
Unveiling Hidden Allies: In Silico Discovery of Prophages in Tenacibaculum Species


Abstract
1. Introduction
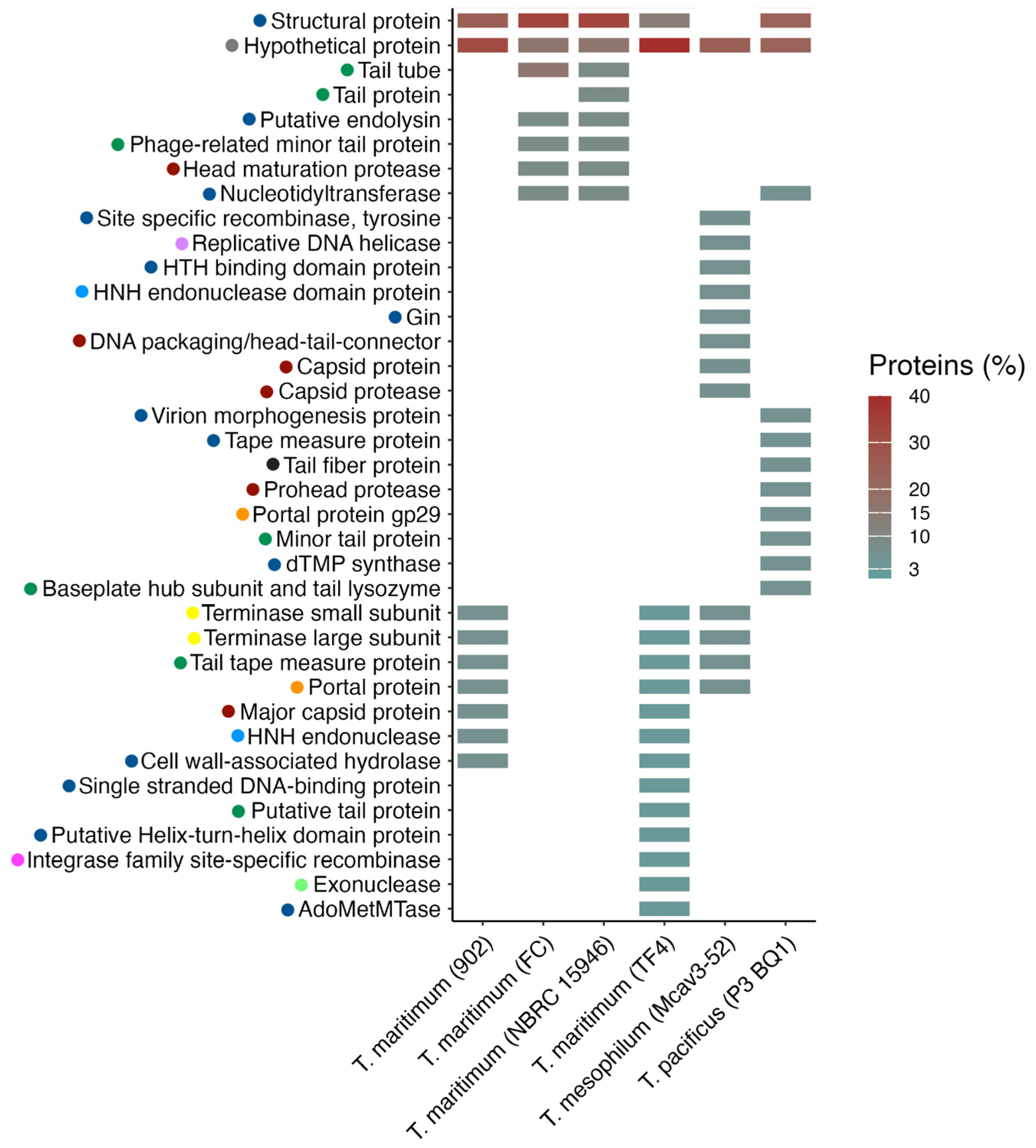
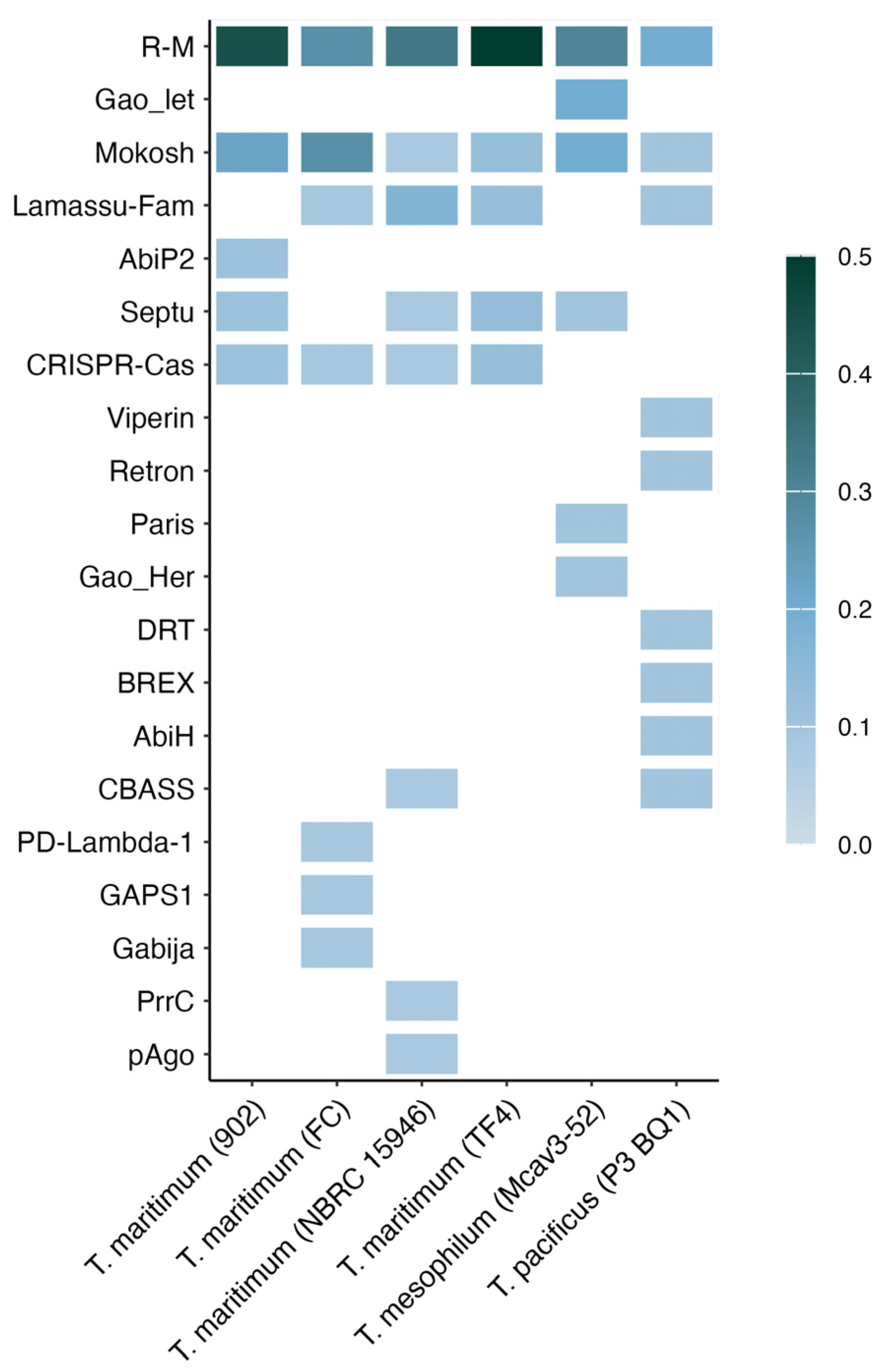
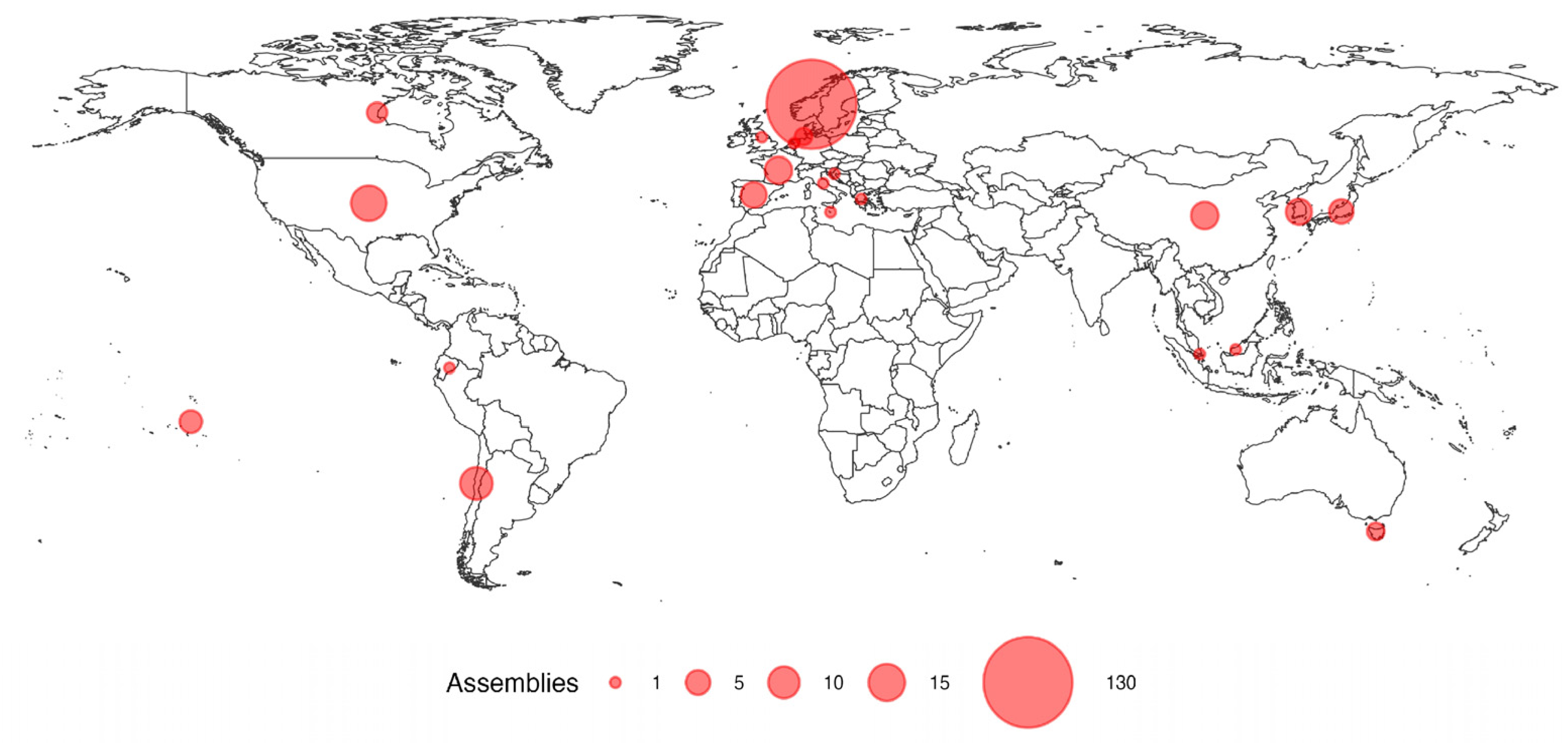
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mabrok, M.; Algammal, A.M.; Sivaramasamy, E.; Hetta, H.F.; Atwah, B.; Alghamdi, S.; Fawzy, A.; Avendaño-Herrera, R.; Rodkhum, C. Tenacibaculosis Caused by Tenacibaculum maritimum: Updated Knowledge of This Marine Bacterial Fish Pathogen. Front. Cell. Infect. Microbiol. 2023, 12, 1068000. [Google Scholar] [CrossRef] [PubMed]
- Nowlan, J.P.; Lumsden, J.S.; Russell, S. Advancements in characterizing Tenacibaculum infections in Canada. Pathogens 2020, 9, 1029. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Herrera, R.; Toranzo, A.E.; Magariños, B. Tenacibaculosis Infection in Marine Fish Caused by Tenacibaculum maritimum: A Review. Dis. Aquat. Organ. 2006, 71, 255–266. [Google Scholar] [CrossRef]
