
Comprehensive Approaches to Combatting Acinetobacter baumannii Biofilms: From Biofilm Structure to Phage-Based Therapies

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Acinetobacter baumannii as a Priority Bacterial Pathogen
3. Mechanisms of Antibiotic Resistance in A. baumannii
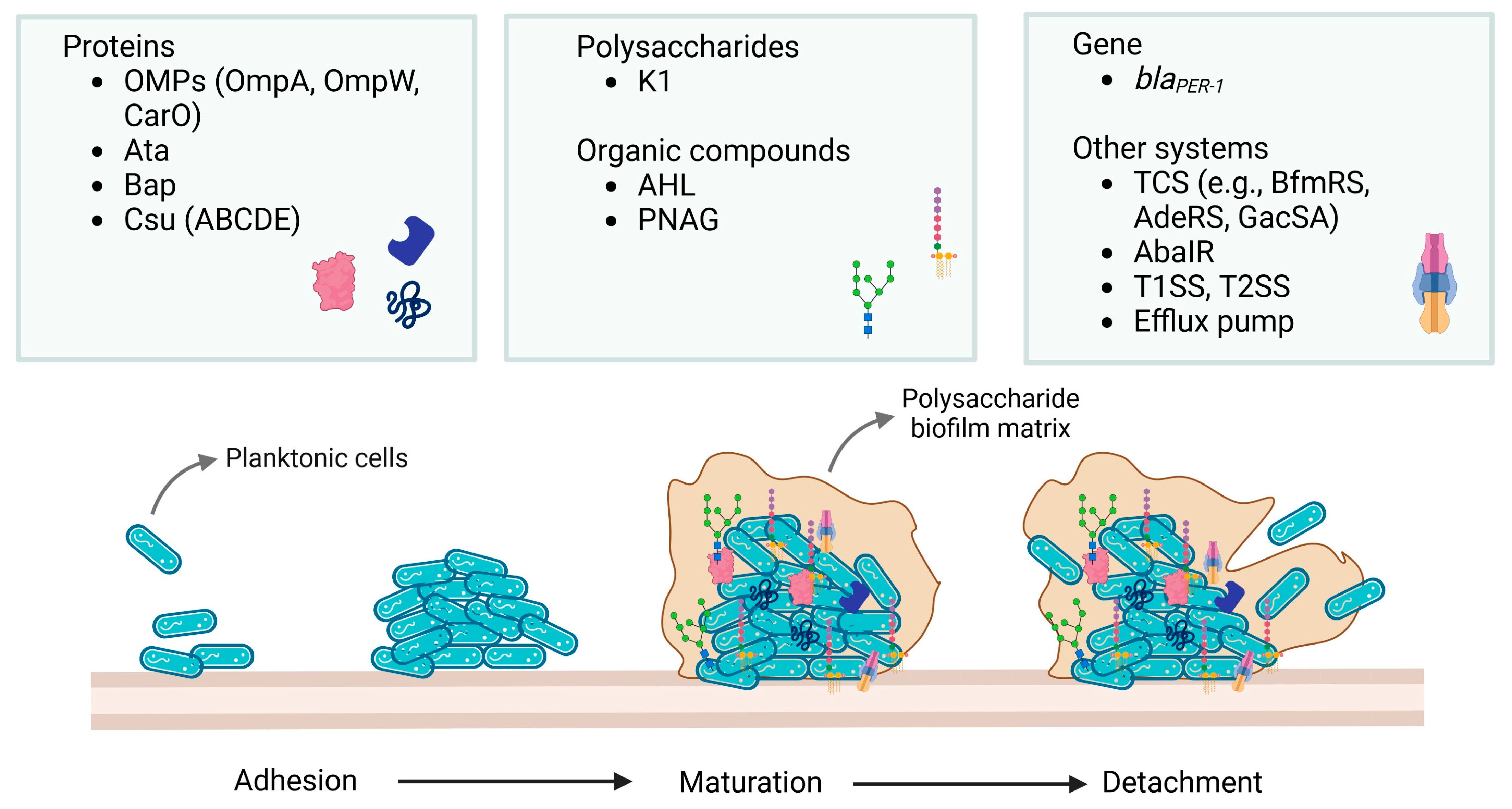
4. Biofilm—Structure and Development
4.1. Factors Responsible for Biofilm Formation by Acinetobacter baumannii Strains
4.2. Physicochemical Factors Influencing Biofilm Formation by A. baumannii
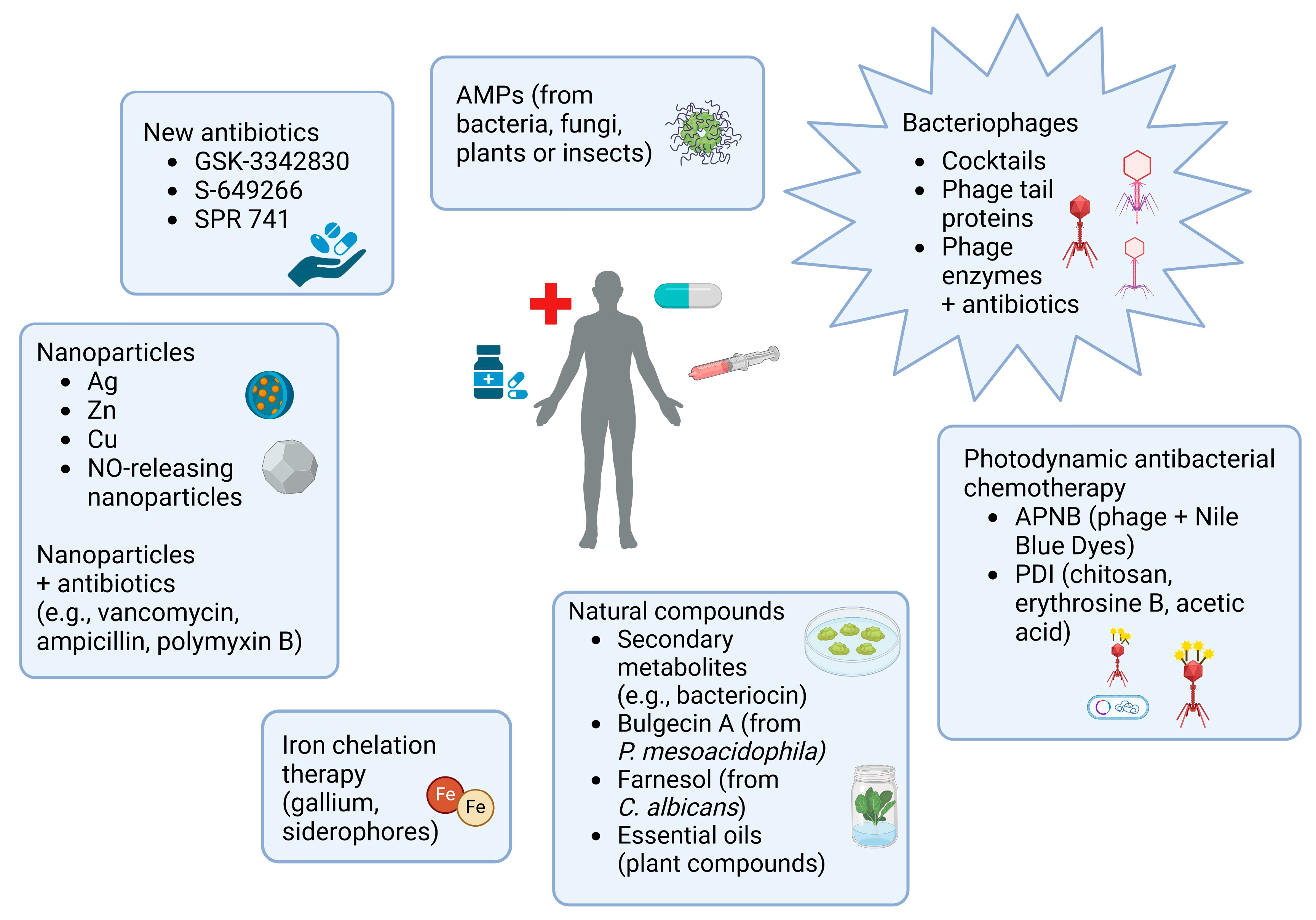
5. Strategies to Inhibit Biofilm Development
6. Phages and Their Use in Targeted Phage Therapy
7. Bacteriophages’ Activity Against Biofilm-Forming by Acinetobacter baumannii
8. Anti-Biofilm Activity of Phage-Derived Enzymes
9. Alternative Treatments for Infections Caused by A. baumannii Biofilm
10. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gedefie, A.; Demsis, W.; Ashagrie, M.; Kassa, Y.; Tesfaye, M.; Tilahun, M.; Bisetegn, H.; Sahle, Z. Acinetobacter baumannii Biofilm Formation and Its Role in Disease Pathogenesis: A Review. Infect. Drug Resist. 2021, 14, 3711. [Google Scholar] [CrossRef]
- Francey, T.; Gaschen, F.; Nicolet, J.; Burnens, A.P. The role of Acinetobacter baumannii as a nosocomial pathogen for dogs and cats in an intensive care unit. J. Vet. Intern. Med. 2000, 14, 177–183. [Google Scholar] [CrossRef]
- Vaneechoutte, M.; Devriese, L.A.; Dijkshoorn, L.; Lamote, B.; Deprez, P.; Verschraegen, G.; Haesebrouck, F. Acinetobacter baumannii-infected vascular catheters collected from horses in an equine clinic. J. Clin. Microbiol. 2000, 38, 4280–4281. [Google Scholar] [CrossRef]
- Allen, J.L. Multifaceted Approach to Understanding Acinetobacter baumannii Biofilm Formation and Drug Resistance. Doctoral Dissertation, University of South Florida, Tampa, FL, USA, 2021. [Google Scholar]
- Colquhoun, J.M.; Rather, P.N. Insights Into Mechanisms of Biofilm Formation in Acinetobacter baumannii and Implications for Uropathogenesis. Front. Cell. Infect. Microbiol. 2020, 10, 253. [Google Scholar] [CrossRef]
- Monem, S.; Furmanek-Blaszk, B.; Łupkowska, A.; Kuczyńska-Wiśnik, D.; Stojowska-Swędrzyńska, K.; Laskowska, E. Mechanisms Protecting Acinetobacter baumannii against Multiple Stresses Triggered by the Host Immune Response, Antibiotics and Outside-Host Environment. Int. J. Mol. Sci. 2020, 21, 5498. [Google Scholar] [CrossRef]
- Eze, E.C.; Chenia, H.Y.; El Zowalaty, M.E. Acinetobacter baumannii biofilms: Effects of physicochemical factors, virulence, antibiotic resistance determinants, gene regulation, and future antimicrobial treatments. Infect. Drug Resist. 2018, 11, 2277–2299. [Google Scholar] [CrossRef]
- Berk, S.L.; McCabe, W.R. Meningitis caused by Acinetobacter calcoaceticus var anitratus. A specific hazard in neurosurgical patients. Arch. Neurol. 1981, 38, 95–98. [Google Scholar] [CrossRef]
- Seifert, H.; Dijkshoorn, L.; Gerner-Smidt, P.; Pelzer, N.; Tjernberg, I.; Vaneechoutte, M. Distribution of Acinetobacter species on human skin: Comparison of phenotypic and genotypic identification methods. J. Clin. Microbiol. 1997, 35, 2819–2825. [Google Scholar] [CrossRef]
- Wintachai, P.; Phaonakrop, N.; Roytrakul, S.; Naknaen, A.; Pomwised, R.; Voravuthikunchai, S.P.; Surachat, K.; Smith, D.R. Enhanced antibacterial effect of a novel Friunavirus phage vWU2001 in combination with colistin against carbapenem-resistant Acinetobacter baumannii. Sci. Rep. 2022, 12, 2633. [Google Scholar] [CrossRef]
- World Health Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance. 2024. Available online: https://www.who.int/publications/i/item/9789240093461 (accessed on 8 October 2024).
