
High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes


 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. DNA Extraction, Library Preparation, and Sequencing
4.3. Genome Assembly and Annotation
4.4. Plasmid Analysis
4.5. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Guidelines for the Prevention and Control of Carbapenem-Resistant Enterobacteriaceae. In Acinetobacter baumannii and Pseudomonas aeruginosa in Health Care Facilities; World Health Organization: Geneva, Switzerland, 2017; ISBN 978-92-4-155017-8. [Google Scholar]
- Rabaan, A.A.; Eljaaly, K.; Alhumaid, S.; Albayat, H.; Al-Adsani, W.; Sabour, A.A.; Alshiekheid, M.A.; Al-Jishi, J.M.; Khamis, F.; Alwarthan, S.; et al. An Overview on Phenotypic and Genotypic Characterisation of Carbapenem-Resistant Enterobacterales. Medicina 2022, 58, 1675. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q.; Yin, Y.; Chen, H.; Jin, L.; Gu, B.; Xie, L.; Yang, C.; Ma, X.; Li, H.; et al. Epidemiology of Carbapenem-Resistant Enterobacteriaceae Infections: Report from the China CRE Network. Antimicrob. Agents Chemother. 2018, 62, e01882-17. [Google Scholar] [CrossRef] [PubMed]
- Di Pilato, V.; Pollini, S.; Miriagou, V.; Rossolini, G.M.; D’Andrea, M.M. Carbapenem-Resistant Klebsiella pneumoniae: The Role of Plasmids in Emergence, Dissemination, and Evolution of a Major Clinical Challenge. Expert Rev. Anti-Infect. Ther. 2024, 22, 25–43. [Google Scholar] [CrossRef]
- Stanton, T.D.; Wyres, K.L. What Defines Hypervirulent Klebsiella pneumoniae? eBioMedicine 2024, 108, 105331. [Google Scholar] [CrossRef]
- Wang, H.; Xu, Q.; Chen, K.; Chan, B.K.W.; Ye, L.; Yang, X.; Xie, M.; Liu, X.; Ni, H.; Chan, E.W.C.; et al. A Siderophore-Encoding Plasmid Encodes High-Level Virulence in Escherichia coli. Microbiol. Spectr. 2022, 10, e02528-21. [Google Scholar] [CrossRef]
- Gu, D.; Dong, N.; Zheng, Z.; Lin, D.; Huang, M.; Wang, L.; Chan, E.W.-C.; Shu, L.; Yu, J.; Zhang, R.; et al. A Fatal Outbreak of ST11 Carbapenem-Resistant Hypervirulent Klebsiella pneumoniae in a Chinese Hospital: A Molecular Epidemiological Study. Lancet Infect. Dis. 2018, 18, 37–46. [Google Scholar] [CrossRef]