- Piñeiro-Vidal, M.; Gijón, D.; Zarza, C.; Santos, Y. Tenacibaculum dicentrarchi sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from European sea bass. Int. J. Syst. Evol. Microbiol. 2012, 62, 425–429. [Google Scholar] [CrossRef]
- Piñeiro-Vidal, M.; Riaza, A.; Santos, Y. Tenacibaculum discolor sp. nov. and Tenacibaculum gallaicum sp. nov., isolated from sole (Solea senegalensis) and turbot (Psetta maxima) culture systems. Int. J. Syst. Evol. Microbiol. 2008, 58, 21–25. [Google Scholar] [CrossRef]
- Suzuki, M.; Nakagawa, Y.; Harayama, S.; Yamamoto, S. Phylogenetic Analysis and Taxonomic Study of Marine Cytophaga-like Bacteria: Proposal for Tenacibaculum gen. nov. with Tenacibaculum maritimum comb. nov. and Tenacibaculum ovolyticum comb. nov., and Description of Tenacibaculum mesophilum sp. nov. and Tenacibaculum amylolyticum sp. nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 1639–1652. [Google Scholar] [CrossRef]
- Samsing, F.; Barnes, A.C. The Rise of the Opportunists: What Are the Drivers of the Increase in Infectious Diseases Caused by Environmental and Commensal Bacteria? Rev. Aquac. 2024, 16, 1787–1797. [Google Scholar] [CrossRef]
- Spilsberg, B.; Nilsen, H.K.; Tavornpanich, S.; Gulla, S.; Jansen, M.D.; Lagesen, K.; Colquhoun, D.J.; Olsen, A. Tenacibaculosis in Norwegian Atlantic Salmon (Salmo salar) Cage-Farmed in Cold Seawater Is Primarily Associated with Tenacibaculum finnmarkense Genomovar finnmarkense. J. Fish Dis. 2022, 45, 523–534. [Google Scholar] [CrossRef]
- Servicio Nacional de Pesca y Acuicultura. Informe con Antecedentes Sanitarios de Agua Dulce y Mar. Available online: https://www.sernapesca.cl/app/uploads/2024/09/Informe-Sanitario-ANO-2023.pdf (accessed on 25 October 2024).
- Available online: https://www.hipra.com/en/icthiovac-tm (accessed on 25 October 2024).
- Higuera, G.; Bastías, R.; Tsertsvadze, G.; Romero, J.; Espejo, R.T. Recently discovered Vibrio anguillarum phages can protect against experimentally induced vibriosis in Atlantic salmon, Salmo salar. Aquaculture 2013, 392–395, 128–133. [Google Scholar] [CrossRef]
- Kalatzis, P.G.; Bastías, R.; Kokkari, C.; Katharios, P. Isolation and Characterization of Two Lytic Bacteriophages, St2 and Grn1; Phage Therapy Application for Biological Control of Vibrio alginolyticus in Aquaculture Live Feeds. PLoS ONE 2016, 11, e0151101. [Google Scholar] [CrossRef]
- Katharios, P.; Kalatzis, P.G.; Kokkari, C.; Sarropoulou, E.; Middelboe, M. Isolation and characterization of a N4-like lytic bacteriophage infecting Vibrio splendidus, a pathogen of fish and bivalves. PLoS ONE 2017, 12, e0190083. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://stim.uk/r-d/bacteriophages/ (accessed on 25 October 2024).
