
Constitutive Activation of RpoH and the Addition of L-arabinose Influence Antibiotic Sensitivity of PHL628 E. coli


Abstract
1. Introduction
2. Results
2.1. Disk Diffusion Assays
2.2. Minimum Inhibitory Concentration (MIC) Determination
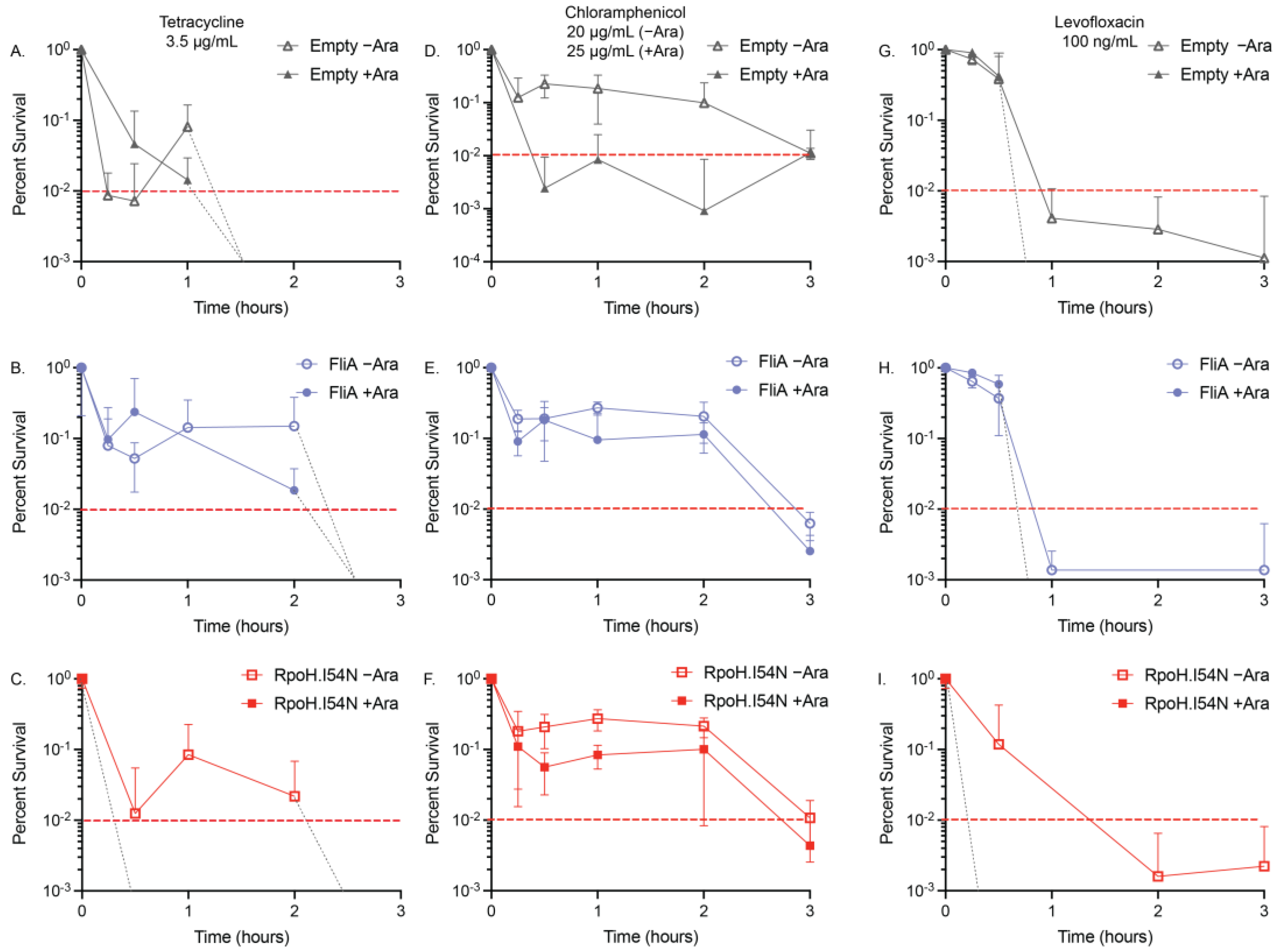
2.3. Minimum Duration of Killing (MDK) Assays
3. Discussion
4. Materials and Methods
4.1. Bacterial Growth and Strains
4.2. Disk Diffusion Assay
4.3. Preparation of Antibiotics for MIC and Tolerance Experiments
4.4. Minimum Inhibitory Concentration (MIC) Determination
4.5. Minimum Duration of Killing (MDK) Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics 2020, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Lazarovits, G.; Gefen, O.; Cahanian, N.; Adler, K.; Fluss, R.; Levin-Reisman, I.; Ronin, I.; Motro, Y.; Moran-Gilad, J.; Balaban, N.Q.; et al. Prevalence of Antibiotic Tolerance and Risk for Reinfection Among Escherichia coli Bloodstream Isolates: A Prospective Cohort Study. Clin. Infect. Dis. 2022, 75, 1706–1713. [Google Scholar] [CrossRef]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic Tolerance Facilitates the Evolution of Resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Zhu, Y.-G.; Zhao, Y.; Zhu, D.; Gillings, M.; Penuelas, J.; Ok, Y.S.; Capon, A.; Banwart, S. Soil Biota, Antimicrobial Resistance and Planetary Health. Environ. Int. 2019, 131, 105059. [Google Scholar] [CrossRef]
- Deter, H.S.; Hossain, T.; Butzin, N.C. Antibiotic Tolerance Is Associated with a Broad and Complex Transcriptional Response in E. coli. Sci. Rep. 2021, 11, 6112. [Google Scholar] [CrossRef]
- Brauner, A.; Shoresh, N.; Fridman, O.; Balaban, N.Q. An Experimental Framework for Quantifying Bacterial Tolerance. Biophys. J. 2017, 112, 2664–2671. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between Resistance, Tolerance and Persistence to Antibiotic Treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef]
- Fridman, O.; Goldberg, A.; Ronin, I.; Shoresh, N.; Balaban, N.Q. Optimization of Lag Time Underlies Antibiotic Tolerance in Evolved Bacterial Populations. Nature 2014, 513, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Levin-Reisman, I.; Brauner, A.; Ronin, I.; Balaban, N.Q. Epistasis between Antibiotic Tolerance, Persistence, and Resistance Mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 14734–14739. [Google Scholar] [CrossRef] [PubMed]
- Santi, I.; Manfredi, P.; Maffei, E.; Egli, A.; Jenal, U. Evolution of Antibiotic Tolerance Shapes Resistance Development in Chronic Pseudomonas aeruginosa Infections. mBio 2021, 12, e03482-20. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gefen, O.; Ronin, I.; Bar-Meir, M.; Balaban, N.Q. Effect of Tolerance on the Evolution of Antibiotic Resistance under Drug Combinations. Science 2020, 367, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, J.E.; Lam, H. Evolution of Bacterial Tolerance Under Antibiotic Treatment and Its Implications on the Development of Resistance. Front. Microbiol. 2021, 12, 617412. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, J.E.; Lam, H. Proteomics in Antibiotic Resistance and Tolerance Research: Mapping the Resistome and the Tolerome of Bacterial Pathogens. Proteomics 2022, 22, 2100409. [Google Scholar] [CrossRef]