- Antimicrobial Resistance, Hypervirulent Klebsiella pneumoniae Global Situation. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2024-DON527 (accessed on 3 November 2024).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Russo, T.A.; Carlino-MacDonald, U.; Drayer, Z.J.; Davies, C.J.; Alvarado, C.L.; Hutson, A.; Luo, T.L.; Martin, M.J.; McGann, P.T.; Lebreton, F. Deciphering the Relative Importance of Genetic Elements in Hypervirulent Klebsiella pneumoniae to Guide Countermeasure Development. eBioMedicine 2024, 107, 105302. [Google Scholar] [CrossRef]
- Russo, T.A.; Alvarado, C.L.; Davies, C.J.; Drayer, Z.J.; Carlino-MacDonald, U.; Hutson, A.; Luo, T.L.; Martin, M.J.; Corey, B.W.; Moser, K.A.; et al. Differentiation of Hypervirulent and Classical Klebsiella pneumoniae with Acquired Drug Resistance. mBio 2024, 15, e02867-23. [Google Scholar] [CrossRef]
- Fursova, N.K.; Astashkin, E.I.; Knyazeva, A.I.; Kartsev, N.N.; Leonova, E.S.; Ershova, O.N.; Alexandrova, I.A.; Kurdyumova, N.V.; Sazikina, S.Y.; Volozhantsev, N.V.; et al. The Spread of Bla OXA-48 and Bla OXA-244 Carbapenemase Genes among Klebsiella pneumoniae, Proteus Mirabilis and Enterobacter Spp. Isolated in Moscow, Russia. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Chudejova, K.; Kraftova, L.; Mattioni Marchetti, V.; Hrabak, J.; Papagiannitsis, C.C.; Bitar, I. Genetic Plurality of OXA/NDM-Encoding Features Characterized From Enterobacterales Recovered From Czech Hospitals. Front. Microbiol. 2021, 12, 641415. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, S.; Hagiya, H.; Gotoh, K.; Tsuji, S.; Iio, K.; Matsushita, O. Detection of Imported Clinical Strain of Bla NDM-1 -Harbouring ST147 Klebsiella pneumoniae from a Ukrainian Immigrant. J. Travel Med. 2024, 31, taae011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, X.; Fan, F.; Wang, X.; Dong, S. The Dynamic Evolution and IS26-Mediated Interspecies Transfer of a blaNDM-1-Bearing Fusion Plasmid Leading to a Hypervirulent Carbapenem-Resistant Klebsiella pneumoniae Strain Harbouring blaKPC-2 in a Single Patient. J. Glob. Antimicrob. Resist. 2023, 35, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bulman, Z.P.; Tan, X.; Chu, T.-Y.; Huang, Y.; Rana, A.P.; Singh, N.; Flowers, S.A.; Kyono, Y.; Kreiswirth, B.N.; Chen, L. Ceftazidime-Avibactam Based Combinations against Carbapenemase Producing Klebsiella pneumoniae Harboring Hypervirulence Plasmids. Comput. Struct. Biotechnol. J. 2022, 20, 3946–3954. [Google Scholar] [CrossRef]