- Tsertou, M.I.; Triga, A.; Droubogiannis, S.; Kokkari, C.; Anasi, G.; Katharios, P. Isolation and Characterization of a Novel Tenacibaculum Species and a Corresponding Bacteriophage from a Mediterranean Fish Hatchery: Description of Tenacibaculum larymnensis sp. nov. and Tenacibaculum Phage Larrie. Front. Microbiol. 2023, 14, 1078669. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Rocha, E.P.C.; Touchon, M. The Adaptation of Temperate Bacteriophages to Their Host Genomes. Mol. Biol. Evol. 2013, 30, 737–751. [Google Scholar] [CrossRef]
- Abad, L.; Gauthier, C.H.; Florian, I.; Jacobs-Sera, D.; Hatfull, G.F. The Heterogeneous and Diverse Population of Prophages in Mycobacterium Genomes. mSystems 2023, 8, e0044623. [Google Scholar] [CrossRef]
- Sharma, V.; Hünnefeld, M.; Luthe, T.; Frunzke, J. Systematic Analysis of Prophage Elements in Actinobacterial Genomes Reveals a Remarkable Phylogenetic Diversity. Sci. Rep. 2023, 13, 4410. [Google Scholar] [CrossRef]
- Butala, M.; Dragoš, A. Unique Relationships between Phages and Endospore-Forming Hosts. Trends Microbiol. 2023, 31, 498–510. [Google Scholar] [CrossRef]
- Wang, Z.; Peng, X.; Hülpüsch, C.; Khan Mirzaei, M.; Reiger, M.; Traidl-Hoffmann, C.; Deng, L.; Schloter, M. Distinct Prophage Gene Profiles of Staphylococcus aureus Strains from Atopic Dermatitis Patients and Healthy Individuals. Microbiol. Spectr. 2024, 12, e00915-24. [Google Scholar] [CrossRef]
- Szafrański, S.P.; Kilian, M.; Yang, I.; Bei der Wieden, G.; Winkel, A.; Hegermann, J.; Stiesch, M.; Szafrański, S.P.; Kilian, M.; Yang, I.; et al. Diversity Patterns of Bacteriophages Infecting Aggregatibacter and Haemophilus Species across Clades and Niches. ISME J. 2019, 13, 2500–2522. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef]
- Castillo, D.; Kauffman, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread Distribution of Prophage-Encoded Virulence Factors in Marine Vibrio Communities. Sci. Rep. 2018, 8, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Resch, G.; Pirnay, J.P.; Blasdel, B.G.; Debarbieux, L.; Gelman, D.; Górski, A.; Hazan, R.; Huys, I.; Kakabadze, E.; et al. Expert Opinion on Three Phage Therapy-Related Topics: Bacterial Phage Resistance, Phage Training, and Prophages in Bacterial Production Strains. Viruses 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- León, M.; Bastías, R. Virulence Reduction in Bacteriophage-Resistant Bacteria. Front. Microbiol. 2015, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- León, M.; Kokkari, C.; García, K.; Castillo, D.; Katharios, P.; Bastías, R. Diversification of Vibrio anguillarum Driven by the Bacteriophage CHOED. Front. Microbiol. 2019, 10, 1396. [Google Scholar] [CrossRef]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef]
- Wu, Y.; Garushyants, S.K.; van den Hurk, A.; Aparicio-Maldonado, C.; Kushwaha, S.K.; King, C.M.; Ou, Y.; Todeschini, T.C.; Clokie, M.R.; Millard, A.D.; et al. Bacterial Defense Systems Exhibit Synergistic Anti-Phage Activity. Cell Host Microbe 2024, 32, 557–572.e6. [Google Scholar] [CrossRef]
