
Source Attribution of Antibiotic Resistance Genes in Estuarine Aquaculture: A Machine Learning Approach

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
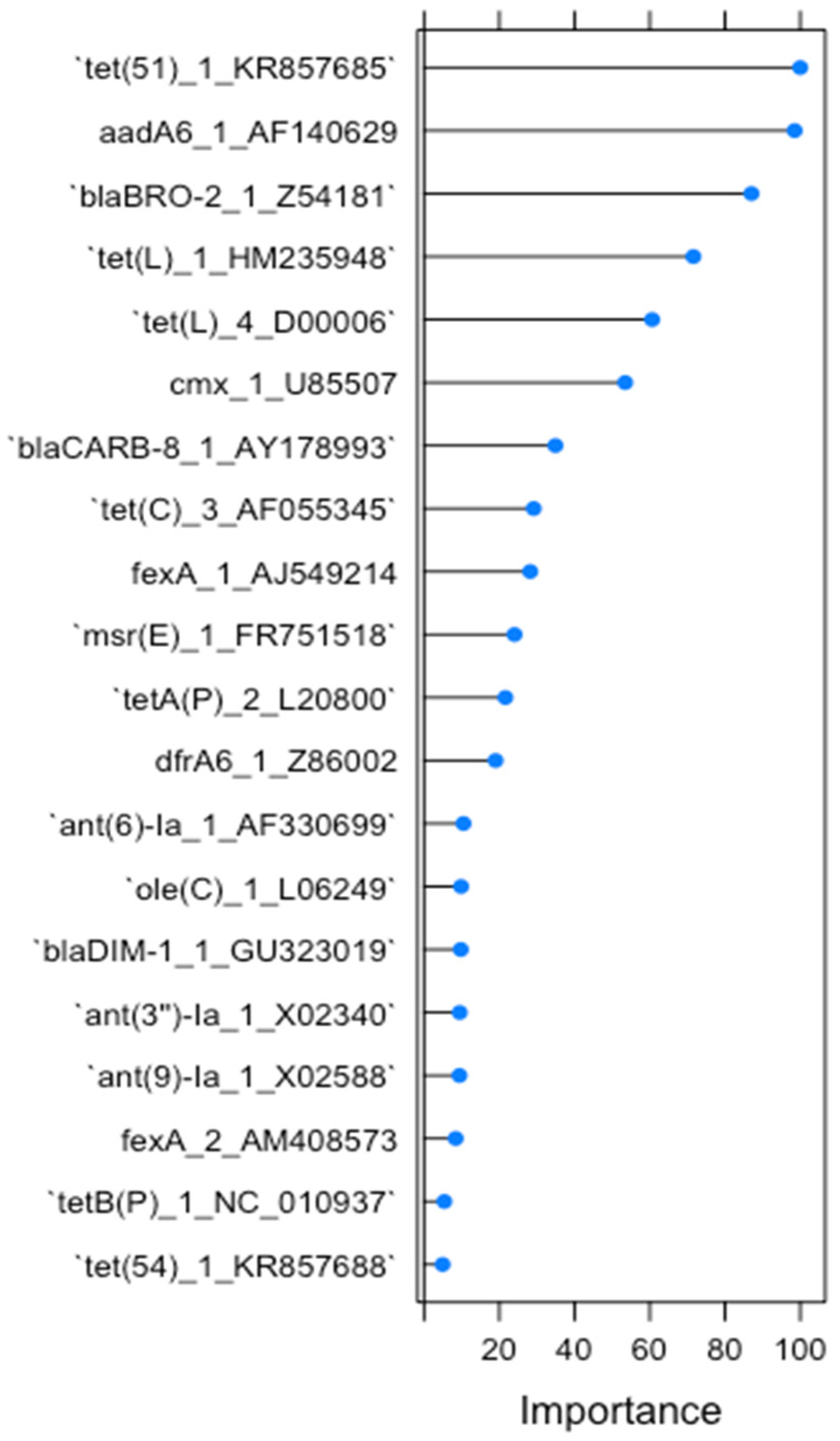
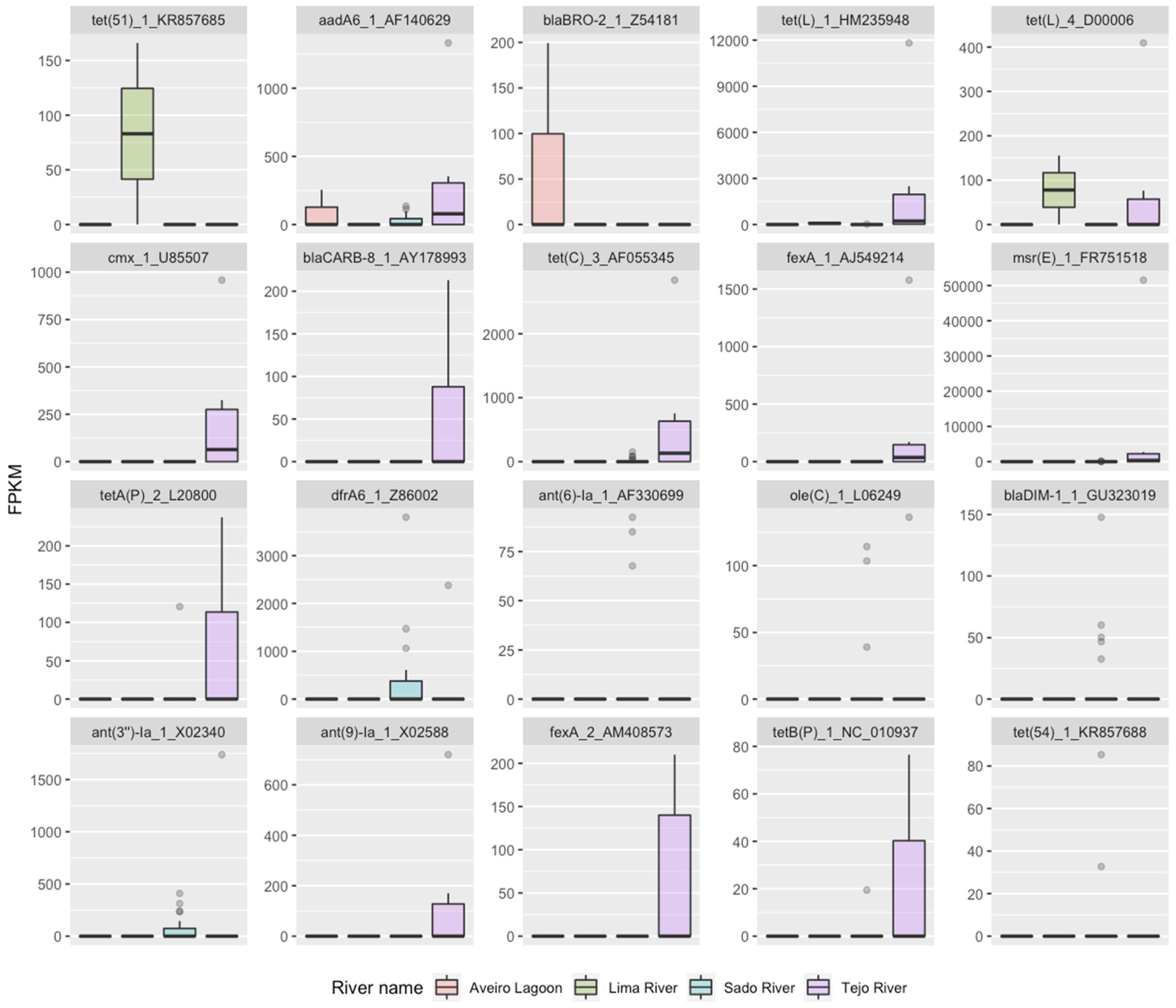