- Ahmed, M.A.E.-G.E.-S.; Yang, Y.; Yang, Y.; Yan, B.; Chen, G.; Hassan, R.M.; Zhong, L.-L.; Chen, Y.; Roberts, A.P.; Wu, Y.; et al. Emergence of Hypervirulent Carbapenem-Resistant Klebsiella pneumoniae Coharboring a Bla NDM-1 -Carrying Virulent Plasmid and a Bla KPC-2 -Carrying Plasmid in an Egyptian Hospital. mSphere 2021, 6, e00088-21. [Google Scholar] [CrossRef]
- Starkova, P.; Lazareva, I.; Avdeeva, A.; Sulian, O.; Likholetova, D.; Ageevets, V.; Lebedeva, M.; Gostev, V.; Sopova, J.; Sidorenko, S. Emergence of Hybrid Resistance and Virulence Plasmids Harboring New Delhi Metallo-β-Lactamase in Klebsiella pneumoniae in Russia. Antibiotics 2021, 10, 691. [Google Scholar] [CrossRef]
- Kuzina, E.S.; Kislichkina, A.A.; Sizova, A.A.; Skryabin, Y.P.; Novikova, T.S.; Ershova, O.N.; Savin, I.A.; Khokhlova, O.E.; Bogun, A.G.; Fursova, N.K. High-Molecular-Weight Plasmids Carrying Carbapenemase Genes blaNDM-1, blaKPC-2, and blaOXA-48 Coexisting in Clinical Klebsiella pneumoniae Strains of ST39. Microorganisms 2023, 11, 459. [Google Scholar] [CrossRef]
- Shelenkov, A.; Mikhaylova, Y.; Yanushevich, Y.; Samoilov, A.; Petrova, L.; Fomina, V.; Gusarov, V.; Zamyatin, M.; Shagin, D.; Akimkin, V. Molecular Typing, Characterization of Antimicrobial Resistance, Virulence Profiling and Analysis of Whole-Genome Sequence of Clinical Klebsiella pneumoniae Isolates. Antibiotics 2020, 9, 261. [Google Scholar] [CrossRef]
- Lipworth, S.; Matlock, W.; Shaw, L.; Vihta, K.-D.; Rodger, G.; Chau, K.; Barker, L.; George, S.; Kavanagh, J.; Davies, T.; et al. The Plasmidome Associated with Gram-Negative Bloodstream Infections: A Large-Scale Observational Study Using Complete Plasmid Assemblies. Nat. Commun. 2024, 15, 1612. [Google Scholar] [CrossRef]
- Wang, B.; Finazzo, M.; Artsimovitch, I. Machine Learning Suggests That Small Size Helps Broaden Plasmid Host Range. Genes 2023, 14, 2044. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.M.C.; Wick, R.R.; Watts, S.C.; Cerdeira, L.T.; Wyres, K.L.; Holt, K.E. A Genomic Surveillance Framework and Genotyping Tool for Klebsiella pneumoniae and Its Related Species Complex. Nat. Commun. 2021, 12, 4188. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dong, N.; Chan, E.W.C.; Chen, S.; Zhang, R. Molecular Epidemiology of Carbapenem-Resistant Klebsiella pneumoniae in China, 2016–2020. Lancet Infect. Dis. 2022, 22, 167–168. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Dong, N.; Lu, J.; Zheng, Z.; Hu, J.; Zeng, W.; Sun, Q.; Chan, E.W.-C.; Zhou, H.; Hu, F.; et al. Emergence of OXA-232 Carbapenemase-Producing Klebsiella pneumoniae That Carries a pLVPK-Like Virulence Plasmid among Elderly Patients in China. Antimicrob. Agents Chemother. 2019, 63, e02246-18. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, C.; Wu, J.; Jin, J.; Xu, T.; Zhou, Y.; Cui, P.; Chen, J.; Chen, S.; Jiang, N.; et al. Sequence-Based Genomic Analysis Reveals Transmission of Antibiotic Resistance and Virulence among Carbapenemase-Producing Klebsiella pneumoniae Strains. mSphere 2022, 7, e00143-22. [Google Scholar] [CrossRef]
- Spadar, A.; Perdigão, J.; Campino, S.; Clark, T.G. Large-Scale Genomic Analysis of Global Klebsiella pneumoniae Plasmids Reveals Multiple Simultaneous Clusters of Carbapenem-Resistant Hypervirulent Strains. Genome Med. 2023, 15, 3. [Google Scholar] [CrossRef]
- Tian, D.; Wang, M.; Zhou, Y.; Hu, D.; Ou, H.-Y.; Jiang, X. Genetic Diversity and Evolution of the Virulence Plasmids Encoding Aerobactin and Salmochelin in Klebsiella pneumoniae. Virulence 2021, 12, 1323–1333. [Google Scholar] [CrossRef]
- Shaidullina, E.R.; Schwabe, M.; Rohde, T.; Shapovalova, V.V.; Dyachkova, M.S.; Matsvay, A.D.; Savochkina, Y.A.; Shelenkov, A.A.; Mikhaylova, Y.V.; Sydow, K.; et al. Genomic Analysis of the International High-Risk Clonal Lineage Klebsiella pneumoniae Sequence Type 395. Genome Med. 2023, 15, 9. [Google Scholar] [CrossRef]
- Fursova, N.K.; Astashkin, E.I.; Gabrielyan, N.I.; Novikova, T.S.; Fedyukina, G.N.; Kubanova, M.K.; Esenova, N.M.; Sharapchenko, S.O.; Volozhantsev, N.V. Emergence of Five Genetic Lines ST395NDM−1, ST13OXA−48, ST3346OXA−48, ST39CTX-M−14, and Novel ST3551OXA−48 of Multidrug-Resistant Clinical Klebsiella pneumoniae in Russia. Microb. Drug Resist. 2020, 26, 924–933. [Google Scholar] [CrossRef]
- Shapovalova, V.; Shaidullina, E.; Azizov, I.; Sheck, E.; Martinovich, A.; Dyachkova, M.; Matsvay, A.; Savochkina, Y.; Khafizov, K.; Kozlov, R.; et al. Molecular Epidemiology of Mcr-1-Positive Escherichia coli and Klebsiella pneumoniae Isolates: Results from Russian Sentinel Surveillance (2013–2018). Microorganisms 2022, 10, 2034. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). In Horizontal Gene Transfer; De La Cruz, F., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2075, pp. 285–294. ISBN 978-1-4939-9876-0. [Google Scholar]