- Kisil, O.V.; Efimenko, T.A.; Gabrielyan, N.I.; Efremenkova, O.V. Development of antimicrobial therapy methods to overcome the antibiotic resistance of Acinetobacter baumannii. Acta Nat. 2020, 12, 34–45. [Google Scholar] [CrossRef]
- Ophir, T.; Gutnick, D.L. A role for exopolysaccharides in the protection of microorganisms from desiccation. Appl. Environ. Microbiol. 1994, 60, 740–745. [Google Scholar] [CrossRef]
- Hassan, K.A.; Jackson, S.M.; Penesyan, A.; Patching, S.G.; Tetu, S.G.; Eijkelkamp, B.A.; Brown, M.H.; Henderson, P.J.F.; Paulsen, I.T. Transcriptomic and biochemical analyses identify a family of chlorhexidine efflux proteins. Proc. Natl. Acad. Sci. USA 2013, 110, 20254–20259. [Google Scholar] [CrossRef]
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef]
- Ma, C.; McClean, S. Mapping Global Prevalence of Acinetobacter baumannii and Recent Vaccine Development to Tackle It. Vaccines 2021, 9, 570. [Google Scholar] [CrossRef]
- Mączyńska, B.; Frej-Mądrzak, M.; Sarowska, J.; Woronowicz, K.; Choroszy-Król, I.; Jama-Kmiecik, A. Evolution of Antibiotic Resistance in Escherichia coli and Klebsiella pneumoniae Clinical Isolates in a Multi-Profile Hospital over 5 Years (2017–2021). J. Clin. Med. 2023, 12, 2414. [Google Scholar] [CrossRef]
- de Oliveira, B.F.D.; Freitas-Silva, J.; Sánchez-Robinet, C.; Laport, M.S. Transmission of the sponge microbiome: Moving towards a unified model. Environ. Microbiol. Rep. 2020, 12, 619–638. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Langford, B.J.; Soucy, J.-P.R.; Leung, V.; So, M.; Kwan, A.T.H.; Portnoff, J.S.; Bertagnolio, S.; Raybardhan, S.; MacFadden, D.R.; Daneman, N. Antibiotic Resistance Associated with the COVID-19 Pandemic: A Systematic Review and Meta-Analysis. Clin. Microbiol. Infect. 2023, 29, 302–309. [Google Scholar] [CrossRef]
- Qiu, H.; Kuolee, R.; Harris, G.; Chen, W. Role of NADPH Phagocyte oxidase in host defense against acute respiratory Acinetobacter baumannii infection in mice. Infect. Immun. 2009, 77, 1015–1021. [Google Scholar] [CrossRef]
- Sanchez-Larrayoz, A.F.; Elhosseiny, N.M.; Chevrette, M.G.; Fu, Y.; Giunta, P.; Spallanzani, R.G.; Ravi, K.; Pier, G.B.; Lory, S.; Maira-Litrán, T. Complexity of Complement Resistance Factors Expressed by Acinetobacter baumannii Needed for Survival in Human Serum. J. Immunol. 2017, 199, 2803–2814. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; van Strijp, J.A. Bacterial complement evasion. Mol. Immunol. 2007, 44, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef]
- Wong, D.; Nielsen, T.B.; Bonomo, R.A.; Pantapalangkoor, P.; Luna, B.; Spellberg, B. Clinical and Pathophysiological Overview of Acinetobacter Infections: A Century of Challenges. Clin. Microbiol. Rev. 2017, 30, 409–447. [Google Scholar] [CrossRef]
- Fournier, P.E.; Vallenet, D.; Barbe, V.; Audic, S.; Ogata, H.; Poirel, L.; Richet, H.; Robert, C.; Mangenot, S.; Abergel, C.; et al. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2006, 2, e7. [Google Scholar] [CrossRef]
- Lai, K.K.; Wang, S.; Kuk, A.K.; Tsang, A.; Tai, J.H.; Ko, C.K. Acinetobacter baumannii–calcoaceticus Complex-associated Orbital Cellulitis: A Case Report and Literature Review. Ocul. Immunol. Inflamm. 2023, 31, 1537–1540. [Google Scholar] [CrossRef] [PubMed]
- Gavrilovska, A.D.; Veljanovski, H.; Jovchevski, R. Successful treatment of pan-drug resistant Acinetobacter baumannii meningitis/ventriculitis following craniotomy and external ventricular drainage: A case report. J. Surg. Case Rep. 2024, 2024, rjae603. [Google Scholar] [CrossRef] [PubMed]
- Dagher, M.; Ruffin, F.; Marshall, S.; Taracila, M.; Bonomo, R.A.; Reilly, R.; Fowler, V.G.; Thaden, J.T. Case Report: Successful Rescue Therapy of Extensively Drug-Resistant Acinetobacter baumannii Osteomyelitis with Cefiderocol. Open Forum Infect. Dis. 2020, 7, ofaa150. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Corvec, S.; Caroff, N.; Espaze, E.; Giraudeau, C.; Drugeon, H.; Reynaud, A. AmpC cephalosporinase hyperproduction in Acinetobacter baumannii clinical strains. J. Antimicrob. Chemother. 2003, 52, 629–635. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef]
- Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Antibiotic resistance mechanisms of clinically important bacteria. Medicina 2011, 47, 137–146. [Google Scholar] [CrossRef]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-beta-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef]
- Philippon, L.N.; Naas, T.; Bouthors, A.T.; Barakett, V.; Nordmann, P. OXA-18, a class D clavulanic acid-inhibited extended-spectrum beta-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1997, 41, 2188–2195. [Google Scholar] [CrossRef]
- Poirel, L.; Nordmann, P. Carbapenem resistance in Acinetobacter baumannii: Mechanisms and epidemiology. Clin. Microbiol. Infect. 2006, 12, 826–836. [Google Scholar] [CrossRef]
- Mussi, M.A.; Limansky, A.S.; Viale, A.M. Acquisition of resistance to carbapenems in multidrug-resistant clinical strains of Acinetobacter baumannii: Natural insertional inactivation of a gene encoding a member of a novel family of β-barrel outer membrane proteins. Antimicrob. Agents Chemother. 2005, 49, 1432–1440. [Google Scholar] [CrossRef]
- Fernández-Cuenca, F.; Martínez-Martínez, L.; Conejo, M.C.; Ayala, J.A.; Perea, E.J.; Pascual, A. Relationship between β-lactamase production, outer membrane protein, and penicillin-binding protein profiles on the activity of carbapenems against clinical isolates of Acinetobacter baumannii. J. Antimicrob. Chemother. 2003, 51, 565–574. [Google Scholar] [CrossRef]
- Sharifzadeh, S.; Brown, N.W.; Shirley, J.D.; Bruce, K.E.; Winkler, M.E.; Carlson, E.E. Chemical tools for selective activity profiling of bacterial penicillin-binding proteins. Methods Enzymol. 2020, 638, 27–55. [Google Scholar]
- Pal, S.; Ghosh, A.S. PBP isolation and DD-carboxypeptidase assay. Methods Mol. Biol. 2019, 1946, 207–225. [Google Scholar]
- Cayô, R.; Rodríguez, M.C.; Espinal, P.; Fernández-Cuenca, F.; Ocampo-Sosa, A.A.; Pascual, Á.; Ayala, J.A.; Vila, J.; Martínez-Martínez, L. Analysis of genes encoding penicillin-binding proteins in clinical isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2011, 55, 5907–5913. [Google Scholar] [CrossRef]
- Nogbou, N.D.; Nkawane, G.M.; Ntshane, K.; Wairuri, C.K.; Phofa, D.T.; Mokgokong, K.K.; Ramashia, M.; Nchabeleng, M.; Obi, L.C.; Musyoki, A.M. Efflux Pump Activity and Mutations Driving Multidrug Resistance in Acinetobacter baumannii at a Tertiary Hospital in Pretoria, South Africa. Int. J. Microbiol. 2021, 2021, 9923816. [Google Scholar] [CrossRef]
- Magnet, S.; Courvalin, P.; Lambert, T. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 2001, 45, 3375–3380. [Google Scholar] [CrossRef]
- Su, X.Z.; Chen, J.; Mizushima, T.; Kuroda, T.; Tsuchiya, T. AbeM, an H+-coupled Acinetobacter baumannii multidrug efflux pump belonging to the MATE family of transporters. Antimicrob. Agents Chemother. 2005, 49, 4362–4364. [Google Scholar] [CrossRef]
- Ribera, A.; Ruiz, J.; Jiminez de Anta, M.T.; Vila, J. Effect of an efflux pump inhibitor on the MIC of nalidixic acid for Acinetobacter baumannii and Stenotrophomonas maltophilia clinical isolates. J. Antimicrob. Chemother. 2002, 49, 697–698. [Google Scholar] [CrossRef]
- Pérez-Varela, M.; Corral, J.; Aranda, J.; Barbé, J. Functional Characterization of AbaQ, a Novel Efflux Pump Mediating Quinolone Resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e00906–e00918. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Potoski, B.A.; Rea, R.; Adams, J.; Sethi, J.; Capitano, B.; Husain, S.; Kwak, E.J.; Bhat, S.V.; Paterson, D.L. Acinetobacter baumannii bloodstream infection while receiving tigecycline: A cautionary report. J. Antimicrob. Chemother. 2007, 59, 128–131. [Google Scholar] [CrossRef]
- Guardabassi, L.; Dijkshoorn, L.; Collard, J.-M.; Olsen, J.; Dalsgaard, A. Distribution and in-vitro transfer of tetracycline resistance determinants in clinical and aquatic Acinetobacter strains. J. Med. Microbiol. 2000, 49, 929–936. [Google Scholar] [CrossRef]
- Dolma, K.G.; Khati, R.; Paul, A.K.; Rahmatullah, M.; de Lourdes Pereira, M.; Wilairatana, P.; Khandelwal, B.; Gupta, C.; Gautam, D.; Gupta, M.; et al. Virulence Characteristics and Emerging Therapies for Biofilm-Forming Acinetobacter baumannii: A Review. Biology 2022, 11, 1343. [Google Scholar] [CrossRef]
- Song, J.Y.; Cheong, H.J.; Noh, J.Y.; Kim, W.J. In Vitro Comparison of Anti-Biofilm Effects against Carbapenem-Resistant Acinetobacter baumannii: Imipenem, Colistin, Tigecycline, Rifampicin and Combinations. Infect. Chemother. 2015, 47, 27. [Google Scholar] [CrossRef]
- Vukotic, G.; Obradovic, M.; Novovic, K.; Di Luca, M.; Jovcic, B.; Fira, D.; Neve, H.; Kojic, M.; McAuliffe, O. Characterization, Antibiofilm, and Depolymerizing Activity of Two Phages Active on Carbapenem-Resistant Acinetobacter baumannii. Front. Med. 2020, 7, 426. [Google Scholar] [CrossRef]
- Królasik, J.; Żakowska, Z.; Krępska, M.; Klimek, L.; Szosland-Fałtyn, A. Dynamika tworzenia biofilmów bakteryjnych na materiałach konstrukcyjnych linii technologicznych w przemyśle spożywczym [The dynamics of biofilm formation on structural materials of processing lines in food industry]. Postępy Nauk. Technol. Przemysłu Rolno-Spożywczego 2011, 66, 74–85. [Google Scholar]
- Shahed-Al-Mahmud, M.; Roy, R.; Sugiokto, F.G.; Islam, M.N.; Lin, M.-D.; Lin, L.-C.; Lin, N.-T. Phage φAB6-borne depolymerase combats Acinetobacter baumannii biofilm formation and infection. Antibiotics 2021, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Shareef, S.M.; Risan, M.H. Molecular identification of epsA and ompA genes for Acinetobacter baumannii Isolated from Burn Wounds. Indian J. Forensic Med. Toxicol. 2021, 15, 2044. [Google Scholar]
- Vidal, R.; Dominguez, M.; Urrutia, H.; Bello, H.; Gonzalez, G.; Garcia, A.; Zemelman, R. Biofilm formation by Acinetobacter baumannii. Microbios 1996, 86, 49–58. [Google Scholar]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival Mechanisms of Clinically Relevant Microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef]
- Marti, S.; Rodriguez-Bano, J.; Catel-Ferreira, M.; Jouenne, T.; Vila, J.; Seifert, H.; Dé, E. Biofilm formation at the solid-liquid and air-liquid interfaces by Acinetobacter species. BMC Res. Notes 2011, 4, 5. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial biofilm and associated infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Kaya, E.; Grassi, L.; Benedetti, A.; Maisetta, G.; Pileggi, C.; Di Luca, M.; Batoni, G.; Esin, S. In vitro interaction of Pseudomonas aeruginosa biofilms with human peripheral blood mononuclear cells. Front. Cell. Infect. Microbiol. 2020, 10, 187. [Google Scholar] [CrossRef]