- Van Elsas, J.D.; Semenov, A.V.; Costa, R.; Trevors, J.T. Survival of Escherichia coli in the Environment: Fundamental and Public Health Aspects. ISME J. 2011, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.L.; Silhavy, T.J. Periplasmic Stress and ECF Sigma Factors. Annu. Rev. Microbiol. 2001, 55, 591–624. [Google Scholar] [CrossRef]
- Chung, H.J.; Bang, W.; Drake, M.A. Stress Response of Escherichia coli. Compr. Rev. Food Sci. Food Saf. 2006, 5, 52–64. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial Biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Fux, C.A.; Costerton, J.W.; Stewart, P.S.; Stoodley, P. Survival Strategies of Infectious Biofilms. Trends Microbiol. 2005, 13, 34–40. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef]
- Yura, T. Regulation of the Heat Shock Response in Escherichia coli: History and Perspectives. Genes Genet. Syst. 2019, 94, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.D.; Straus, D.B.; Walter, W.A.; Gross, C.A. Sigma 32 Synthesis Can Regulate the Synthesis of Heat Shock Proteins in Escherichia coli. Genes Dev. 1987, 1, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Arsène, F.; Tomoyasu, T.; Bukau, B. The Heat Shock Response of Escherichia coli. Int. J. Food Microbiol. 2000, 55, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.A.; Zahrl, D.; Rieser, G.; Koraimann, G. Growth Phase- and Cell Division-Dependent Activation and Inactivation of the σ32 Regulon in Escherichia coli. J. Bacteriol. 2008, 191, 1695–1702. [Google Scholar] [CrossRef]
- Morita, M.T.; Kanemori, M.; Yanagi, H.; Yura, T. Dynamic Interplay between Antagonistic Pathways Controlling the Σ32 Level in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5860–5865. [Google Scholar] [CrossRef] [PubMed]
- Narberhaus, F.; Balsiger, S. Structure-Function Studies of Escherichia coli RpoH (Sigma32) by in Vitro Linker Insertion Mutagenesis. J. Bacteriol. 2003, 185, 2731–2738. [Google Scholar] [CrossRef]
- Pesavento, C.; Becker, G.; Sommerfeldt, N.; Possling, A.; Tschowri, N.; Mehlis, A. Hengge, RInverse Regulatory Coordination of Motility and Curli-Mediated Adhesion in Escherichia coli. Genes Dev. 2008, 22, 2434–2446. [Google Scholar] [CrossRef]
- Claret, L.; Miquel, S.; Vieille, N.; Ryjenkov, D.A.; Gomelsky, M.; Darfeuille-Michaud, A. The Flagellar Sigma Factor FliA Regulates Adhesion and Invasion of Crohn Disease-Associated Escherichia coli via a Cyclic Dimeric GMP-Dependent Pathway. J. Biol. Chem. 2007, 282, 33275–33283. [Google Scholar] [CrossRef]
- Smith, T.G.; Hoover, T.R. Deciphering Bacterial Flagellar Gene Regulatory Networks in the Genomic Era. Adv. Appl. Microbiol. 2009, 67, 257–295. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, I.; Ni, B.; Glatter, T.; Sourjik, V. Inefficient Secretion of Anti-Sigma Factor FlgM Inhibits Bacterial Motility at High Temperature. iScience 2019, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kutsukake, K.; Iino, T. Role of the FliA-FlgM Regulatory System on the Transcriptional Control of the Flagellar Regulon and Flagellar Formation in Salmonella typhimurium. J. Bacteriol. 