- Robertson, J.; Nash, J.H.E. MOB-Suite: Software Tools for Clustering, Reconstruction and Typing of Plasmids from Draft Assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- Arredondo-Alonso, S.; Gladstone, R.A.; Pöntinen, A.K.; Gama, J.A.; Schürch, A.C.; Lanza, V.F.; Johnsen, P.J.; Samuelsen, Ø.; Tonkin-Hill, G.; Corander, J. Mge-Cluster: A Reference-Free Approach for Typing Bacterial Plasmids. NAR Genom. Bioinform. 2023, 5, lqad066. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Holley, G.; Melsted, P. Bifrost: Highly Parallel Construction and Indexing of Colored and Compacted de Bruijn Graphs. Genome Biol. 2020, 21, 249. [Google Scholar] [CrossRef]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and Sharing Data for Genomic Epidemiology and Phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & Clustermap.Js: Automatic Generation of Gene Cluster Comparison Figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Plasmids | Species | Replicon | Virulence Factors | Carbapenemase Genes | Number of Plasmids with Genes | Hybrid Plasmids | Predicted Mobility | Median Length, bp | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Carbapenemase | AMR | Virulence | |||||||||
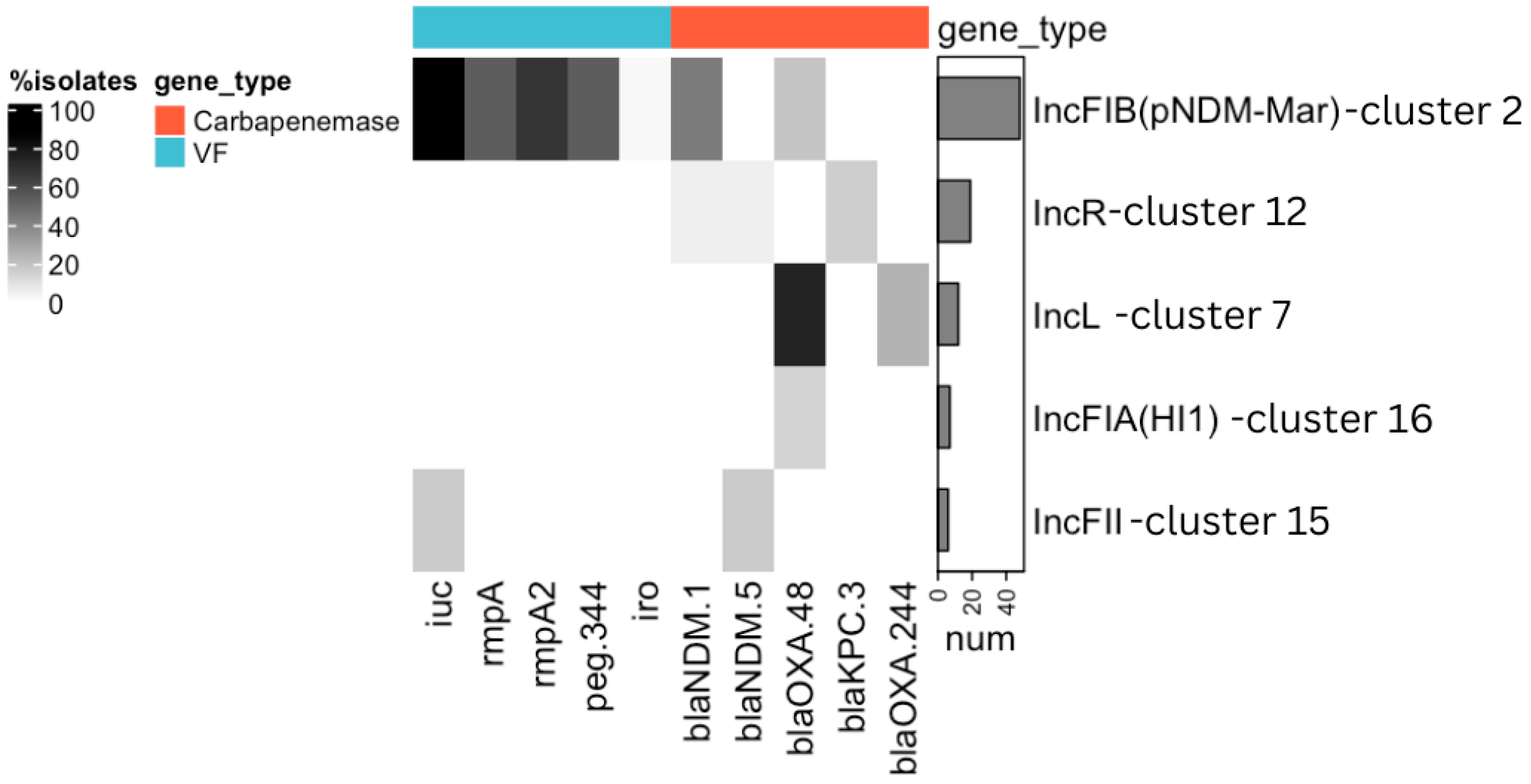
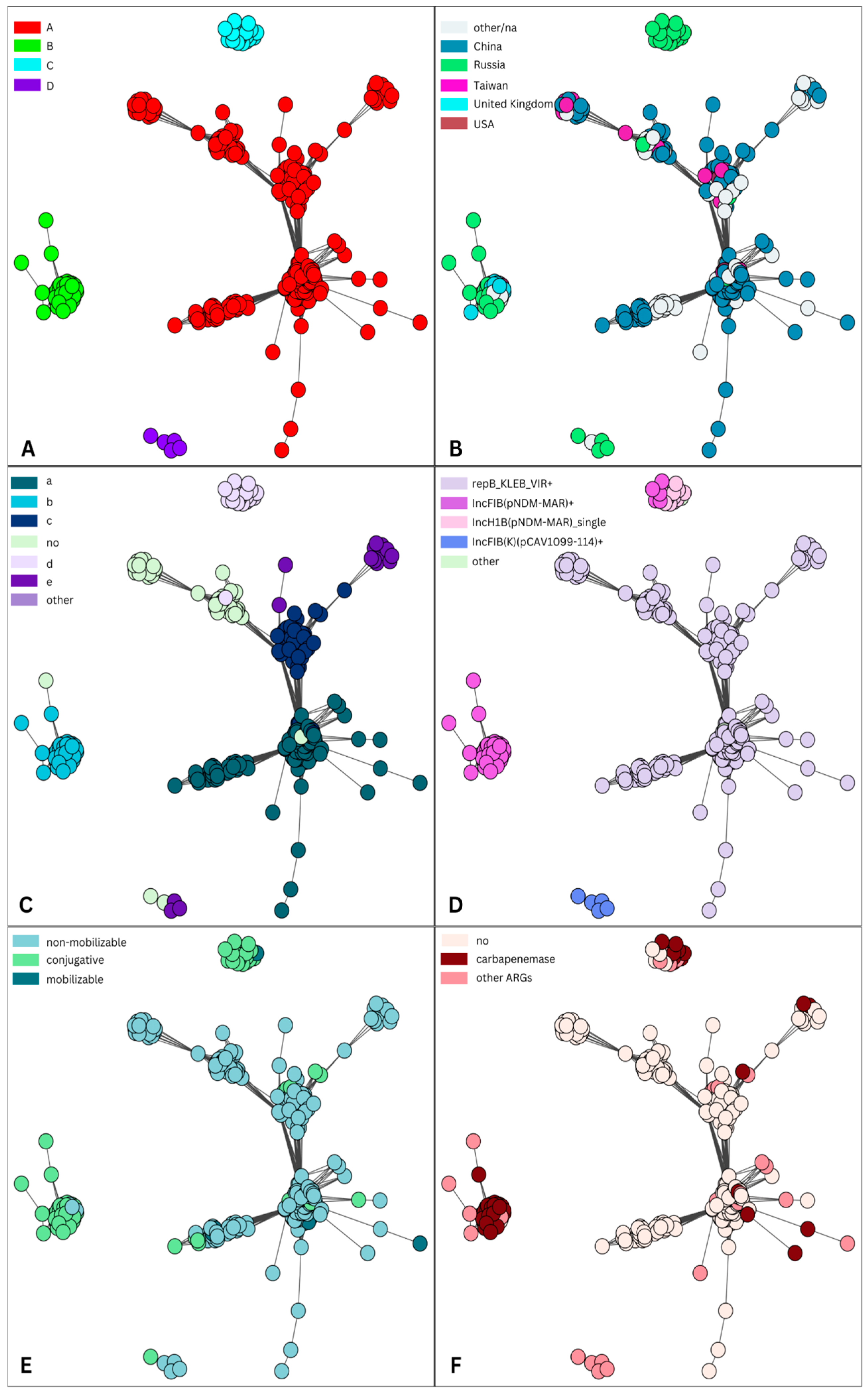
| 2 | 48 | Kpn, E. coli | IncFIB(pNDM-Mar), IncHI1B(pNDM-MAR) | iutA (44), iucA (44), iucB (44), iucD (44), iucC (43), rmpA2 (33), rmpA (26), peg-344 (26), iroBCDN (1) | blaNDM-1 (21), blaOXA-48 (9), blaNDM (1) | 31 | 41 | 44 | 38 | conjugative | 321,406 |
| 12 | 19 | Kpn, E. coli | IncR | - | blaNDM-5 (1), blaNDM-1 (2), blaKPC-3 (3) | 6 | 18 | 0 | 0 | conjugative | 51,882 |
| 5 | 13 | Kpn | ColRNAI | - | - | 0 | 0 | 0 | 0 | mobilizable | 9294 |
| 11 | 13 | Kpn, E. coli | Col(pHAD28) | - | - | 0 | 1 | 0 | 0 | non-mobilizable | 4915 |
| 7 | 12 | Kpn | IncL | - | blaOXA-48 (9), blaOXA-244 (3) | 12 | 12 | 0 | 0 | conjugative | 65,474 |
| 13 | 10 | Kpn | IncFIB(K) | - | - | 0 | 10 | 0 | 0 | conjugative | 196,937 |
| 4 | 9 | Kpn | IncFIB | - | - | 0 | 0 | 0 | 0 | non-mobilizable | 109,650 |
| 8 | 8 | Kpn, E. coli | Col(pHAD28), Col440II | - | - | 0 | 0 | 0 | 0 | mobilizable | 3511 |