- Hathroubi, S.; Mekni, M.A.; Domenico, P.; Nguyen, D.; Jacques, M. Biofilms: Microbial shelters against antibiotics. Microb. Drug Resist. 2017, 23, 147–156. [Google Scholar] [CrossRef]
- Choudhary, M.; Shrivastava, R.; Vashistt, J. Acinetobacter baumannii Biofilm Formation: Association with Antimicrobial Resistance and Prolonged Survival under Desiccation. Curr. Microbiol. 2022, 79, 361. [Google Scholar] [CrossRef] [PubMed]
- Mendes, S.G.; Combo, S.I.; Allain, T.; Domingues, S.; Buret, A.G.; Da Silva, G.J. Co-regulation of biofilm formation and antimicrobial resistance in Acinetobacter baumannii: From mechanisms to therapeutic strategies. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 1405–1423. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.S.; Burmolle, M.; Hansen, L.H.; Sorensen, S.J. The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol. 2012, 65, 183–195. [Google Scholar] [CrossRef]
- Penesyan, A.; Nagy, S.S.; Kjelleberg, S.; Gillings, M.R.; Paulsen, I.T. Rapid microevolution of biofilm cells in response to antibiotics. NPJ Biofilms Microbiomes 2019, 5, 34. [Google Scholar] [CrossRef]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Stone, G.; Wood, P.; Dixon, L.; Keyhan, M.; Matin, A. Tetracycline rapidly reaches all the constituent cells of uropathogenic Escherichia coli biofilms. Antimicrob. Agents Chemother. 2002, 46, 2458–2461. [Google Scholar] [CrossRef]
- Shigeta, M.; Tanaka, G.; Komatsuzawa, H.; Sugai, M.; Suginaka, H.; Usui, T. Permeation of antimicrobial agents through Pseudomonas aeruginosa biofilms: A simple method. Chemotherapy 1997, 43, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Spitz, O.; Erenburg, I.N.; Beer, T.; Kanonenberg, K.; Holland, I.B.; Schmitt, L. Type I Secretion Systems-One Mechanism for All? Microbiol. Spectr. 2019, 7, 81–102. [Google Scholar]
- Morris, F.C.; Dexter, C.; Kostoulias, X.; Uddin, M.I.; Peleg, A.Y. The Mechanisms of Disease Caused by Acinetobacter baumannii. Front. Microbiol. 2019, 10, 1601. [Google Scholar] [CrossRef]
- Loehfelm, T.W.; Luke, N.R.; Campagnari, A.A. Identification and characterization of an Acinetobacter baumannii biofilm-associated protein. J. Bacteriol. 2008, 190, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.M.; Pulido, M.R.; Di Venanzio, G.; Kinsella, R.L.; Webb, A.I.; Scott, N.E.; Pachón, J.; Feldman, M.F. Pathogenic Acinetobacter species have a functional type I secretion system and contact-dependent inhibition systems. J. Biol. Chem. 2017, 292, 9075–9087. [Google Scholar] [CrossRef]
- Goh, H.M.S.; Beatson, S.A.; Totsika, M.; Moriel, D.G.; Phan, M.-D.; Szubert, J.; Runnegar, N.; Sidjabat, H.E.; Paterson, D.L.; Nimmo, G.R.; et al. Molecular analysis of the Acinetobacter baumannii biofilm-associated protein. Appl. Environ. Microbiol. 2013, 79, 6535–6543. [Google Scholar] [CrossRef]
- De Gregorio, E.; Del Franco, M.; Roscetto, M.; Zarrilli, R.; Di Nocera, P.P. Biofilm-associated proteins: News from Acinetobacter. BMC Genom. 2015, 16, 933. [Google Scholar] [CrossRef]
- Bentancor, L.V.; Camacho-Peiro, A.; Bozkurt-Guzel, C.; Pier, G.B.; Maira-Litrán, T. Identification of Ata, a multifunctional trimeric autotransporter of Acinetobacter baumannii. J. Bacteriol. 2012, 194, 3950–3960. [Google Scholar] [CrossRef]
- Jacoby, G.A. β-Lactamase Nomenclature. Antimicrob. Agents Chemother. 2006, 50, 1123–1129. [Google Scholar] [CrossRef]
- Lai, S.J.; Tu, I.F.; Tseng, T.S.; Tsai, Y.H.; Wu, S.H. The deficiency of poly-β-1, 6-N-acetyl-glucosamine deacetylase trigger A. baumannii to convert to biofilm-independent colistin-tolerant cells. Sci. Rep. 2023, 13, 2800. [Google Scholar] [CrossRef]
- Richmond, G.E.; Evans, L.P.; Anderson, M.J.; Wand, M.E.; Bonney, L.C.; Ivens, A.; Chua, K.L.; Webber, M.A.; Mark Sutton, J.; Peterson, M.L.; et al. The Acinetobacter baumannii two-component system AdeRS regulates genes required for multidrug efflux, biofilm formation, and virulence in a strain-specific manner. mBio 2016, 7, e00430-16. [Google Scholar] [CrossRef]
- Cerqueira, G.M.; Kostoulias, X.; Khoo, C.; Aibinu, I.; Qu, Y.; Traven, A.; Peleg, A.Y. A global virulence regulator in Acinetobacter baumannii and its control of the phenylacetic acid catabolic pathway. J. Infect. Dis. 2014, 210, 46–55. [Google Scholar] [CrossRef]
- Tomaras, A.P.; Flagler, M.J.; Dorsey, C.W.; Gaddy, J.A.; Actis, L.A. Characterization of a two-component regulatory system from Acinetobacter baumannii that controls biofilm formation and cellular morphology. Microbiol. Read. Engl. 2008, 154, 3398–3409. [Google Scholar] [CrossRef]
- Krasauskas, R.; Skerniškytė, J.; Armalytė, J.; Sužiedėlienė, E. The role of Acinetobacter baumannii response regulator BfmR in pellicle formation and competitiveness via contact-dependent inhibition system. BMC Microbiol. 2019, 19, 241. [Google Scholar] [CrossRef] [PubMed]
- Perez Mora, B.; Giordano, R.; Permingeat, V.; Calderone, M.; Arana, N.; Müller, G.; Rodríguez, R.E.; Krasauskas, R.; Mussi, M.A. BfmRS encodes a regulatory system involved in light signal transduction modulating motility and desiccation tolerance in the human pathogen Acinetobacter baumannii. Sci. Rep. 2023, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ni, Z.; Tang, J.; Ding, Y.; Wang, X.; Li, F. The abaI/abaR Quorum Sensing System Effects on Pathogenicity in Acinetobacter baumannii. Front. Microbiol. 2021, 12, 679241. [Google Scholar] [CrossRef] [PubMed]
- Modarresi, F.; Azizi, O.; Shakibaie, M.R.; Motamedifar, M.; Mosadegh, E.; Mansouri, S. Iron limitation enhances acyl homoserine lactone (AHL) production and biofilm formation in clinical isolates of Acinetobacter baumannii. Virulence 2015, 6, 152–161. [Google Scholar] [CrossRef]
- Liou, M.-L.; Soo, P.-C.; Ling, S.-R.; Kuo, H.-Y.; Tang, C.Y.; Chang, K.-C. The sensor kinase BfmS mediates virulence in Acinetobacter baumannii. J. Microbiol. Immunol. Infect. 2014, 47, 275–281. [Google Scholar] [CrossRef]
- Smani, Y.; Fàbrega, A.; Roca, I.; Sańchez-Encinales, V.; Vila, J.; Pachón, J. Role of OmpA in the multidrug resistance phenotype of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2014, 58, 1806–1808. [Google Scholar] [CrossRef]
- Nie, D.; Hu, Y.; Chen, Z.; Li, M.; Hou, Z.; Luo, X.; Mao, X.; Xue, X. Outer membrane protein A (OmpA) as a potential therapeutic target for Acinetobacter baumannii infection. J. Biomed. Sci. 2020, 27, 26. [Google Scholar] [CrossRef]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2022, 46, fuab048. [Google Scholar] [CrossRef]
- Shih, P.C.; Huang, C.T. Effects of quorum-sensing deficiency on Pseudomonas aeruginosa biofilm formation and antibiotic resistance. J. Antimicrob. Chemother. 2002, 49, 309–314. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Park, W. Contribution of quorum-sensing system to hexadecane degradation and biofilm formation in Acinetobacter sp. strain DR1. J. Appl. Microbiol. 2010, 109, 1650–1659. [Google Scholar] [CrossRef]
- Chow, J.Y.; Yang, Y.; Tay, S.B.; Chua, K.L.; Yew, W.S. Disruption of biofilm formation by the human pathogen Acinetobacter baumannii using engineered quorum-quenching lactonases. Antimicrob. Agents Chemother. 2014, 58, 1802–1805. [Google Scholar] [CrossRef]
- De Silva, P.M.; Chong, P.; Fernando, D.M.; Westmacott, G.; Kumar, A. Effect of incubation temperature on antibiotic resistance and virulence factors of Acinetobacter baumannii ATCC 17978. Antimicrob. Agents Chemother. 2018, 62, e01514–e01517. [Google Scholar] [CrossRef]
- Sato, Y.; Ubagai, T.; Tansho-Nagakawa, S.; Yoshino, Y.; Ono, Y. Effects of colistin and tigecycline on multidrug-resistant Acinetobacter baumannii biofilms: Advantages and disadvantages of their combination. Sci. Rep. 2021, 11, 11700. [Google Scholar] [CrossRef]
- Gentile, V.; Frangipani, E.; Bonchi, C.; Minandri, F.; Runci, F.; Visca, P. Iron and Acinetobacter baumannii Biofilm Formation. Pathogens 2014, 3, 704–719. [Google Scholar] [CrossRef]
- Smitran, A.; Lukovic, B.; Bozic, L.; Jelic, D.; Jovicevic, M.; Kabic, J.; Kekic, D.; Ranin, J.; Opavski, N.; Gajic, I. Carbapenem-Resistant Acinetobacter baumannii: Biofilm-Associated Genes, Biofilm-Eradication Potential of Disinfectants, and Biofilm-Inhibitory Effects of Selenium Nanoparticles. Microorganisms 2023, 11, 171. [Google Scholar] [CrossRef]
- Mussi, M.A.; Gaddy, J.A.; Cabruja, M.; Arivett, B.A.; Viale, A.M.; Rasia, R.; Actis, L.A. The Opportunistic Human Pathogen Acinetobacter baumannii Senses and Responds to Light. J. Bacteriol. 2010, 192, 6336–6345. [Google Scholar] [CrossRef]
- Greene, C.; Wu, J.; Rickard, A.H.; Xi, C. Evaluation of the ability of Acinetobacter baumannii to form biofilms on six different biomedical relevant surfaces. Lett. Appl. Microbiol. 2016, 63, 233–239. [Google Scholar] [CrossRef]
- Hendiani, S.; Ahya Abdi, A.; Mohammadi, P.; Kharrazi, S. Synthesis of silver nanoparticles and its synergistic effects in combination with imipenem and two biocides against biofilm producing Acinetobacter baumannii. Nanomed. J. 2015, 2, 291–298. [Google Scholar]
- Jawad, A.; Seifert, H.; Snelling, A.M.; Heritage, J.; Hawkey, P.M. Survival of Acinetobacter baumannii in Dry Surfaces: Comparison of Outbreak and Sporadic Isolates. J. Clin. Microbiol. 1998, 36, 1938–1941. [Google Scholar] [CrossRef]
- Subhadra, B.; Kim, D.H.; Woo, K.; Surendran, S.; Choi, C.H. Control of Biofilm Formation in Healthcare: Recent Advances Exploiting Quorum-Sensing Interference Strategies and Multidrug Efflux Pump Inhibitors. Materials 2018, 11, 1676. [Google Scholar] [CrossRef]
- Uppu, D.S.S.M.; Samaddar, S.; Ghosh, C.; Paramanandham, K.; Shome, B.R.; Haldar, J. Amide side chain amphiphilic polymers disrupt surface established bacterial bio-films and protect mice from chronic Acinetobacter baumannii infection. Biomaterials 2016, 74, 131–143. [Google Scholar] [CrossRef]
- Moon, K.H.; Weber, B.S.; Feldman, M.F. Subinhibitory concentrations of trimethoprim and sulfamethoxazole prevent biofilm formation by Acinetobacter baumannii through inhibition of Csu pilus expression. Antimicrob. Agents Chemoth. 2017, 61, e00778-17. [Google Scholar] [CrossRef]
- Zhang, Y.; Brackman, G.; Coenye, T. Pitfalls associated with evaluating enzymatic quorum quenching activity: The case of MomL and its effect on Pseudomonas aeruginosa and Acinetobacter baumannii biofilms. PeerJ 2017, 5, e3251. [Google Scholar] [CrossRef]
