.M.; Alshammari, F.A.; Alrouji, M.; Termignoni, C.; Vaz, I.d.S.; Tanaka, T. Host immune responses to salivary components—A critical facet of tick-host interactions. Front. Cell. Infect. Microbiol. 2022, 12, 809052. [Google Scholar] [CrossRef]
- Torina, A.; Villari, S.; Blanda, V.; Vullo, S.; La Manna, M.P.; Azgomi, M.S.; Di Liberto, D.; de la Fuente, J.; Sireci, G. Innate immune response to tick-borne pathogens: Cellular and molecular mechanisms induced in the hosts. Int. J. Mol. Sci. 2020, 21, 5437. [Google Scholar] [CrossRef]
- Wikel, S.K. Tick modulation of host immunity: An important factor in pathogen transmission. Int. J. Parasitol. 1999, 29, 851–859. [Google Scholar] [CrossRef]
- Haddad, V., Jr.; Haddad, M.R.; Santos, M.; Cardoso, J.L.C. Skin manifestations of tick bites in humans. An. Bras. Dermatol. 2018, 93, 251–255. [Google Scholar]
- Preston, S.G.; Majtán, J.; Kouremenou, C.; Rysnik, O.; Burger, L.F.; Cruz, A.C.; Guzman, M.C.; Nunn, M.A.; Paesen, G.C.; Nuttall, P.A.; et al. Novel immunomodulators from hard ticks selectively reprogramme human dendritic cell responses. PLoS Pathog. 2013, 9, e1003450. [Google Scholar]
- Carletti, T.; Zakaria, M.K.; Marcello, A. The host cell response to tick-borne encephalitis virus. Biochem. Biophys. Res. Commun. 2017, 492, 533–540. [Google Scholar]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Płóciennikowska, A.; Hromada-Judycka, A.; Borzęcka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. CMLS 2015, 72, 557–581. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. CMLS 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Durso, D.F.; Bryant, C.E.; Kurt-Jones, E.A.; Silverman, N.; Golenbock, D.T.; Gazzinelli, R.T. The IRAK4 scaffold integrates TLR4-driven TRIF and MYD88 signaling pathways. Cell Rep. 2022, 40, 111225. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, L.; Lin, Q.; Liu, H.; Ye, G.; Liu, X.; Jiao, S.; Li, J.; Tang, Y.; Shi, D.; et al. Pseudorabies virus infection activates the TLR-NF-κB axis and AIM2 inflammasome to enhance inflammatory responses in mice. J. Virol. 2023, 97, e0000323. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Deng, W.; Chen, S.; Qin, B.; Yao, Y.; Zhou, C.; Guo, M. Strongylocentrotus nudus egg polysaccharide (SEP) suppresses HBV replication via activation of TLR4-induced immune pathway. Int. J. Biol. Macromol. 2023, 245, 125539. [Google Scholar] [CrossRef]
- Yamada, S.; Shimojima, M.; Narita, R.; Tsukamoto, Y.; Kato, H.; Saijo, M.; Fujita, T. RIG-I-like receptor and Toll-like receptor signaling pathways cause aberrant production of inflammatory cytokines/chemokines in a severe fever with thrombocytopenia syndrome virus infection mouse model. J. Virol. 2018, 92, e02246-17. [Google Scholar] [CrossRef]
- Tang, X.; Arora, G.; Matias, J.; Hart, T.; Cui, Y.; Fikrig, E. A tick C1q protein alters infectivity of the Lyme disease agent by modulating interferon γ. Cell Rep. 2022, 41, 111673. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, R.; Jin, H.; Xie, J.; Gu, Q.; Yang, X. A novel peptide derived from the mannose binding lectin inhibits LPS-activated TLR4/NF-κB signaling and suppresses ocular inflammation. Cell Biol. Int. 2023, 47, 1614–1626. [Google Scholar] [CrossRef]
- Lysakova-Devine, T.; Keogh, B.; Harrington, B.; Nagpal, K.; Halle, A.; Golenbock, D.T.; Monie, T.; Bowie, A.G. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein A46, specifically inhibits TLR4 by directly targeting MyD88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 2010, 185, 4261–4271. [Google Scholar] [CrossRef]
- Wang, L.; Sun, F.; Hu, J.; Zuo, W.; Zheng, Y.; Wu, Y.; Kwok, H.F.; Cao, Z. The tick saliva peptide HIDfsin2 promotes the tick-borne virus SFTSV replication in vitro by enhancing p38 signal pathway. Arch. Toxicol. 2023, 97, 1783–1794. [Google Scholar] [CrossRef]