1994, 176, 3598–3605. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, P.; Hughes, K.T. Regulation of Flagellar Assembly. Curr. Opin. Microbiol. 2002, 5, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Buck, L.D.; Paladino, M.M.; Nagashima, K.; Brezel, E.R.; Holtzman, J.S.; Urso, S.J.; Ryno, L.M. Temperature-Dependent Influence of FliA Overexpression on PHL628 E. coli Biofilm Growth and Composition. Front. Cell. Infect. Microbiol. 2021, 11, 775270. [Google Scholar] [CrossRef]
- Wood, T.K.; Barrios, A.F.G.; Herzberg, M.; Lee, J. Motility Influences Biofilm Architecture in Escherichia coli. Appl. Microbiol. Biotechnol. 2006, 72, 361–367. [Google Scholar] [CrossRef]
- Amores, G.R.; Heras, A.d.L.; Sanches-Medeiros, A.; Elfick, A.; Silva-Rocha, R. Systematic Identification of Novel Regulatory Interactions Controlling Biofilm Formation in the Bacterium Escherichia coli. Sci. Rep. 2017, 7, 16768. [Google Scholar] [CrossRef]
- Duatepe, F.P.G. FliA-Dependent Surface Macromolecules Promote Initial Biofilm Development of Escherichia coli by Influencing the Bacterial Surface Properties. Hittite J. Sci. Eng. 2023, 10, 83–90. [Google Scholar] [CrossRef]
- Xiao, Y.; Nie, L.; Chen, H.; He, M.; Liang, Q.; Nie, H.; Chen, W.; Huang, Q. The Two-component System TarR–TarS Is Regulated by C-di-GMP/FleQ and FliA and Modulates Antibiotic Susceptibility in Pseudomonas putida. Environ. Microbiol. 2021, 23, 5239–5257. [Google Scholar] [CrossRef]
- Pinto, L.; Shastry, R.P.; Alva, S.; Rao, R.S.P.; Ghate, S.D. Functional Network Analysis Identifies Multiple Virulence and Antibiotic Resistance Targets in Stenotrophomonas maltophilia. Microb. Pathog. 2023, 183, 106314. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Genereux, J.C.; Nolan, C.; Singh, M.; Kelly, J.W. Heat-Shock Response Transcriptional Program Enables High-Yield and High-Quality Recombinant Protein Production in Escherichia coli. ACS Chem. Biol. 2014, 9, 1945–1949. [Google Scholar] [CrossRef]
- Rusu, A.; Lungu, I.-A.; Moldovan, O.-L.; Tanase, C.; Hancu, G. Structural Characterization of the Millennial Antibacterial (Fluoro)Quinolones—Shaping the Fifth Generation. Pharmaceutics 2021, 13, 1289. [Google Scholar] [CrossRef]
- Vidal, O.; Longin, R.; Prigent-Combaret, C.; Dorel, C.; Hooreman, M.; Lejeune, P. Isolation of an Escherichia coli K-12 Mutant Strain Able to Form Biofilms on Inert Surfaces: Involvement of a New ompR Allele That Increases Curli Expression. J. Bacteriol. 1998, 180, 2442–2449. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Prigent-Combaret, C.; Brombacher, E.; Vidal, O.; Ambert, A.; Lejeune, P.; Landini, P.; Dorel, C. Complex Regulatory Network Controls Initial Adhesion and Biofilm Formation in Escherichia coli via Regulation of the csgD Gene. J. Bacteriol. 2001, 183, 7213–7223. [Google Scholar] [CrossRef]