- Blasco, L.; Bleriot, I.; González de Aledo, M.; Fernández-García, L.; Pacios, O.; Oliveira, H.; López, M.; Ortiz-Cartagena, C.; Fernández-Cuenca, F.; Pascual, Á.; et al. Development of an Anti-Acinetobacter baumannii Biofilm Phage Cocktail: Genomic Adaptation to the Host. Antimicrob. Agents Chemother. 2022, 66, e0192321. [Google Scholar] [CrossRef]
- de Leeuw, M.; Baron, M.; Ben David, O.; Kushmaro, A. Molecular Insights into Bacteriophage Evolution toward Its Host. Viruses 2020, 12, 1132. [Google Scholar] [CrossRef]
- Ackermann, H.W. Bacteriophage Observations and Evolution. Res. Microbiol. 2003, 154, 245–251. [Google Scholar] [CrossRef]
- Rehman, S.; Ali, Z.; Khan, M.; Bostan, N.; Naseem, S. The dawn of phage therapy. Rev. Med. Virol. 2019, 29, e2041. [Google Scholar] [CrossRef]
- Połaska, M.; Sokołowska, B. Bacteriophages—A new hope or a huge problem in the food industry. AIMS Microbiol. 2019, 5, 324–346. [Google Scholar] [CrossRef]
- Hu, J.; Chen, J.; Nie, Y.; Zhou, C.; Hou, Q.; Yan, X. Characterizing the gut phageome and phage-borne antimicrobial resistance genes in pigs. Microbiome 2024, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.Z.; Fokine, A.; Mahalingam, M.; Zhang, Z.; Garcia-Doval, C.; van Raaij, M.J.; Rossmann, M.G.; Rao, V.B. Molecular anatomy of the receptor binding module of a bacteriophage long tail fiber. PLoS Pathog. 2019, 15, e1008193. [Google Scholar] [CrossRef] [PubMed]
- Letarov, A.V.; Kulikov, E.E. Adsorption of bacteriophages on bacterial cells. Biochemistry 2017, 82, 1632–1658. [Google Scholar] [CrossRef]
- Weigel, C.; Seitz, H. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006, 30, 321–381. [Google Scholar] [CrossRef]
- Benkovic, S.J.; Spiering, M.M. Understanding DNA replication by the bacteriophage T4 replisome. J. Biol. Chem. 2017, 292, 18434–18442. [Google Scholar] [CrossRef]
- Oliveira, H.; Domingues, R.; Evans, B.; Sutton, J.M.; Adriaenssens, E.M.; Turner, D. Genomic Diversity of Bacteriophages Infecting the Genus Acinetobacter. Viruses 2022, 14, 181. [Google Scholar] [CrossRef]
- Fokine, A.; Leiman, P.G.; Shneider, M.M.; Ahvazi, B.; Boeshans, K.M.; Steven, A.C.; Black, L.W.; Mesyanzhinov, V.V.; Rossmann, M.G. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc. Natl. Acad. Sci. USA 2005, 102, 7163–7168. [Google Scholar] [CrossRef]
- Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M.A.; Drulis-Kawa, Z. Phage Life Cycles Behind Bacterial Biodiversity. Curr. Med. Chem. 2017, 24, 3987–4001. [Google Scholar] [CrossRef]
- Cieślik, M.; Bagińska, N.; Jończyk-Matysiak, E.; Węgrzyn, A.; Węgrzyn, G.; Górski, A. Temperate Bacteriophages—The Powerful Indirect Modulators of Eukaryotic Cells and Immune Functions. Viruses 2021, 13, 1013. [Google Scholar] [CrossRef]
- Cieślik, M.; Harhala, M.; Orwat, F.; Dąbrowska, K.; Górski, A.; Jończyk-Matysiak, E. Two Newly Isolated Enterobacter-Specific Bacteriophages: Biological Properties and Stability Studies. Viruses 2022, 14, 1518. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Międzybrodzki, R.; Drulis-Kawa, Z.; Cater, K.; Knežević, P.; Winogradow, C.; Amaro, K.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Rękas, J.; et al. Bacteriophages and antibiotic interactions in clinical practice: What we have learned so far. J. Biomed. Sci. 2022, 29, 23. [Google Scholar] [CrossRef]
- Międzybrodzki, R.; Borysowski, J.; Weber-Dąbrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóż, P.; Kłak, M.; Wojtasik, E.; et al. Clinical Aspects of Phage Therapy. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2012; Volume 83, pp. 73–121. [Google Scholar]
- Guo, Z.; Yuan, M.; Chai, J. Mini review Advantages and limitations of lytic phages compared with chemical antibiotics to combat bacterial infections. Heliyon 2024, 10, e34849. [Google Scholar] [CrossRef]
- Wójcicki, M.; Błażejak, S.; Gientka, I.; Brzezicka, K. The concept of using bacteriophages to improve the microbiological quality of minimally processed foods. Acta Sci. Pol. Technol. Aliment. 2019, 18, 373–383. [Google Scholar]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef]
- Hughes, K.; Sutherland, I.; Clark, J.; Jones, M. Bacteriophage and associated polysaccharide depolymerases–novel tools for study of bacterial biofilms. J. Appl. Microbiol. 1998, 85, 583–590. [Google Scholar] [CrossRef]
- Pires, D.P.; Oliveira, H.; Melo, L.D.R.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef]
- Prokhorov, N.S.; Riccio, C.; Zdorovenko, E.L.; Shneider, M.M.; Browning, C.; Knirel, Y.A.; Leiman, P.G.; Letarov, A.V. Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol. Microbiol. 2017, 105, 385–398. [Google Scholar] [CrossRef]
- Greenfield, J.; Shang, X.; Luo, H.; Zhou, Y.; Heselpoth, R.D.; Nelson, D.C.; Herzberg, O. Structure and tailspike glycosidase machinery of ORF212 from E. coli O157:H7 phage CBA120 (TSP3). Sci. Rep. 2019, 9, 7349. [Google Scholar] [CrossRef]
- Soontarach, R.; Srimanote, P.; Enright, M.C.; Blundell-Hunter, G.; Dorman, M.J.; Thomson, N.R.; Taylor, P.W.; Voravuthikunchai, S.P. Isolation and Characterization of Bacteriophage Selective for Key Acinetobacter baumannii Capsule Chemotypes. Pharmaceuticals 2022, 15, 443. [Google Scholar] [CrossRef]
- Doolittle, M.M.; Cooney, J.J.; Caldwell, D.E. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Ind. Microbiol. Biotechnol. 1996, 16, 331–341. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Bacteriophages and their enzymes in biofilm control. Curr. Pharm. Des. 2015, 21, 85–99. [Google Scholar] [CrossRef]
- Luo, J.; Xie, L.; Yang, M.; Liu, M.; Li, Q.; Wang, P.; Fan, J.; Jin, J.; Luo, C. Synergistic Antibacterial Effect of Phage pB3074 in Combination with Antibiotics Targeting Cell Wall against Multidrug-Resistant Acinetobacter baumannii In Vitro and Ex Vivo. Microbiol. Spectr. 2023, 11, e0034123. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Yu, X.; Guo, W.; Guo, C.; Guo, X.; Li, Q.; Zhu, Y. Bacteriophage-Mediated Control of Biofilm: A Promising New Dawn for the Future. Front. Microbiol. 2022, 13, 825828. [Google Scholar] [CrossRef] [PubMed]
- Huss, P.; Raman, S. Engineered bacteriophages as programmable biocontrol agents. Curr. Opin. Biotechnol. 2020, 61, 116–121. [Google Scholar] [CrossRef]
- Bagińska, N.; Harhala, M.A.; Cieślik, M.; Orwat, F.; Weber-Dąbrowska, B.; Dąbrowska, K.; Górski, A.; Jończyk-Matysiak, E. Biological Properties of 12 Newly Isolated Acinetobacter baumannii-Specific Bacteriophages. Viruses 2023, 15, 231. [Google Scholar] [CrossRef]
- Gliźniewicz, M.; Miłek, D.; Olszewska, P.; Czajkowski, A.; Serwin, N.; Cecerska-Heryć, E.; Dołęgowska, B.; Grygorcewicz, B. Advances in bacteriophage-mediated strategies for combating polymicrobial biofilms. Front. Microbiol. 2024, 14, 1320345. [Google Scholar] [CrossRef]
- Popescu, M.; Van Belleghem, J.D.; Khosravi, A.; Bollyky, P.L. Bacteriophages and the Immune System. Annu. Rev. Virol. 2021, 8, 415–435. [Google Scholar] [CrossRef]
- Kleiner, M.; Bushnell, B.; Sanderson, K.E.; Hooper, L.V.; Duerkop, B.A. Transductomics: Sequencing-based detection and analysis of transduced DNA in pure cultures and microbial communities. Microbiome 2020, 8, 158. [Google Scholar] [CrossRef]
- Borodovich, T.; Shkoporov, A.N.; Ross, R.P.; Hill, C. Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterol. Rep. 2022, 10, goac012. [Google Scholar] [CrossRef]
- Sisakhtpour, B.; Mirzaei, A.; Karbasizadeh, V.; Hosseini, N.; Shabani, M.; Moghim, S. The characteristic and potential therapeutic effect of isolated multidrug-resistant Acinetobacter baumannii lytic phage. Ann. Clin. Microbiol. Antimicrob. 2022, 21, 1. [Google Scholar] [CrossRef]
- Suh, G.A.; Lodise, T.P.; Tamma, P.D.; Knisely, J.M.; Alexander, J.; Aslam, S.; Barton, K.D.; Bizzell, E.; Totten, K.M.C.; Campbell, J.L.; et al. Considerations for the Use of Phage Therapy in Clinical Practice. Antimicrob. Agents Chemother. 2022, 66, e0207121. [Google Scholar] [CrossRef]
- Unnikrishnan, V.K.; Sundaramoorthy, N.S.; Nair, V.G.; Ramaiah, K.B.; Roy, J.S.; Rajendran, M.; Srinath, S.; Kumar, S.; Sankaran, P.; Mohan, S.; et al. Genome analysis of triple phages that curtails MDR E. coli with ML based host receptor prediction and its evaluation. Sci. Rep. 2023, 13, 23040. [Google Scholar] [CrossRef]
- Alqahtani, A. Bacteriophage treatment as an alternative therapy for multidrug-resistant bacteria. Saudi Med. J. 2023, 44, 1222. [Google Scholar] [CrossRef]
- Nawaz, A.; Khalid, N.A.; Zafar, S.; Majid, A.; Shahzadi, M.; Saleem, S.; Shah, A.A.; Badshah, M.; Khan, S. Phage Therapy as a revolutionary treatment for Multidrug-resistant Pseudomonas aeruginosa infections: A Narrative review. Microbe 2024, 2, 100030. [Google Scholar] [CrossRef]
- Aranaga, C.; Pantoja, L.D.; Martínez, E.A.; Falco, A. Phage Therapy in the Era of Multidrug Resistance in Bacteria: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 4577. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Vesal, S.; Moradabadi, A.; Hashemi, A.; Shariati, A. Bacteriophage therapy for inhibition of multi drug-resistant uropathogenic bacteria: A narrative review. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 30. [Google Scholar] [CrossRef]
- Ahamed, S.T.; Rai, S.; Guin, C.; Jameela, R.M.; Dam, S.; Muthuirulandi Sethuvel, D.P.; Balaji, V.; Giri, N. Characterizations of novel broad-spectrum lytic bacteriophages Sfin-2 and Sfin-6 infecting MDR Shigella spp. with their application on raw chicken to reduce the Shigella load. Front. Microbiol. 2023, 14, 1240570. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Kim, H.; Cho, E.; Park, H.; Jeon, B.; Ryu, S. Characterization of the lytic phage MSP1 for the inhibition of multidrug-resistant Salmonella enterica serovars Thompson and its biofilm. Int. J. Food Microbiol. 2023, 385, 110010. [Google Scholar] [CrossRef]
- Wójcicki, M.; Świder, O.; Średnicka, P.; Shymialevich, D.; Ilczuk, T.; Koperski, Ł.; Cieślak, H.; Sokołowska, B.; Juszczuk-Kubiak, E. Newly Isolated Virulent Salmophages for Biocontrol of Multidrug-Resistant Salmonella in Ready-to-Eat Plant-Based Food. Int. J. Mol. Sci. 2023, 24, 10134. [Google Scholar] [CrossRef]