- Nasrullah, H.A.; Ahmed, S.; Rasool, M.; Shah, A.J. DNA Methylation across the Tree of Life, from Micro to Macro-Organism. Bioengineered 2022, 13, 1666–1685. [Google Scholar] [CrossRef]
- Millman, A.; Melamed, S.; Leavitt, A.; Doron, S.; Bernheim, A.; Hör, J.; Garb, J.; Bechon, N.; Brandis, A.; Lopatina, A.; et al. An Expanded Arsenal of Immune Systems That Protect Bacteria from Phages. Cell Host Microbe 2022, 30, 1556–1569.e5. [Google Scholar] [CrossRef]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs Guide Antiviral Defense in Prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef]
- Zhang, T.; Cepauskas, A.; Nadieina, A.; Thureau, A.; Coppieters ‘t Wallant, K.; Martens, C.; Lim, D.C.; Garcia-Pino, A.; Laub, M.T. A Bacterial Immunity Protein Directly Senses Two Disparate Phage Proteins. Nature 2024, 635, 728–735. [Google Scholar] [CrossRef] [PubMed]
- de Freitas Almeida, G.M.; Hoikkala, V.; Ravantti, J.; Rantanen, N.; Sundberg, L.R. Mucin Induces CRISPR-Cas Defense in an Opportunistic Pathogen. Nat. Commun. 2022, 13, 3653. [Google Scholar] [CrossRef] [PubMed]
- Almeida, G.M.F.; Laanto, E.; Ashrafi, R.; Sundberg, L.-R. Bacteriophage adherence to mucus mediates preventive protection against pathogenic bacteria. mBio 2019, 10, e01984-19. [Google Scholar] [CrossRef]
- Romero, J.; Blas-Chumacero, S.; Urzúa, V.; Villasante, A.; Opazo, R.; Gajardo, F.; Miranda, C.D.; Rojas, R. Lysin and Lytic Phages Reduce Vibrio Counts in Live Feed and Fish Larvae. Microorganisms 2024, 12, 904. [Google Scholar] [CrossRef]
- Cheng, G.; Hao, H.; Xie, S.; Wang, X.; Dai, M.; Huang, L.; Yuan, Z. Antibiotic alternatives: The substitution of antibiotics in animal husbandry? Front. Microbiol. 2014, 5, 217. [Google Scholar] [CrossRef]
- Nachimuthu, R.; Madurantakam Royam, M.; Manohar, P.; Leptihn, S. Application of bacteriophages and endolysins in aquaculture as a biocontrol measure. In Biological Control; Academic Press Inc.: Cambridge, MA, USA, 2021; Volume 160. [Google Scholar] [CrossRef]
- Horgan, M.; O’Flynn, G.; Garry, J.; Cooney, J.; Coffey, A.; Fitzgerald, G.; Ross, P.; McAuliffe, O. Phage Lysin LysK Can Be Truncated to Its CHAP Domain and Retain Lytic Activity against Live Antibiotic-Resistant Staphylococci. Appl. Environ. Microbiol. 2009, 75, 872–874. [Google Scholar] [CrossRef]
- Hjelm, L.C.; Nilvebrant, J.; Nygren, P.-Å.; Nilsson, A.S.; Seijsing, J. Lysis of Staphylococcal Cells by Modular Lysin Domains Linked via an Interaction Bridge. Front. Microbiol. 2019, 10, 558. [Google Scholar] [CrossRef]