| 16 | 7 | Kpn | IncFIA(HI1) | - | blaOXA-48 (1) | 1 | 1 | 0 | 0 | conjugative | 81,374 |
| 3 | 7 | Kpn, E. coli | Col(MG828) | - | - | 0 | 0 | 0 | 0 | non-mobilizable | 1549 |
| 15 | 6 | Kpn, E. coli | IncFII | iucABCD, iutA (1) | blaNDM-5 (1) | 1 | 6 | 1 | 1 | conjugative | 77,238 |
| 1 | 6 | Kpn | - | - | - | 0 | 0 | 0 | 0 | non-mobilizable | 5010 |
| 6 | 5 | Kpn | ColpVC | - | - | 0 | 0 | 0 | 0 | non-mobilizable | 1934 |
| 17 | 5 | Kpn | IncFIB(pQil) | - | - | 0 | 4 | 0 | 0 | conjugative | 141,904 |
| 0 | 5 | Kpn, E. coli | ColRNAI | - | - | 0 | 0 | 0 | 0 | mobilizable | 9716 |
| 14 | 4 | E. coli | IncFIA, IncI1-I(Alpha) | - | - | 0 | 1 | 0 | 0 | conjugative | 91,854 |
| 10 | 3 | Kpn, E. coli | Col(pHAD28) | - | - | 0 | 1 | 0 | 0 | mobilizable | 4428 |
| 9 | 3 | Kpn | IncFII | - | - | 0 | 3 | 0 | 0 | conjugative | 81,641 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shapovalova, V.V.; Chulkova, P.S.; Ageevets, V.A.; Nurmukanova, V.; Verentsova, I.V.; Girina, A.A.; Protasova, I.N.; Bezbido, V.S.; Sergevnin, V.I.; Feldblum, I.V.; et al. High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes. Antibiotics 2024, 13, 1224. https://doi.org/10.3390/antibiotics13121224
Shapovalova VV, Chulkova PS, Ageevets VA, Nurmukanova V, Verentsova IV, Girina AA, Protasova IN, Bezbido VS, Sergevnin VI, Feldblum IV, et al. High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes. Antibiotics. 2024; 13(12):1224. https://doi.org/10.3390/antibiotics13121224
Chicago/Turabian StyleShapovalova, Valeria V., Polina S. Chulkova, Vladimir A. Ageevets, Varvara Nurmukanova, Irina V. Verentsova, Asya A. Girina, Irina N. Protasova, Victoria S. Bezbido, Victor I. Sergevnin, Irina V. Feldblum, and et al. 2024. "High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes" Antibiotics 13, no. 12: 1224. https://doi.org/10.3390/antibiotics13121224
APA StyleShapovalova, V. V., Chulkova, P. S., Ageevets, V. A., Nurmukanova, V., Verentsova, I. V., Girina, A. A., Protasova, I. N., Bezbido, V. S., Sergevnin, V. I., Feldblum, I. V., Kudryavtseva, L. G., Sharafan, S. N., Semerikov, V. V., Babushkina, M. L., Valiullina, I. R., Chumarev, N. S., Isaeva, G. S., Belyanina, N. A., Shirokova, I. U., ... Sidorenko, S. V. (2024). High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes. Antibiotics, 13(12), 1224. https://doi.org/10.3390/antibiotics13121224