- Rakov, C.; Ben Porat, S.; Alkalay-Oren, S.; Yerushalmy, O.; Abdalrhman, M.; Gronovich, N.; Huang, L.; Pride, D.; Coppenhagen-Glazer, S.; Nir-Paz, R.; et al. Targeting Biofilm of MDR Providencia stuartii by Phages Using a Catheter Model. Antibiotics 2021, 10, 375. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.-F.; Krisch, H.M. Phage-Antibiotic Synergy (PAS): β-Lactam and Quinolone Antibiotics Stimulate Virulent Phage Growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loh, B.; Altamirano, F.G.; Yu, Y.; Hua, X.; Leptihn, S. Colistin-phage combinations decrease antibiotic resistance in Acinetobacter baumannii via changes in envelope architecture. Emerg. Microbes Infect. 2021, 10, 2205–2219. [Google Scholar] [CrossRef]
- Liu, G.C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-antibiotic synergy is driven by a unique combination of antibacterial mechanism of action and stoichiometry. mBio 2020, 11, e01462-20. [Google Scholar]
- Diallo, K.; Dublanchet, A. Benefits of Combined Phage–Antibiotic Therapy for the Control of Antibiotic-Resistant Bacteria: A Literature Review. Antibiotics 2022, 11, 839. [Google Scholar] [CrossRef] [PubMed]
- Bulssico, J.; Papukashvili, I.; Espinosa, L.; Gandon, S.; Ansaldi, M. Phage-antibiotic synergy: Cell filamentation is a key driver of successful phage predation. PLoS Pathog. 2023, 19, e1011602. [Google Scholar] [CrossRef]
- Loganathan, A.; Manohar, P.; Nachimuthu, R. Phage-Antibiotic Combination: An Effective Method for Eradication of Staphylococcus aureus. bioRxiv 2023. [Google Scholar] [CrossRef]
- Manohar, P.; Madurantakam Royam, M.; Loh, B.; Bozdogan, B.; Nachimuthu, R.; Leptihn, S. Synergistic Effects of Phage-Antibiotic Combinations against Citrobacter amalonaticus. ACS Infect. Dis. 2022, 8, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Manohar, P.; Loh, B.; Nachimuthu, R.; Leptihn, S. Phage-antibiotic combinations to control Pseudomonas aeruginosa-Candida two-species biofilms. Sci. Rep. 2024, 14, 9354. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Wang, Z.; Wei, J.; Hu, T.; Si, J.; Tao, G.; Zhang, L.; Xie, L.; Abdalla, A.E. A combination therapy of phages and antibiotics: Two is better than one. Int. J. Biol. Sci. 2021, 17, 3573. [Google Scholar] [CrossRef]
- Srinivasan, R.; Santhakumari, S.; Poonguzhali, P.; Geetha, M.; Dyavaiah, M.; Lin, X. Bacterial biofilm inhibition: A focused review on recent therapeutic strategies for combating the biofilm mediated infections. Front. Microbiol. 2021, 12, 676458. [Google Scholar] [CrossRef]
- Dijkshoorn, L.; Aucken, H.; Gerner-Smidt, P.; Janssen, P.; Kaufmann, M.E.; Garaizar, J.; Ursing, J.; Pitt, T.L. Comparison of outbreak and nonoutbreak Acinetobacter baumannii strains by genotypic and phenotypic methods. J. Clin. Microbiol. 1996, 34, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Pereira, C.; Romalde, J.L.; Almeida, A. A Game of Resistance: War Between Bacteria and Phages and How Phage Cocktails can be the Solution. Virology 2024, 599, 110209. [Google Scholar] [CrossRef] [PubMed]
- Mancilla-Rojano, J.; Flores, V.; Cevallos, M.A.; Ochoa, S.A.; Parra-Flores, J.; Arellano-Galindo, J.; Xicohtencatl-Cortes, J.; Cruz-Córdova, A. A bioinformatic approach to identify confirmed and probable CRISPR–Cas systems in the Acinetobacter calcoaceticus–Acinetobacter baumannii complex genomes. Front. Microbiol. 2024, 15, 1335997. [Google Scholar] [CrossRef]
- Yu, T.; Huang, J.; Huang, X.; Hao, J.C.; Zhang, P.; Guo, T.; Bao, G.; Li, G. Sub-MIC antibiotics increased the fitness cost of CRISPR-Cas in Acinetobacter baumannii. Front. Microbiol. 2024, 15, 1381749. [Google Scholar] [CrossRef]
- Høyland-Kroghsbo, N.M.; Maerkedahl, R.B.; Svenningsen, S.L. A quorum-sensing-induced bacteriophage defense mechanism. mBio 2013, 4, e00362. [Google Scholar] [CrossRef]
- Patterson, A.G.; Jackson, S.A.; Taylor, C.; Evans, G.B.; Salmond, G.P.C.; Przybilski, R.; Staals, R.H.J.; Fineran, P.C. Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol. Cell 2016, 64, 1102–1108. [Google Scholar] [CrossRef]
- Lopatina, A.; Tal, N.; Sorek, R. Abortive infection: Bacterial suicide as an antiviral immune strategy. Annu. Rev. Virol. 2020, 7, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Ambroa, A.; Blasco, L.; Lopez, M.; Pacios, O.; Bleriot, I.; Fernandez-Garcia, L.; Gonzalez de Aledo, M.; Ortiz-Cartagena, C.; Millard, A.; Tomas, M. Genomic analysis of molecular bacterial mechanisms of resistance to phage infection. Front. Microbiol. 2021, 12, 784949. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Su, X.; Wang, C.; Xu, H.; Hu, D.; Wang, J.; Pei, K.; Sun, M.; Zou, T. Bacterial MazF/MazE toxin-antitoxin suppresses lytic propagation of arbitrium-containing phages. Cell Rep. 2022, 41, 111752. [Google Scholar] [CrossRef]
- Malone, L.M.; Birkholz, N.; Fineran, P.C. Conquering CRISPR: How phages overcome bacterial adaptive immunity. Curr. Opin. Biotechnol. 2021, 68, 30–36. [Google Scholar] [CrossRef]
- Morris, J.; Kelly, N.; Elliott, L.; Grant, A.; Wilkinson, M.; Hazratwala, K.; McEwen, P. Evaluation of Bacteriophage Anti-Biofilm Activity for Potential Control of Orthopedic Implant-Related Infections Caused by Staphylococcus aureus. Surg. Infect. 2019, 20, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Moryl, M.; Różalski, A.; de Figueiredo, J.A.P.; Palatyńska-Ulatowska, A. How Do Phages Disrupt the Structure of Enterococcus faecalis Biofilm? Int. J. Mol. Sci. 2023, 24, 17260. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Chun, J.; Son, B.; Ryu, S. Characterization and Genome Analysis of Staphylococcus aureus Podovirus CSA13 and Its Anti-Biofilm Capacity. Viruses 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Zurabov, F.; Glazunov, E.; Kochetova, T.; Uskevich, V.; Popova, V. Bacteriophages with depolymerase activity in the control of antibiotic resistant Klebsiella pneumoniae biofilms. Sci. Rep. 2023, 13, 15188. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Taati Moghadam, M.; Farahani, I.; Jazireian, P.; Shariati, A. Bacteriophage therapy against Pseudomonas aeruginosa biofilms: A review. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 45. [Google Scholar] [CrossRef]
- Jamal, M.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rahman, S.U.; Das, C.R. Isolation, Characterization and Efficacy of Phage Mj2 against Biofilm Forming Multi-Drug Resistant Enterobacter cloacae. Folia Microbiol. 2019, 64, 101–111. [Google Scholar] [CrossRef]
- Dubrovin, E.V.; Popova, A.V.; Kraevskiy, S.V.; Ignatov, S.G.; Ignatyuk, T.E.; Yaminsky, I.V.; Volozhantsev, N.V. Atomic Force Microscopy Analysis of the Acinetobacter baumannii Bacteriophage AP22 Lytic Cycle. PLoS ONE 2012, 7, e47348. [Google Scholar] [CrossRef]
- Thawal, N.D.; Yele, A.B.; Sahu, P.; Chopade, B.A. Effect of a Novel Podophage AB7-IBB2 on Acinetobacter baumannii Biofilm. Curr. Microbiol. 2012, 65, 66–72. [Google Scholar] [CrossRef]
- Yele, A.B.; Thawal, N.D.; Sahu, P.K.; Chopade, B.A. Novel lytic bacteriophage AB7-IBB1 of Acinetobacter baumannii: Isolation, characterization and its effect on biofilm. Arch. Virol. 2012, 157, 1441–1450. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Z.; Niu, W.; An, X.; Yuan, X.; Liu, H.; Wang, Y.; Feng, Y.; Huang, Y.; Zhang, X.; et al. Potential of a lytic bacteriophage to disrupt Acinetobacter baumannii biofilms in vitro. Future Microbiol. 2016, 11, 1383–1393. [Google Scholar] [CrossRef]
- Wintachai, P.; Naknaen, A.; Pomwised, R.; Voravuthikunchai, S.P.; Smith, D.R. Isolation and characterization of Siphoviridae phage infecting extensively drug-resistant Acinetobacter baumannii and evaluation of therapeutic efficacy in vitro and in vivo. J. Med. Microbiol. 2019, 68, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Tan, J.; Hao, Y.; Wang, Q.; Yan, X.; Wang, D.; Tuo, L.; Wei, Z.; Huang, G. Isolation and Characterization of a Novel Myophage Abp9 against Pandrug Resistant Acinetobacter baumannii. Front. Microbiol. 2020, 11, 506068. [Google Scholar]
- Grygorcewicz, B.; Wojciuk, B.; Roszak, M.; Łubowska, N.; Błażejczak, P.; Jursa-Kulesza, J.; Rakoczy, R.; Masiuk, H.; Dołęgowska, B. Environmental Phage-Based Cocktail and Antibiotic Combination Effects on Acinetobacter baumannii Biofilm in a Human Urine Model. Microb. Drug Resist. 2021, 27, 25–35. [Google Scholar] [CrossRef]
- Wintachai, P.; Voravuthikunchai, S.P. Characterization of Novel Lytic Myoviridae Phage Infecting Multidrug-Resistant Acinetobacter baumannii and Synergistic Antimicrobial Efficacy between Phage and Sacha Inchi Oil. Pharmaceuticals 2022, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Erol, H.B.; Kaskatepe, B.; Yildiz, S.; Altanlar, N. The effect of phage-antibiotic combination strategy on multidrug-resistant Acinetobacter baumannii biofilms. J. Microbiol. Met. 2023, 210, 106752. [Google Scholar] [CrossRef]
- Erol, H.B.; Kaskatepe, B.; Yildiz, S.; Altanlar, N.; Bayrakdar, F. Characterization of two bacteriophages specific to Acinetobacter baumannii and their effects on catheters biofilm. Cell Biochem. Funct. 2024, 42, e3966. [Google Scholar] [CrossRef]
- Rastegar, S.; Skurnik, M.; Tadjrobehkar, O.; Samareh, A.; Samare-Najaf, M.; Lotfian, Z.; Khajedadian, M.; Hosseini-Nave, H.; Sabouri, S. Synergistic effects of bacteriophage cocktail and antibiotics combinations against extensively drug-resistant Acinetobacter baumannii. BMC Infect. Dis. 2024, 24, 1208. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Sisakhtpour, B.; Mirzaei, A.; Karbasizadeh, V.; Moghim, S. Efficacy of isolated bacteriophage against biofilm embedded colistin-resistant Acinetobacter baumanni. Gene Rep. 2021, 22, 100984. [Google Scholar] [CrossRef]
- Vashisth, M.; Yashveer, S.; Jaglan, A.B.; Virmani, N.; Bera, B.C.; Vaid, R.K.; Anand, T. Synergy of a virulent phage (φAB182) with antibiotics leading to successful elimination of biofilms formed by MDR Acinetobacter baumannii. Can. J. Microbiol. 2022, 68, 731–746. [Google Scholar] [CrossRef]
- Soontarach, R.; Nwabor, O.F.; Voravuthikunchai, S.P. Interaction of lytic phage T1245 with antibiotics for enhancement of antibacterial and anti-biofilm efficacy against multidrug-resistant Acinetobacter baumannii. Biofouling 2022, 38, 994–1005. [Google Scholar] [CrossRef]
- Hailemichael, T.; Girma, L.; Fissiha, P.; Geteneh, A.; Kassa, T. Isolation of virulent phages against multidrug-resistant Acinetobacter baumannii recovered from inanimate objects of Jimma Medical Center, Southwest Ethiopia. BMC Infect. Dis. 2023, 23, 820. [Google Scholar]
- Narancic, J.; Gavric, D.; Kostanjsek, R.; Knezevic, P. First Characterization of Acinetobacter baumannii-Specific Filamentous Phages. Viruses 2024, 16, 857. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Zhang, P.; To, K.K.; Liu, Y.; Bai, C.; Leung, S.S. Sequential treatment effects on phage–antibiotic synergistic application against multi-drug-resistant Acinetobacter baumannii. Int. J. Antimicrob. Agent. 2023, 62, 106951. [Google Scholar] [CrossRef] [PubMed]