- Smith, D.R.; Price, J.E.; Burby, P.E.; Blanco, L.P.; Chamberlain, J.; Chapman, M.R. The Production of Curli Amyloid Fibers Is Deeply Integrated into the Biology of Escherichia coli. Biomolecules 2017, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Hammar, M.; Arnqvist, A.; Bian, Z.; Olsén, A.; Normark, S. Expression of Two Csg Operons Is Required for Production of Fi-bronectin- and Congo Red-Binding Curli Polymers in Escherichia coli K-12. Mol. Microbiol. 1995, 18, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Leech, J.; Golub, S.; Allan, W.; Simmons, M.J.H.; Overton, T.W. Non-Pathogenic Escherichia coli Biofilms: Effects of Growth Conditions and Surface Properties on Structure and Curli Gene Expression. Arch. Microbiol. 2020, 202, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Junker, L.M.; Peters, J.E.; Hay, A.G. Global Analysis of Candidate Genes Important for Fitness in a Competitive Biofilm Using DNA-Array-Based Transposon Mapping. Microbiology 2006, 152, 2233–2245. [Google Scholar] [CrossRef][Green Version]
- Junker, L.M.; Toba, F.A.; Hay, A.G. Transcription in Escherichia coli PHL628 Biofilms. FEMS Microbiol. Lett. 2007, 268, 237–243. [Google Scholar] [CrossRef][Green Version]
- Yura, T.; Guisbert, E.; Poritz, M.; Lu, C.Z.; Campbell, E.; Gross, C.A. Analysis of Sigma32 Mutants Defective in Chaperone-Mediated Feedback Control Reveals Unexpected Complexity of the Heat Shock Response. Proc. Natl. Acad. Sci. USA 2007, 104, 17638–17643. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberg, W.E. Kirby-Bauer Disk Diffusion Method. Am. J. Clin. Pathol. 1981, 75, 273. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 33rd ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2023; ISBN 978-1-68440-170-3. [Google Scholar]
- Shimizu, K. Regulation Systems of Bacteria such as Escherichia coli in Response to Nutrient Limitation and Environmental Stresses. Metabolites 2013, 4, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; Yang, C.; Oshima, T.; Mori, H.; Shimizu, K. Analysis of Gene Expression in Escherichia coli in Response to Changes of Growth-Limiting Nutrient in Chemostat Cultures. Appl. Environ. Microbiol. 2004, 70, 2354–2366. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K. Metabolic Regulation and Coordination of the Metabolism in Bacteria in Response to a Variety of Growth Con-ditions. Adv. Biochem. Eng. Biotechnol. 2016, 155, 1–54. [Google Scholar] [CrossRef]
- Seshasayee, A.S.; Bertone, P.; Fraser, G.M.; Luscombe, N.M. Transcriptional Regulatory Networks in Bacteria: From Input Signals to Output Responses. Curr. Opin. Microbiol. 2006, 9, 511–519. [Google Scholar] [CrossRef]
- Guan, N.; Li, J.; Shin, H.-D.; Du, G.; Chen, J.; Liu, L. Microbial Response to Environmental Stresses: From Fundamental Mechanisms to Practical Applications. Appl. Microbiol. Biotechnol. 2017, 101, 3991–4008. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Metabolic Control of Cell Death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef]
- Wolska, K.I.; Bugajska, E.; Jurkiewicz, D.; Kuć, M.; Jóźwik, A. Antibiotic Susceptibility of Escherichia coli DnaK and DnaJ Mutants. Microb. Drug Resist. 2000, 6, 119–126. [Google Scholar] [CrossRef]
- Wolfe, A.D.; Hahn, F.E. Mode of Action of Chloramphenicol IX. Effects of Chloramphenicol upon a Ribosomal Amino Acid Polymerization System and Its Binding to Bacterial Ribosome. Biochim. Biophys. Acta BBA-Nucleic Acids Protein Synth. 1965, 95, 146–155. [Google Scholar] [CrossRef]