- Li, M.; Jin, Y.; Lin, H.; Wang, J.; Jiang, X. Complete Genome of a Novel Lytic Vibrio parahaemolyticus Phage VPp1 and Characterization of Its Endolysin for Antibacterial Activities. J. Food Prot. 2018, 81, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Lal, T.M.; Sano, M.; Ransangan, J. Genome characterization of a novel vibriophage VpKK5 (Siphoviridae) specific to fish pathogenic strain of Vibrio parahaemolyticus. J. Basic Microbiol. 2016, 56, 872–888. [Google Scholar] [CrossRef]
- Matamp, N.; Bhat, S.G. Phage Endolysins as Potential Antimicrobials against Multidrug Resistant Vibrio alginolyticus and Vibrio parahaemolyticus: Current Status of Research and Challenges Ahead. Microorganisms 2019, 7, 84. [Google Scholar] [CrossRef]
- Ning, H.; Cong, Y.; Lin, H.; Wang, J. Development of cationic peptide chimeric lysins based on phage lysin Lysqdvp001 and their antibacterial effects against Vibrio parahaemolyticus: A preliminary study. Int. J. Food Microbiol. 2021, 358, 109396. [Google Scholar] [CrossRef]
- Wang, L.; Ju, X.; Cong, Y.; Lin, H.; Wang, J. A Single Catalytic Endolysin Domain Plychap001: Characterization and Application to Control Vibrio parahaemolyticus and Its Biofilm Directly. Foods 2022, 11, 1578. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage Endolysins as Novel Antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef]
- Briers, Y.; Walmagh, M.; Van Puyenbroeck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliveira, H.; Azeredo, J.; Verween, G.; Pirnay, J.-P.; et al. Engineered Endolysin-Based “Artilysins” to Combat Multidrug-Resistant Gram-Negative Pathogens. mBio 2014, 5, e01379-14. [Google Scholar] [CrossRef]
- Wishart, D.S.; Han, S.; Saha, S.; Oler, E.; Peters, H.; Grant, J.; Stothard, P.; Gautam, V. PHASTEST: Faster than PHASTER, Better than PHAST. Nucleic Acids Res. 2023, 51, W443–W450. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome Sequence-Based Species Delimitation with Confidence Intervals and Improved Distance Functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. bioRxiv 2022. [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2022, 50, D1–D13. [Google Scholar] [CrossRef]
- Kelley, L.; Mezulis, S.; Yates, C.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Tesson, F.; Hervé, A.; Mordret, E.; Touchon, M.; D’humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. Nat. Commun. 2022, 13, 2561. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species (Strain) | Region | Region Length | Completeness | Score | Total Proteins | Region Position | Most Common Phage | GC% |
|---|---|---|---|---|---|---|---|---|
| T. maritimum (TF4) | 1 | 50 Kb | Intact | 140 | 55 | 3079–53,132 | Cellul_phi19:1 | 30.29 |
| T. maritimum (FC) | 1 | 15.8 Kb | Intact | 100 | 22 | 13,262–29,093 | Flavob_vB_FspM_pippi8_1 | 33.2 |
| T. maritimum (NBRC 15946) | 1 | 15.8 Kb | Intact | 100 | 21 | 35,460–51,291 | Flavob_vB_FspM_lotta8_1 | 33.22 |
| T. maritimum (902) | 1 2 | 21.5 Kb 11.5 Kb | Intact Incomplete | 100 50 | 20 18 | 12,829–34,424 18,576–40,083 | Cellul_phi19:1 Flavob_vB_FspM_lotta8_1 | 29.99 31.6 |