- Thummeepak, R.; Kitti, T.; Kunthalert, D.; Sitthisak, S. Enhanced antibacterial activity of Acinetobacter baumannii bacteriophage øABP-01 endolysin (LysABP-01) in combination with colistin. Front. Microbiol. 2016, 7, 1402. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Li, X.; Wang, L.; Li, G.; Cong, C.; Li, R.; Cui, H.; Murtaza, B.; Xu, Y. The endolysin of the Acinetobacter baumannii phage vB_AbaP_D2 shows broad antibacterial activity. Microb. Biotechnol. 2021, 14, 403–418. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, Y.; Galgano, S.; Houdijk, J.; Xie, W.; Jin, Y.; Lin, J.; Song, W.; Fu, Y.; Li, X.; et al. Recent Progress in Phage Therapy to Modulate Multidrug-Resistant Acinetobacter baumannii, including in Human and Poultry. Antibiotics 2022, 11, 1406. [Google Scholar] [CrossRef]
- Antonova, N.P.; Vasina, D.V.; Lendel, A.M.; Usachev, E.V.; Makarov, V.V.; Gintsburg, A.L.; Tkachuk, A.P.; Gushchin, V.A. Broad Bactericidal Activity of the Myoviridae Bacteriophage Lysins LysAm24, LysECD7, and LysSi3 against Gram-Negative ESKAPE Pathogens. Viruses 2019, 11, 284. [Google Scholar] [CrossRef]
- Vasina, D.V.; Antonova, N.P.; Grigoriev, I.V.; Yakimakha, V.S.; Lendel, A.M.; Nikiforova, M.A.; Pochtovyi, A.A.; Remizov, T.A.; Usachev, E.V.; Shevlyagina, N.V.; et al. Discovering the Potentials of Four Phage Endolysins to Combat Gram-Negative Infections. Front. Microbiol. 2021, 12, 748718. [Google Scholar] [CrossRef]
- Vorob’ev, A.M.; Anurova, M.N.; Aleshkin, A.V.; Gushchin, V.A.; Vasina, D.V.; Antonova, N.P.; Kiseleva, I.A.; Rubalskii, E.O.; Zul’karneev, E.R.; Laishevtsev, A.I.; et al. Determination of bactericidal activity spectrum of recombinant endolysins of ECD7, Am24, Ap22, Si3, and St11 Bacteriophages. Bull. Exp. Biol. Med. 2021, 170, 636–639. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and phage-derived proteins—application approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef]
- Lee, K.J.; Kim, J.A.; Hwang, W.; Park, S.J.; Lee, K.H. Role of capsular polysaccharide (CPS) in biofilm formation and regulation of CPS production by quorum-sensing in Vibrio vulnificus. Mol. Microbiol. 2013, 90, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Danis-Wlodarczyk, K.M.; Wozniak, D.J.; Abedon, S.T. Treating Bacterial Infections with Bacteriophage-Based Enzybiotics: In Vitro, In Vivo and Clinical Application. Antibiotics 2021, 10, 1497. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Rogelj, S.; Zhang, P. Rose Bengal-Decorated Silica Nanoparticles as Photosensitizers for Inactivation of Gram-Positive Bacteria. Nanotechnology 2010, 21, 065102. [Google Scholar] [CrossRef]
- Topka-Bielecka, G.; Dydecka, A.; Necel, A.; Bloch, S.; Nejman-Faleńczyk, B.; Węgrzyn, G.; Węgrzyn, A. Bacteriophage-Derived Depolymerases against Bacterial Biofilm. Antibiotics 2021, 10, 175. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Bai, C.; Leung, S.S.Y. Translating bacteriophage-derived depolymerases into antibacterial therapeutics: Challenges and prospects. Acta Pharm. Sin. B 2024, 14, 155–169. [Google Scholar] [CrossRef]
- Lood, R.; Winer, B.Y.; Pelzek, A.J.; Diez-Martinez, R.; Thandar, M.; Euler, C.W.; Schuch, R.; Fischetti, V.A. Novel Phage Lysin Capable of Killing the Multidrug-Resistant Gram-Negative Bacterium Acinetobacter baumannii in a Mouse Bacteremia Model. Antimicrob. Agents Chemother. 2015, 59, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, L.L.; Gan, D.; Zhang, X. In vitro study of bacteriophage AB3 endolysin LysAB3 activity against Acinetobacter baumannii biofilm and biofilm-bound A. baumannii. Clin. Lab. 2018, 64, 1021–1030. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Zhang, P.F.; Leung, S.S.Y.; Xia, J. Membrane-permeable antibacterial enzyme against multidrug-resistant Acinetobacter baumannii. ACS Infect. Dis. 2021, 7, 2192–2204. [Google Scholar] [CrossRef]
- Chu, J.J.K.; Poh, W.H.; Hasnuddin, N.T.B.; Hew, E.Y.; Dam, L.C.; Sahili, A.E.; Rice, S.A.; Goh, B.C. Novel Phage Lysin Abp013 against Acinetobacter baumannii. Antibiotics 2022, 11, 169. [Google Scholar] [CrossRef]
- Nandi, A.; Yadav, R.; Singh, A. Phage derived lytic peptides, a secret weapon against Acinetobacter baumannii—An in silico approach. Front. Med. 2022, 9, 1047752. [Google Scholar] [CrossRef]
- Sitthisak, S.; Manrueang, S.; Khongfak, S.; Leungtongkam, U.; Thummeepak, R.; Thanwisai, A.; Burton, N.; Dhanoa, G.K.; Tsapras, P.; Sagona, A.P. Antibacterial activity of vB_AbaM_PhT2 phage hydrophobic amino acid fusion endolysin, combined with colistin against Acinetobacter baumannii. Sci. Rep. 2023, 13, 7470. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Han, L.M.; Chen, X.Z.; Yi, P.C.; Li, H.; Ren, Y.Y.; Gao, J.H.; Zhang, C.Y.; Huang, J.; Wang, W.X.; et al. Engineered endolysin of Klebsiella pneumoniae phage is a potent and broad-spectrum bactericidal agent against “ESKAPEE” pathogens. Front. Microbiol. 2024, 15, 1397830. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Morales, A.C.; Lessor, L.L.; Wood, T.L.; Migl, D.; Mijalis, E.M.; Cahill, J.; Russell, W.K.; Young, R.F.; Gill, J.J. Genomic and Biochemical Characterization of Acinetobacter Podophage Petty Reveals a Novel Lysis Mechanism and Tail-Associated Depolymerase Activity. J. Virol. 2018, 92, e01064-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.; Zhang, P.; Xu, M.; Yuan, W.; Bian, L.; Liu, Y.; Xia, J.; Leung, S.S.Y. Phage-Derived Depolymerase as an Antibiotic Adjuvant Against Multidrug-Resistant Acinetobacter baumannii. Front. Microbiol. 2022, 13, 845500. [Google Scholar] [CrossRef] [PubMed]
- Kachur, K.; Suntres, Z. The antibacterial properties of phenolic isomers, carvacrol and thymol. Crit. Rev. Food Sci. Nutr. 2020, 60, 3042–3053. [Google Scholar] [CrossRef]
- Tapia-Rodriguez, M.R.; Cantu-Soto, E.U.; Vazquez-Armenta, F.J.; Bernal-Mercado, A.T.; Ayala-Zavala, J.F. Inhibition of Acinetobacter baumannii Biofilm Formation by Terpenes from Oregano (Lippia graveolens) Essential Oil. Antibiotics 2023, 12, 1539. [Google Scholar] [CrossRef]
- Fimbres-García, J.O.; Flores-Sauceda, M.; Othon-Díaz, E.D.; García-Galaz, A.; Tapia-Rodríguez, M.R.; Silva-Espinoza, B.A.; Ayala-Zavala, J.F. Facing Resistant Bacteria with Plant Essential Oils: Reviewing the Oregano Case. Antibiotics 2022, 11, 1777. [Google Scholar] [CrossRef]
- Dutta, P.; Das, S. Mammalian antimicrobial peptides: Promising therapeutic targets against infection and chronic inflammation. Curr. Top. Med. Chem. 2016, 16, 99–129. [Google Scholar] [CrossRef]
- Galdiero, S.; Falanga, A.; Berisio, R.; Grieco, P.; Morelli, G.; Galdiero, M. Antimicrobial Peptides as an Opportunity Against Bacterial Diseases. Curr. Med. Chem. 2015, 22, 1665–1677. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- e la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef]
- Wieczorek, M.; Jenssen, H.; Kindrachuk, J.; Scott, W.R.; Elliott, M.; Hilpert, K.; Cheng, J.T.; Hancock, R.E.; Straus, S.K. Structural studies of a peptide with immune modulating and direct antimicrobial activity. Chem. Biol. 2010, 17, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Friedman, J. New biomaterials for the sustained release of nitric oxide: Past, present and future. Expert Opin. Drug Deliv. 2009, 6, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Mihu, M.R.; Sandkovsky, U.; Han, G.; Friedman, J.M.; Nosanchuk, J.D.; Martinez, L.R. The use of nitric oxide releasing nanoparticles as a treatment against Acinetobacter baumannii in wound infections. Virulence 2010, 1, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef]
- Antunes, L.S.C.; Imperi, F.; Mindandri, F.; Visca, P. In Vitro and In Vivo Antimicrobial Activities of Gallium Nitrate against Multidrug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 5961–5970. [Google Scholar] [CrossRef]
- Arivett, B.A.; Fiester, S.E.; Ohneck, E.J.; Penwell, W.F.; Kaufman, C.M.; Relich, R.F.; Actis, L.A. Antimicrobial Activity of Gallium Protoporphyrin IX against Acinetobacter baumannii Strains Displaying Different Antibiotic Resistance Phenotypes. Antimicrob. Agents Chemother. 2015, 59, 7657–7665. [Google Scholar] [CrossRef]
- Bernstein, L. Mechanisms of Therapeutic Activity for Gallium. Pharmacol. Rev. 1998, 50, 665–682. [Google Scholar]
- Hemeg, H.A. Nanomaterials for Alternative Antibacterial Therapy. Int. J. Nanomed. 2017, 12, 8211–8225. [Google Scholar] [CrossRef]
- Banoub, N.G.; Saleh, S.E.; Helal, H.S.; Aboshanab, K.M. Antibiotics Combinations and Chitosan Nanoparticles for Combating Multidrug Resistance Acinetobacter baumannii. Infect. Drug Resist. 2021, 14, 3327–3339. [Google Scholar] [CrossRef]
- Shaker, M.A.; Shaaban, M.I. Synthesis of silver nanoparticles with antimicrobial and anti-adherence activities against multidrug-resistant isolates from Acinetobacter baumannii. J. Taibah Univ. Med. Sci. 2017, 12, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Workman, D.G.; Hunter, M.; Wang, S.; Brandel, J.; Hubscher, V.; Dover, L.G.; Tétard, D. The influence of linkages between 1-hydroxy-2(1H)-pyridinone coordinating groups and a tris(2-aminoethyl)amine core in a novel series of synthetic hexadentate iron(III) chelators on antimicrobial activity. Bioorg. Chem. 2020, 95, 103465. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [PubMed]
- Ran, B.; Yuan, Y.Y.; Xia, W.X.; Li, M.L.; Yao, Q.C.; Wang, Z.K.; Wang, L.L.; Li, X.Y.; Xu, Y.P.; Peng, X.J. A photo-sensitizable phage for multidrug-resistant Acinetobacter baumannii therapy and biofilm ablation. Chem. Sci. 2021, 12, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.; Metcalf, D.; Devine, D.; Robinson, C. Erythrosine is a potential photosensitizer for the photodynamic therapy of oral plaque biofilms. J. Antimicrob. Chemother. 2006, 57, 680–684. [Google Scholar] [CrossRef]
- Fekrirad, Z.; Darabpour, E.; Kashef, N. Eradication of Acinetobacter baumannii Planktonic and Biofilm Cells Through Erythro-sine-Mediated Photodynamic Inactivation Augmented by Acetic Acid and Chitosan. Curr. Microbiol. 2021, 78, 879–886. [Google Scholar] [CrossRef]
- Frade, M.L.; De Annunzio, S.R.; Calixto, G.M.F.; Victorelli, F.D.; Chorilli, M.; Fontana, C.R. Assessment of Chitosan-Based Hydrogel and Photodynamic Inactivation against Propionibacterium acnes. Molecules 2018, 23, 473. [Google Scholar] [CrossRef]