- Hahn, F.E.; Wisseman, C.L.; Hopps, H.E. Mode of Action of Chloramphenicol III. Action of Chloramphenicol on Bacterial Energy Metabolism. J. Bacteriol. 1955, 69, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Koita, K.; Rao, C.V. Identification and Analysis of the Putative Pentose Sugar Efflux Transporters in Escherichia coli. PLoS ONE 2012, 7, e43700. [Google Scholar] [CrossRef] [PubMed]
- Saiful, A.J.; Mastura, M.; Zarizal, S.; Mazurah, M.I.; Shuhaimi, M.; Ali, A.M. Efflux Genes and Active Efflux Activity Detection in Malaysian Clinical Isolates of Methicillin-resistant Staphylococcus aureus (MRSA). J. Basic Microbiol. 2008, 48, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Linnell, S.K.; Boyd, L.B.; Steffen, D.; Zechiedrich, L. Mechanisms Accounting for Fluoroquinolone Resistance in Escherichia coli Clinical Isolates. Antimicrob. Agents Chemother. 2009, 53, 235–241. [Google Scholar] [CrossRef]
- Van der Putten, B.C.L.; Remondini, D.; Pasquini, G.; Janes, V.A.; Matamoros, S.; Schultsz, C. Quantifying the Contribution of Four Resistance Mechanisms to Ciprofloxacin MIC in Escherichia coli: A Systematic Review. J. Antimicrob. Chemother. 2019, 74, 298–310. [Google Scholar] [CrossRef]
- Mazzariol, A.; Zuliani, J.; Cornaglia, G.; Rossolini, G.M.; Fontana, R. AcrAB Efflux System: Expression and Contribution to Fluoroquinolone Resistance in Klebsiella spp. Antimicrob. Agents Chemother. 2002, 46, 3984–3986. [Google Scholar] [CrossRef]
- Spagnolo, F.; Rinaldi, C.; Sajorda, D.R.; Dykhuizen, D.E. Evolution of Resistance to Continuously Increasing Streptomycin Concentrations in Populations of Escherichia coli. Antimicrob. Agents Chemother. 2015, 60, 1336–1342. [Google Scholar] [CrossRef]
- Boulanger, E.F.; Sabag-Daigle, A.; Thirugnanasambantham, P.; Gopalan, V.; Ahmer, B.M.M. Sugar-Phosphate Toxicities. Microbiol. Mol. Biol. Rev. 2021, 85, e00123-21. [Google Scholar] [CrossRef]
- Englesberg, E.; Anderson, R.L.; Weinberg, R.; Lee, N.; Hoffee, P.; Huttenhauer, G.; Boyer, H. L-Arabinose-Sensitivt, 1-Ribulose-5-Phosphate 4-Epimerase-Deficient Mutants of Escherichia coli. J. Bacteriol. 1962, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.-W.; Lee, J.-J.; Ptak, C.P.; Wu, Y.-C.; Hsuan, S.-L.; Kuo, C.-J.; Chen, T.-H. Effects of L-Arabinose Efflux on λ Red Recombination-Mediated Gene Knockout in Multiple-Antimicrobial-Resistant Salmonella enterica Serovar Choleraesuis. Arch. Microbiol. 2018, 200, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Thorsing, M.; Bentin, T.; Givskov, M.; Tolker-Nielsen, T.; Goltermann, L. The Bactericidal Activity of β-Lactam Antibiotics Is Increased by Metabolizable Sugar Species. Microbiology 2015, 161, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Goltermann, L.; Good, L.; Bentin, T. Chaperonins Fight Aminoglycoside-Induced Protein Misfolding and Promote Short-Term Tolerance in Escherichia coli. J. Biol. Chem. 2013, 288, 10483–10489. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-Enabled Eradication of Bacterial Persisters by Aminoglycosides. Nature 2011, 473, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hare, P.J.; Gonzalez, J.R.; Quelle, R.M.; Wu, Y.I.; Mok, W.W.K. Metabolic and Transcriptional Activities Underlie Stationary-Phase Pseudomonas aeruginosa Sensitivity to Levofloxacin. Microbiol. Spectr. 2024, 12, e0356723. [Google Scholar] [CrossRef]