- Liu, Y.; You, Y.; Lu, Z.; Yang, J.; Li, P.; Liu, L.; Xu, H.; Niu, Y.; Cao, X. N (6)-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 2019, 365, 1171–1176. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.; Lee, G.J.; Hur, Y.; Keum, J.; Jo, J.K.; Yun, S.-E.; Park, S.J.; Park, Y.J.; Choi, M.J.; et al. A novel kinase inhibitor AX-0085 inhibits interferon-γ-mediated induction of PD-L1 expression and promotes immune reaction to lung adenocarcinoma cells. Cells 2021, 11, 19. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Domadia, P.N.; Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: Role of the helical hairpin conformation in outer-membrane permeabilization. J. Am. Chem. Soc. 2010, 132, 18417–18428. [Google Scholar] [CrossRef]
- Hammer, M.U.; Brauser, A.; Olak, C.; Brezesinski, G.; Goldmann, T.; Gutsmann, T.; Andrä, J. Lipopolysaccharide interaction is decisive for the activity of the antimicrobial peptide NK-2 against Escherichia coli and Proteus mirabilis. Biochem. J. 2010, 427, 477–488. [Google Scholar] [CrossRef]
- Schromm, A.B.; Brandenburg, K.; Rietschel, E.T.; Flad, H.D.; Carroll, S.F.; Seydel, U. Lipopolysaccharide-binding protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS Lett. 1996, 399, 267–271. [Google Scholar] [CrossRef]
- Shaykhiev, R.; Sierigk, J.; Herr, C.; Krasteva, G.; Kummer, W.; Bals, R. The antimicrobial peptide cathelicidin enhances activation of lung epithelial cells by LPS. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 4756–4766. [Google Scholar]
- Liu, Y.; Zhao, C.; Meng, J.; Li, N.; Xu, Z.; Liu, X.; Hou, S. Galectin-3 regulates microglial activation and promotes inflammation through TLR4/MyD88/NF-kB in experimental autoimmune uveitis. Clin. Immunol. 2022, 236, 108939. [Google Scholar] [CrossRef]
- Kotál, J.; Langhansová, H.; Lieskovská, J.; Andersen, J.F.; Francischetti, I.M.; Chavakis, T.; Kopecký, J.; Pedra, J.H.; Kotsyfakis, M.; Chmelař, J. Modulation of host immunity by tick saliva. J. Proteom. 2015, 128, 58–68. [Google Scholar] [CrossRef]
- Francischetti, I.M.; Sa-Nunes, A.; Mans, B.J.; Santos, I.M.; Ribeiro, J.M. The role of saliva in tick feeding. Front. Biosci. 2009, 14, 2051–2088. [Google Scholar] [CrossRef]
- Li, P.; Sun, M.; Wohland, T.; Ho, B.; Ding, J.L. The molecular mechanism of interaction between sushi peptide and pseudomonas endotoxin. Cell. Mol. Immunol. 2006, 3, 21–28. [Google Scholar]
- Nagaoka, I.; Tamura, H.; Reich, J. Therapeutic potential of cathelicidin peptide LL-37, an antimicrobial agent, in a murine sepsis model. Int. J. Mol. Sci. 2020, 21, 5973. [Google Scholar] [CrossRef]
- Giuliani, A.; Pirri, G.; Rinaldi, A.C. Antimicrobial peptides: The LPS connection. Methods Mol. Biol. 2010, 618, 137–154. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primers (5′–3′) | Reverse Primers (5′–3′) |
|---|---|---|
| SFTSV | ATGTCAGAGTGGTCCAGGA | TCTCCACCTGTCTCCTTCAG |
| β-actin | CCGTGAAAAGATGACCCAGA | TACGACCAGAGGCATACAG |
| TNF-α | TGATCCGCGACGTGGAA | ACCGCCTGGAGTTCTGGAA |
| IL-1β | ACTCCTTAGTCCTCGGCCA | CCATCAGAGGCAAGGAGGAA |
| IL-6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Liu, Y.; Pang, R.; Guo, Y.; Ren, Y.; Wu, Y.; Cao, Z. The Tick Saliva Peptide HIDfsin2 TLR4-Dependently Inhibits the Tick-Borne Severe Fever with Thrombocytopenia Syndrome Virus in Mouse Macrophages. Antibiotics 2024, 13, 449. https://doi.org/10.3390/antibiotics13050449
Wang L, Liu Y, Pang R, Guo Y, Ren Y, Wu Y, Cao Z. The Tick Saliva Peptide HIDfsin2 TLR4-Dependently Inhibits the Tick-Borne Severe Fever with Thrombocytopenia Syndrome Virus in Mouse Macrophages. Antibiotics. 2024; 13(5):449. https://doi.org/10.3390/antibiotics13050449
Chicago/Turabian StyleWang, Luyao, Yishuo Liu, Rui Pang, Yiyuan Guo, Yingying Ren, Yingliang Wu, and Zhijian Cao. 2024. "The Tick Saliva Peptide HIDfsin2 TLR4-Dependently Inhibits the Tick-Borne Severe Fever with Thrombocytopenia Syndrome Virus in Mouse Macrophages" Antibiotics 13, no. 5: 449. https://doi.org/10.3390/antibiotics13050449
APA StyleWang, L., Liu, Y., Pang, R., Guo, Y., Ren, Y., Wu, Y., & Cao, Z. (2024). The Tick Saliva Peptide HIDfsin2 TLR4-Dependently Inhibits the Tick-Borne Severe Fever with Thrombocytopenia Syndrome Virus in Mouse Macrophages. Antibiotics, 13(5), 449. https://doi.org/10.3390/antibiotics13050449