- Kardumyan, V.V.; Aksenova, N.A.; Glagolev, N.N.; Timashev, P.S.; Solovieva, A.B. Influence of acetic acid on the photocatalytic activity of photosensitiser–amphiphilic polymer complexes in the oxidation reaction of tryptophan. J. Chem. Phys. 2020, 152, 194901. [Google Scholar] [CrossRef]
- Wainwright, M.; Maisch, T.; Nonell, S.; Plaetzer, K.; Almeida, A.; Tegos, G.P.; Hamblin, M.R. Photoantimicrobials—Are We Afraid of the Light? Lancet Infect. Dis. 2017, 17, e49–e55. [Google Scholar] [CrossRef]
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic adjuvants: Identification and clinical use. Microb. Biotechnol. 2013, 6, 445–449. [Google Scholar] [CrossRef]
- Rhomberg, P.R.; Shortridge, D.; Huband, M.D.; Butler, D.; West, J.; Flamm, R.K. Multilaboratory broth microdilution MIC reproducibility study for GSK3342830, a novel catechol-cephem, abstr SATURDAY287. In Proceedings of the ASM Microbe, New Orleans, LA, USA, 1–5 June 2017; GlaxoSmithKline: Brentford, UK, 2017. [Google Scholar]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In vitro antibacterial properties of cefiderocol, a novel siderophore cephalosporin, against Gram-negative bacteria. Antimicrob. Agents Chemother. 2017, 62, e01454-17. [Google Scholar] [CrossRef] [PubMed]
- Isler, B.; Doi, Y.; Bonomo, R.A.; Paterson, D.L. New treatment options against carbapenem-resistant Acinetobacter baumannii infections. Antimicrob. Agents Chemother. 2019, 63, e01110-18. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Miller, P.A.; Möllmann, U.; Claypool, W.D.; Schroeder, V.A.; Wolter, W.R.; Suckow, M.; Yu, H.; Li, S.; Huang, W.; et al. Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity against Multidrug Resistant Acinetobacter baumannii Both in Vitro and in Vivo. J. Med. Chem. 2017, 60, 4577–4583. [Google Scholar] [CrossRef] [PubMed]
- Reid, G. The importance of guidelines in the development and application of probiotics. Curr. Pharm. Des. 2005, 11, 11–16. [Google Scholar] [CrossRef]
- Parra Millán, R.; Jiménez Mejías, M.E.; Sánchez Encinales, V.; Ayerbe Algaba, R.; Gutiérrez Valencia, A.; Pachón Ibáñez, M.E.; Díaz, C.; Pérez Del Palacio, J.; López Cortés, L.F.; Pachón, J.; et al. Efficacy of lysophosphatidylcholine in combination with antimicrobial agents against Acinetobacter baumannii in experimental murine peritoneal sepsis and pneumonia models. Antimicrob. Agents Chemother. 2016, 60, 4464–4470. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| The Factor Responsible for Biofilm Formation in A. baumannii | Function | Reference |
|---|---|---|
| OMPs (including OmpA, OmpW and CarO) | Contributes to drug resistance, adhesion to epithelial cells, mediates their invasion, enables cell membrane integrity, promotes cell death, contributes to serum resistance, biofilm formation, transposon-mediated inactivation of bfmS in bacteria, regulates iron uptake | [4,5,7,51] |
| Bap | Involved in resistance mechanisms, and required for biofilm reduction, maintains a stable mature biofilm structure, mediates adherence to bronchial cells, interbacterial cell adhesion, enhances hydrophobicity, structural integrity as well as water channel formation, up-regulation is linked to low iron concentration | [5,7,51] |
| Bla(PER-1) | Increases cell adhesiveness and biofilm formation | [5] |
| Ata | Assists in biofilm adherence to the host | [4] |
| PNAG (and its pgaABCD gene) | Substrate-specific transmembrane transporter, increases biofilm formation, enhances drug resistance, protection against innate host defense, cell–cell adherence, and production of the extracellular matrix | [1,7] |
| K1 capsule | Mediates resistance to cationic AMPs and serum; K1 locus regulates the production, modification, and export of capsular polysaccharides | [1,25] |
| Csu (ABCDE) | Biofilm formation on abiotic surfaces, controls pili biogenesis | [51] |
| Two-component systems (BfmRS, AdeRS, and GacSA) | Biofilm formation, involved in biogenesis of pili, regulates csu operon (including motility) and K-locus to obtain capsule production, regulates quorum sensing (QS), amino acid metabolism, resistance to human serum, involved in tolerance to desiccation | [4,5,7,51] |
| Efflux pump (5 families) | Overexpression leads to multidrug resistance, decreased biofilm production, synthesis and transport of autoinducer molecules, and altered membrane composition | [7] |
| AbaIR (and aba genes) | Regulates the QS system, involved in reducing biofilm formation | [5,7] |
| AHL | Increases expression of Csu pili and stimulates biofilm formation | [5] |
| BlsA | Influences virulence through iron metabolism via direct interactions with Fur | [4] |
| T1SS, T2SS | Maintains biofilm stability, exports Bap beyond the OM, involved in virulence of A. baumannii, responsible for secretion of extracellular enzymes—lipases, such as lipoyl synthases LipA, LipH, and proteases, such as CpaA | [4,71] |
| Phage Symbol (Morphotype/Type of Replication Cycle) | Bacterial Strain(s) Used in the Experiment | Type of Experiment (In Vitro/In Vivo)/Tested Surface | The Main Result(s) of the Experiment | Reference |
|---|---|---|---|---|
| vB_AbaP_WU2001 (P/lytic) | ABPW052 | in vitro/96-well plate/abiotic | A 48.72% inhibition of biofilm formation, and 78.82% degradation of mature biofilm at 108 PFU/well | [10] |
| vB_AbaM_ISTD (M/lytic) | 6077/12 | in vitro/porous glass beads/abiotic | A 30% degradation of mature biofilm at MOI 100 | [53] |
| vB_AbaM_NOVI (M/lytic) | in vitro/porous glass beads/abiotic | A 30% degradation of mature biofilm at MOI 100 | ||
| vB_AbaP_B3 (P/lytic) | Ab404_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | [106] |
| Ab019_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab034_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| Ab007_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab008_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| NIPH2061 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| Ab105-2phiΔCI404ad (S/created lytic mutant) | Ab404_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | |
| Ab019_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab034_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| Ab007_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab008_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| NIPH2061 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
|
vB_AbaP_B3 (P/lytic) and Ab105-2phiΔCI404ad (S/created lytic mutant) | Ab404_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | |
| Ab019_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab034_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| Ab007_GEIH-2010 | in vitro/96-well plate/abiotic | A low degradation of mature biofilm at MOI 10 | ||
| Ab008_GEIH-2010 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
| NIPH2061 | in vitro/96-well plate/abiotic | A high degradation of mature biofilm at MOI 10 | ||
|
AB7-IBB2 (P/lytic) | AIIMS 7 | in vitro/96-well plate/abiotic | An 80% inhibition of biofilm formation at MOIs 1000 and 100,000 | [181] |
| AB7-IBB1 (S/lytic) | AIIMS 7 | in vitro/human embryonic kidney 293 cell line/biotic | A 50% inhibition of biofilm formation at all tested MOIs (i.e., 0.1, 10, 100, and 1000) | [182] |
| in vitro/polystyrene/abiotic | A 75% inhibition of biofilm formation at MOI 100,000 | |||
| vB_AbaM_IME_AB2 (M/lytic) | MDR-AB2 | in vitro/96-well plate/abiotic | An 88.5% degradation of mature biofilm at MOI 0.1 | [183] |
| in vitro/metal surfaces (with protocol mimicking clinical settings; European standards EN 13727:2012)/abiotic | A 93.33% degradation of mature biofilm at MOI 10 | |||
|
AB1801 (S/lytic) | NPRC AB11 | in vitro/96-well plate/abiotic | A 66% inhibition of biofilm formation, and a 70% degradation of performed biofilm at MOI 1 | [184] |
|
Abp9 (M/lytic) | ABZY9 | in vitro/96-well plate/abiotic | A 72.22% degradation of mature biofilm at MOI 10 | [185] |
|
Aba-1 (nd/lytic) | AB3 | in vitro/96-well plate/abiotic | A 77.69% inhibition of biofilm formation at MOI 0.5 | [186] |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 67.45% inhibition of biofilm formation at MOI 0.5 | |||
|
Aba-2 (nd/lytic) | AB8 | in vitro/96-well plate/abiotic | A 55% inhibition of biofilm formation at MOI 0.5 | |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 34% inhibition of biofilm formation at MOI 0.5 | |||
|
Aba-3 (nd/lytic) | AB14 | in vitro/96-well plate/abiotic | A 53% inhibition of biofilm formation at MOI 0.5 | |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 44% inhibition of biofilm formation at MOI 0.5 | |||
|
Aba-4 (nd/lytic) | AB23 | in vitro/96-well plate/abiotic | A 61% inhibition of biofilm formation at MOI 0.5 | |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 52% inhibition of biofilm formation at MOI 0.5 | |||
|
Aba-5 (nd/lytic) | AB11 | in vitro/96-well plate/abiotic | A 35% inhibition of biofilm formation at MOI 0.5 | |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 35% inhibition of biofilm formation at MOI 0.5 | |||
|
Aba-6 (nd/lytic) | AB10 | in vitro/96-well plate/abiotic | A 62% inhibition of biofilm formation at MOI 0.5 | |
| in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic | A 91% inhibition of biofilm formation at MOI 0.5 | |||
| vWUPSU (M/lytic) | NPRCOE 160519 | in vitro/96-well plate/abiotic | A 68.3% inhibition of biofilm formation, and 53.3% degradation of mature biofilm at 108 PFU/well | [187] |
| vB_AbaP_HB01 (P/lytic) | 24 different strains: C1, C3, C5, C7, C10, C18, C19, C22, A1, A4, A7, A8, SU1, SU2, SU3, ATCC, M1, M2, M6, M7, M20, M24, M27, M34 | in vitro/96-well plate/abiotic | After 6 h—degradation of mature biofilm at the level of 40–86%; after 24 h—degradation of mature biofilm at the level of 18–84%; after 48 h—degradation of mature biofilm at the level of 1–81% among all tested strains | [188] |
| vB_AbaM_HB02 (M/lytic) | in vitro/96-well plate/abiotic | After 6 h—degradation of mature biofilm at the level of 35–85%; after 24 h—degradation of mature biofilm at the level of 26–85%; after 48 h—degradation of mature biofilm at the level of 1–80% among all tested strains | ||
| vB_AbaP_HB01 (P/lytic) | 78 different strains | in vitro/biofilm cultured on a catheter/abiotic | A ~68% inhibition of biofilm formation, and ~78.57% degradation of mature biofilm | [189] |
| vB_AbaM_HB02 (M/lytic) | in vitro/biofilm cultured on a catheter/abiotic | A ~60% inhibition of biofilm formation, and ~71.43% degradation of mature biofilm | ||