- Zhou, Y.; Yong, Y.; Zhu, C.; Yang, H.; Fang, B. Exogenous D-Ribose Promotes Gentamicin Treatment of Several Drug-Resistant Salmonella. Front. Microbiol. 2022, 13, 1053330. [Google Scholar] [CrossRef]
- Chung, C.T.; Miller, R.H. Preparation and Storage of Competent Escherichia coli Cells. Meth. Enzymol. 1993, 218, 621–627. [Google Scholar] [CrossRef]
- Lüders, S.; Fallet, C.; Franco-Lara, E. Proteome Analysis of the Escherichia coli Heat Shock Response under Steady-State Conditions. Proteome Sci. 2009, 7, 36. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MIC (µg/mL) at 37 °C | ||||||
|---|---|---|---|---|---|---|
| pBAD.empty | pBAD.fliA | pBAD.rpoH.I54N | ||||
| −Ara | +Ara | −Ara | +Ara | −Ara | +Ara | |
| Tetracycline | 1.0 | 1.0 | 1.5 | 1.5 | 1.0 | 0.75 |
| Chloramphenicol | 7.0 | 7.0 | 7.0 | 7.0 | 7.0 | 4.0 |
| Levofloxacin | 0.05 | 0.15 | 0.05 | 0.10 | 0.05 | 0.15 |
| MIC (µg/mL) at 37 °C | ||||
|---|---|---|---|---|
| PHL628 | Heat Shock | |||
| −Ara | +Ara | −Ara | +Ara | |
| Tetracycline | 0.75 | 1.75 | 0.75 | 1.75 |
| Chloramphenicol | 7.0 | 7.0 | 5.0 | 25 |
| Levofloxacin | 0.05 | 0.15 | 0.05 | 0.15 |
| MDK99 (µg/mL) at 37 °C | ||||||
|---|---|---|---|---|---|---|
| pBAD.empty | pBAD.fliA | pBAD.rpoH.I54N | ||||
| −Ara | +Ara | −Ara | +Ara | −Ara | +Ara | |
| Tetracycline | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| Chloramphenicol | 20 | 25 | 20 | 25 * | 20 | 25 |
| Levofloxacin | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 | 0.10 |
| MDK99 (µg/mL) at 37 °C | ||||
|---|---|---|---|---|
| PHL628 | Heat Shock | |||
| −Ara | +Ara | −Ara | +Ara | |
| Tetracycline | 3.5 | 6.0 | 3.5 | 6.0 * |
| Chloramphenicol | 12.5 | 25 ** | 12.5 | 25 ** |
| Levofloxacin | 0.125 | 0.50 ** | 0.125 | 0.50 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frizzell, J.K.; Taylor, R.L.; Ryno, L.M. Constitutive Activation of RpoH and the Addition of L-arabinose Influence Antibiotic Sensitivity of PHL628 E. coli. Antibiotics 2024, 13, 143. https://doi.org/10.3390/antibiotics13020143
Frizzell JK, Taylor RL, Ryno LM. Constitutive Activation of RpoH and the Addition of L-arabinose Influence Antibiotic Sensitivity of PHL628 E. coli. Antibiotics. 2024; 13(2):143. https://doi.org/10.3390/antibiotics13020143
Chicago/Turabian StyleFrizzell, Jenna K., Ryan L. Taylor, and Lisa M. Ryno. 2024. "Constitutive Activation of RpoH and the Addition of L-arabinose Influence Antibiotic Sensitivity of PHL628 E. coli" Antibiotics 13, no. 2: 143. https://doi.org/10.3390/antibiotics13020143
APA StyleFrizzell, J. K., Taylor, R. L., & Ryno, L. M. (2024). Constitutive Activation of RpoH and the Addition of L-arabinose Influence Antibiotic Sensitivity of PHL628 E. coli. Antibiotics, 13(2), 143. https://doi.org/10.3390/antibiotics13020143