| T. maritimum (JIP 32/91-4) | 1 | 17.2 Kb | Questionable | 80 | 12 | 31,735–48,966 | Cellul_phi19:1 | 30.34 |
| T. maritimum (P4-45) | 1 | 22.2 Kb | Questionable | 90 | 31 | 139–22,399 | Flavob_vB_FspM_pippi8_1 | 34.09 |
| T. maritimum (NVIB-2097) | 1 | 11.3 Kb | Incomplete | 50 | 18 | 1–11,355 | Flavob_vB_FspM_lotta8_1 | 31.61 |
| T. maritimum (NVIB-2096) | 1 | 11.3 Kb | Incomplete | 50 | 18 | 1–11,355 | Flavob_vB_FspM_pippi8_1 | 31.61 |
| T. maritimum (DPIF 89/0239-1) | 1 | 11 Kb | Incomplete | 50 | 16 | 74,959–86,020 | Cellul_phi46:1 | 31.79 |
| T. mesophilum (Mcav3-52) | 1 | 38 Kb | Intact | 110 | 45 | 360,314–398,338 | Cellul_phi39:1 | 31.95 |
| T. mesophilum (bac2) | 1 | 12.7 Kb | Questionable | 70 | 13 | 1,752,433–1,765,200 | Strept_BRock | 33.26 |
| T. mesophilum (DSM 13764) | 1 | 9.3 Kb | Questionable | 70 | 9 | 1,339,122–1,348,490 | Bacill_BCD7 | 33.93 |
| T. mesophilum (DSM 13764) | 1 | 9.3 Kb | Questionable | 70 | 9 | 2,013,166–2,022,534 | Strept_BRock | 33.93 |
| T. mesophilum (546.fasta) | 1 | 9.3 Kb | Incomplete | 60 | 9 | 31,278–40,645 | Strept_BRock | 33.85 |
| T. mesophilum (XPcli2-G) | 1 | 9.3 Kb | Incomplete | 60 | 9 | 84,611–93,978 | Bacill_BCD7 | 34 |
| T. mesophilum (SSh1-16) | 1 | 9.3 Kb | Incomplete | 60 | 9 | 31,113–40,481 | Bacill_BCD7 | 34 |
| T. pacificus (P3-BQ1) | 1 | 22 Kb | Intact | 110 | 31 | 2,677,157–2,699,185 | Flavob_vB_FspM_lotta8_1 | 30.2 |
| T. ovolyticum (20-4135-2) | 1 | 9.3 Kb | Questionable | 80 | 9 | 3,744,277–3,753,604 | Bacill_BCD7 | 31.89 |
| T. ovolyticum (To-7Br) | 1 | 9.3 Kb | Questionable | 70 | 9 | 21,823–31,151 | Strept_BRock | 31.79 |
| T. ovolyticum (da5A-8) | 1 | 9.3 Kb | Questionable | 70 | 9 | 80,624–89,952 | Strept_BRock | 31.76 |
| T. discolor (LAR25) | 1 | 7.4 Kb | Incomplete | 60 | 8 | 256,719–264,196 | Serrat_phiMAM1 | 32.23 |
| T. discolor (IMLK18) | 1 | 5.6 Kb | Incomplete | 60 | 6 | 338,872–344,494 | Synech_S_CBP3 | 30.82 |
| T. finnmarkense (NVIB-3194) | 1 | 16.7 Kb | Incomplete | 40 | 22 | 3–16,707 | Cellul_phiSM | 30.55 |
| T. dicentrarchi (TD3509T) | 1 | 9.3 Kb | Incomplete | 50 | 8 | 853,075–862,413 | Flavob_vB_FspM_pippi8_1 | 33.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, C.; Romero, J. Unveiling Hidden Allies: In Silico Discovery of Prophages in Tenacibaculum Species. Antibiotics 2024, 13, 1184. https://doi.org/10.3390/antibiotics13121184
Ramírez C, Romero J. Unveiling Hidden Allies: In Silico Discovery of Prophages in Tenacibaculum Species. Antibiotics. 2024; 13(12):1184. https://doi.org/10.3390/antibiotics13121184
Chicago/Turabian StyleRamírez, Carolina, and Jaime Romero. 2024. "Unveiling Hidden Allies: In Silico Discovery of Prophages in Tenacibaculum Species" Antibiotics 13, no. 12: 1184. https://doi.org/10.3390/antibiotics13121184
APA StyleRamírez, C., & Romero, J. (2024). Unveiling Hidden Allies: In Silico Discovery of Prophages in Tenacibaculum Species. Antibiotics, 13(12), 1184. https://doi.org/10.3390/antibiotics13121184