|
vB_AbaP_HB01 (P/lytic) and vB_AbaM_HB02 (M/lytic) | in vitro/96-well plate/abiotic | A ~76% inhibition of biofilm formation, and ~83.33% degradation of mature biofilm | ||
| vB_AbaS_SA1 (S/temperate); vB_AbaS_Ftm (S/temperate); vB_AbaS_Eva (S/temperate); vB_AbaS_Gln (S/temperate) | 30 different XDR strains | in vitro/96-well plate/abiotic | A ~78% inhibition of biofilm formation, and ~66% degradation of mature biofilm at MOI 1000 | [190] |
| Phage(s) Symbol (Morphotype/Type of Replication Cycle) | Tested Antibiotic(s) | Bacterial Strain(s) Used in the Experiment | Type of Experiment (In Vitro/In Vivo)/Tested Surface/PAS and/or Phage Cocktail | The Main Result(s) of the Experiment | Reference |
|---|---|---|---|---|---|
| pB3074 (nd/lytic) | cefotaxime (2 × MIC) or meropenem (0.5 × MIC) | Bm3074 | in vitro/96-well plate/abiotic | The highest degradation of mature biofilm was observed with phage and meropenem | [134] |
| Aba-1(nd/lytic); Aba-2 (nd/lytic); Aba-3 (nd/lytic); Aba-4 (nd/lytic); Aba-6 (nd/lytic) | amikacin, gentamicin, tobramycin, colistin, imipenem, meropenem, trimethoprim/sulfamethoxazole, ciprofloxacin, levofloxacin | AB20 | in vitro/urine model—standing for the natural environment within the human’s urinary tract/biotic/phage cocktail and PAS | A 98.6% biofilm degradation considering phages (107 PFU/mL) and 0.5 × MIC trimethoprim/sulfamethoxazole | [186] |
|
vB_AbaP_HB01 (P/lytic) and vB_AbaM_HB02 (M/lytic) | colistin | 24 different strains | in vitro/96-well plate/abiotic | After 6 h—degradation of mature biofilm at the level of 42–87%; after 24 h—degradation of mature biofilm at the level of 28–85%; after 48 h—degradation of mature biofilm at the level of 0–81% among all tested strains | [188] |
| φAB182 (M/lytic) | ceftazidime, polymyxin B, cefotaxime, colistin | MDR strains | in vitro/96-well plate/ abiotic/PAS | The highest degradation of mature biofilm with combining phages and colistin, followed by polymixin B, ceftazidime, and cefotaxime | [192] |
| T1245 (P/lytic) | imipenem or colistin, meropenem or ceftazidime | MDR strains | in vitro/96-well plate/ abiotic/PAS | An ~80% degradation of mature biofilm; synergistic effect of phage in combination with all tested antibiotics was observed | [193] |
| vB_AbaM-IME-AB2 (M/lytic) | colistin (2 × MIC) | MDR-AB2 | in vitro/96-well plate/abiotic/first phage then colistin | A ~90% degradation of mature biofilm | [196] |
| in vitro/96-well plate/abiotic/first colistin then phage | A ~72% degradation of mature biofilm | ||||
| in vitro/96-well plate/abiotic/colistin and phage simultaneously | A ~88% degradation of mature biofilm | ||||
| vB_AbaS_SA1 (S/temperate); vB_AbaS_Ftm (S/temperate); vB_AbaS_Eva (S/temperate); vB_AbaS_Gln (S/temperate) | ampicillin/sulbactam | 30 different XDR strains | in vitro/96-well plate/ abiotic/PAS | At 10×, 5×, and 1 × MIC, the PAS effect on biofilm degradation was observed | [190] |
| meropenem | At 1 × MIC, the PAS effect on biofilm degradation was observed | ||||
| colistin | At 15 × MIC, the PAS effect on biofilm degradation, and at 2 × MIC, the PAS effect on inhibition of biofilm formation was observed |
| Type of Enzyme(s) | Phage Which Is the Source of the Enzyme | Description of Observed Activity | Reference |
|---|---|---|---|
| polysaccharide depolymerase | tailspike protein of phage φAB6 | A 48 h biofilm formed in vitro by the A. baumannii strain Ab-54149 was treated with three different concentrations of the tailspike protein (10, 50, and 100 ng/well) for 4 h. The biofilm removal was ~9%, ~35%, and ~38%, whereas the biofilm inhibition demonstrated significant results: ~34%, ~44%, and ~56%, respectively. | [55] |
| Abtn-4 | phage vB_AbaP | The lowest biofilm inhibition (~35%) was observed within the A. baumannii strains ATCC 17978 and ATCC 19606. The highest results (almost 70% biofilm reduction) were observed for A. baumanii strains AB7, AB10, and AB16. The inhibiting activity of Abtn-4 was also evaluated on a biofilm formed by bacterial strains belonging to other species (K. pneumoniae, P. aeruginosa, Salmonella sp., E. faecium, E. faecalis, and S. aureus); however, within all tested pathogens, it did not reach 50%. | [198] |
| endolysin PlyF307 | a prophage induced from A. baumannii strain 2198 | The enzyme activity was tested on biofilm formed by the A. baumannii strain 1791 in vitro (on catheter) and in vivo (in mice; infected by a small incision and later an insertion of a 3 cm catheter containing a 2-day preformed biofilm). After 2 h of the treatment, the bacterial density was decreased by 1.6 log units (in vitro; 1 dose—300 μL of 1 mg/mL solution of endolysin PlyF307), and after 3 h of the treatment by 2 log units (in vivo; 2 doses—250 μL of 4 mg/mL solution of endolysin PlyF307). The anti-biofilm effect of proteins P307 and P307SQ-8C (P307 with additional disulfide bond, C-terminal proteins derived from PlyF307 endolysin) was assessed. After 2 h, a 3-log-unit, and a 4-log unit decrease in bacterial density was noted for P307 andP307SQ-8C, respectively. | [209] |
| LysAB3 | phage AB3, and LysAB3 was subjected to a knock-out of a structural amphiphilic peptide region (forming LysAB3-D) | The decrease in antibacterial activity (between intact LysAB3 and LysAB3-D) was statistically relevant—from 95.8% to 33.3% of inhibiting activity. | [210] |
| modified lysin LysAB2-KWK | nd | LysAB2-KWK, with additional CeA peptide octamer, potentially enhanced lytic activity against an MDR-AB2 strain. The use of this enzyme reduced biofilm by ~40%. There was no difference after increasing the concentration of this enzyme. | [211] |
| endolysins: LysAm24, LysAp22, LysECD7, and LysSi3 | myovirus | The anti-biofilm activity of endolysins was assessed on mature biofilms formed by the A. baumannii strain Ts 50–16. Inhibition of biofilms formed by different pathogens (K. pneumoniae and P. aeruginosa) was observed; the biofilm formed by A. baumannii was the most susceptible to these endolysins. The highest anti-biofilm activity was observed for the LysECD7 at 1 mg/mL concentration (decrease in OD600 from 1 to ~0.15). LysAm24 and LysAp22 also demonstrated great biofilm reduction (decrease in OD600 from 1 to ~0.2). The lowest anti-biofilm activity was observed fot LysSi3 (decrease in OD600 from 1 to ~0.5), suggesting that it was the least optimal enzyme in the eradication of biofilm. | [201] |
| lysin Abp013 | phage φAbp2 | A 3 h biofilm formed by the A. baumannii strain ATCC 17961 was reduced by 2.65 log units (99.78%), 2.23 log units (99.42%), and 1.51 log units (96.93%)—for the Abp013 concentration of 400, 800, and 1600 µg/mL, respectively. Although the mature (24 h) biofilm formation was more resistant to Abp013, the endolysin was able to reduce the CFU by 0.827 log units (85.13%) and 0.777 log units (83.32%)—for the Abp013 concentrations of 800 and 1600 µg/mL, respectively. | [212] |
| virion-associated lysins (VAL), PG_binding_3 domain-containing protein, putative endolysin, putative chitinase, carboxypeptidase, 1,4-beta-N-acetylmuramidase, putative lytic murein transglycosylase, and lysozyme | nd | Of the VAL proteins, only 1 of 3 was able to inhibit biofilm formation (with antimicrobial peptide MTKIGKRFRTKN). Furthermore, putative endolysin, putative chitinase, carboxypeptidase, and putative lytic murein transglycosylase were all 1 of 2 in terms of acting as an inhibitor of A. baumannii biofilms—GFRRKRPVSKYNKQQYIA (putative endolysin), GFRRKRPVSKYNKQQYIA (putative chitinase), FRRKRPVSKYNKQQYIA (carboxypeptidase), GWKHQRGALYSRNVLKKANY (putative lytic murein transglycosylase). All three members (PG_binding_3 domain-containing protein (LVRVLNIMQGQR), 1,4-beta-N-acetylmuramidase (KGRKSKVINSKGL), and lysozyme (LKYKYVAKRDCS)) had anti-biofilm activity. Importantly, all these lytic peptides were defined as not toxic to other cells (e.g., lacking poisonous placements of amino acids like His, Cys, Pro, Asn). | [213] |
| lysAB-vT2 fused with the hydrophobic amino acid at the C-terminus | phage vB_AbaM_PhT2 | The highest density decrease (by 1.5 log unit) of mature biofilm formed by A. baumannii strain AB183 by lysAB-vT2-fusion was observed at 4 µg/mL concentration. It was similar to the activity of the vPhT2 (Φ2) phage (from which the endolysin was derived) with the titer 108 PFU/mL. 2 µg/mL, and lower concentrations of the lysAB-vT2-fusion showed increased biofilm formation by the A. baumannii strain AB183 compared to the control sample. | [214] |
| endolysin Kp84B, linked to ApoE23 and COG133 peptides | phage ФKp84B | Chimeric protein significantly decreased the biofilm bacterial OD600 from 0.7 to 0.2. Moreover, it reduced the log unit of persister cells from 6.0 to 4.5 compared to the non-treated control sample. | [215] |
| depolymerase Dpo1 | phage Petty (podovirus) | Dpo1 was able to disrupt biofilms formed by many tested A. baumannii strains; the results were not satisfying, since this protein degraded biofilm maximally by ~20%. Interestingly, the biofilm formed by the host of a Petty phage (AU0783) was the most resistant to the activity of Dpo1. | [216] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grygiel, I.; Bajrak, O.; Wójcicki, M.; Krusiec, K.; Jończyk-Matysiak, E.; Górski, A.; Majewska, J.; Letkiewicz, S. Comprehensive Approaches to Combatting Acinetobacter baumannii Biofilms: From Biofilm Structure to Phage-Based Therapies. Antibiotics 2024, 13, 1064. https://doi.org/10.3390/antibiotics13111064
Grygiel I, Bajrak O, Wójcicki M, Krusiec K, Jończyk-Matysiak E, Górski A, Majewska J, Letkiewicz S. Comprehensive Approaches to Combatting Acinetobacter baumannii Biofilms: From Biofilm Structure to Phage-Based Therapies. Antibiotics. 2024; 13(11):1064. https://doi.org/10.3390/antibiotics13111064
Chicago/Turabian StyleGrygiel, Ilona, Olaf Bajrak, Michał Wójcicki, Klaudia Krusiec, Ewa Jończyk-Matysiak, Andrzej Górski, Joanna Majewska, and Sławomir Letkiewicz. 2024. "Comprehensive Approaches to Combatting Acinetobacter baumannii Biofilms: From Biofilm Structure to Phage-Based Therapies" Antibiotics 13, no. 11: 1064. https://doi.org/10.3390/antibiotics13111064
APA StyleGrygiel, I., Bajrak, O., Wójcicki, M., Krusiec, K., Jończyk-Matysiak, E., Górski, A., Majewska, J., & Letkiewicz, S. (2024). Comprehensive Approaches to Combatting Acinetobacter baumannii Biofilms: From Biofilm Structure to Phage-Based Therapies. Antibiotics, 13(11), 1064. https://doi.org/10.3390/antibiotics13111064