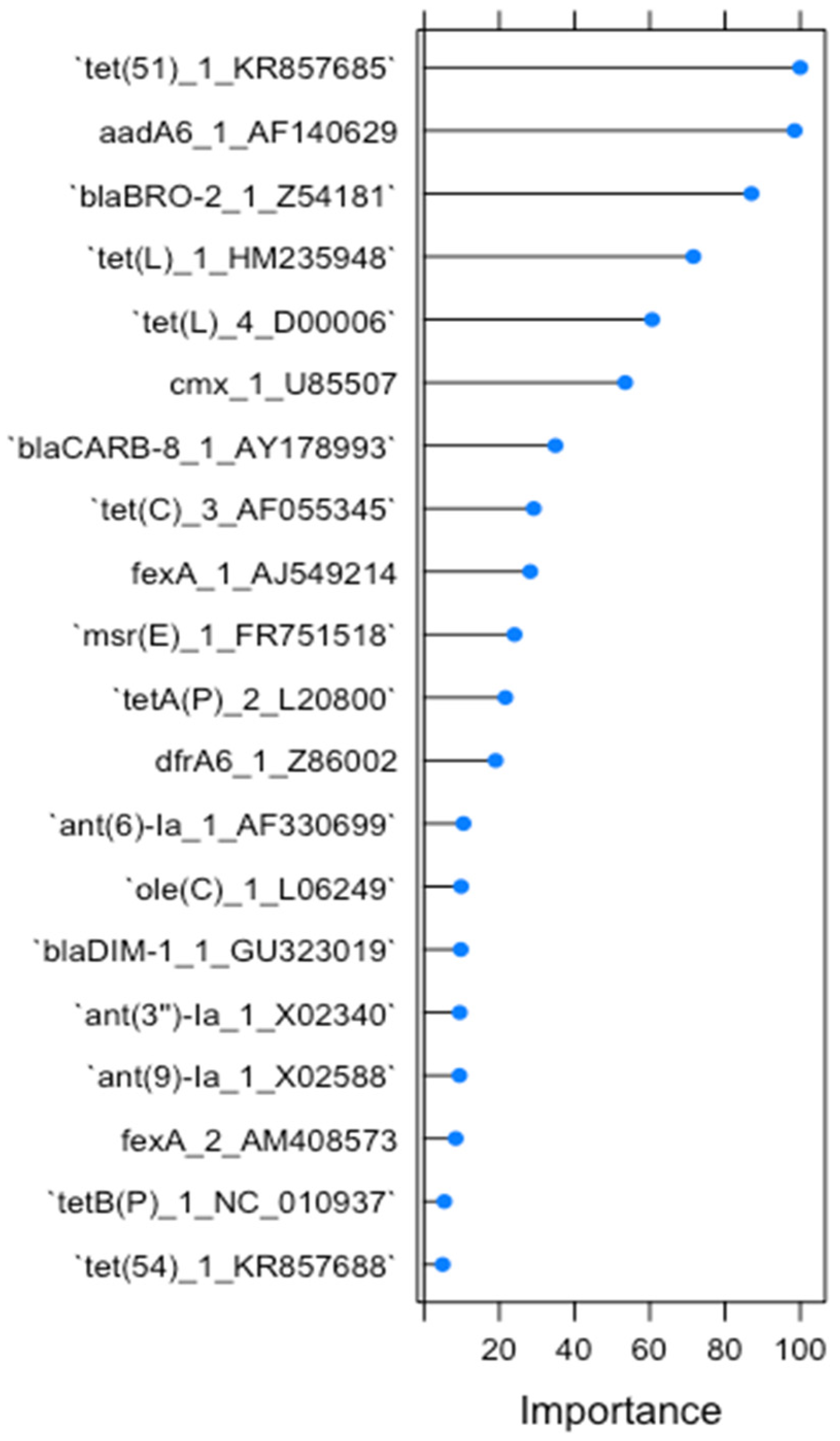
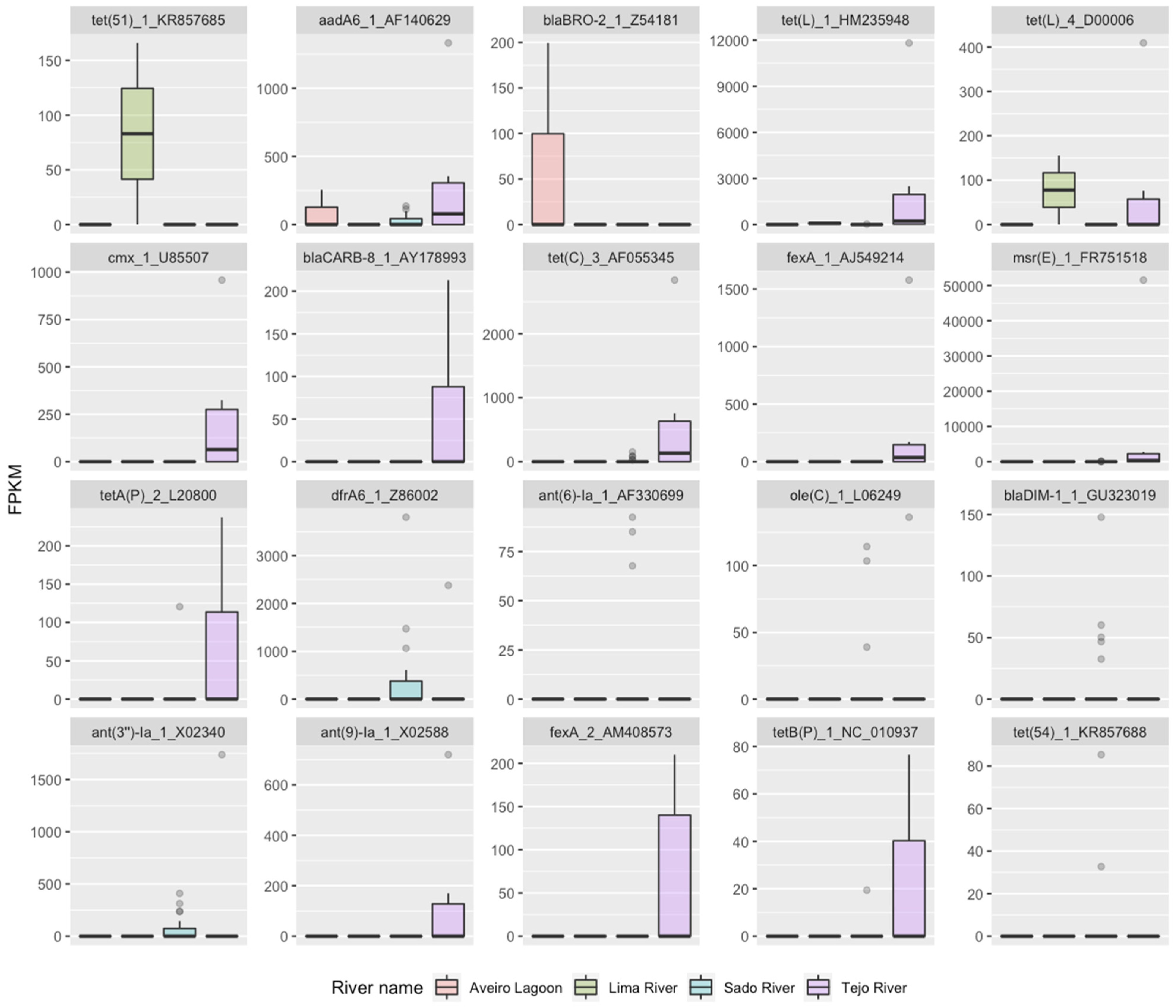
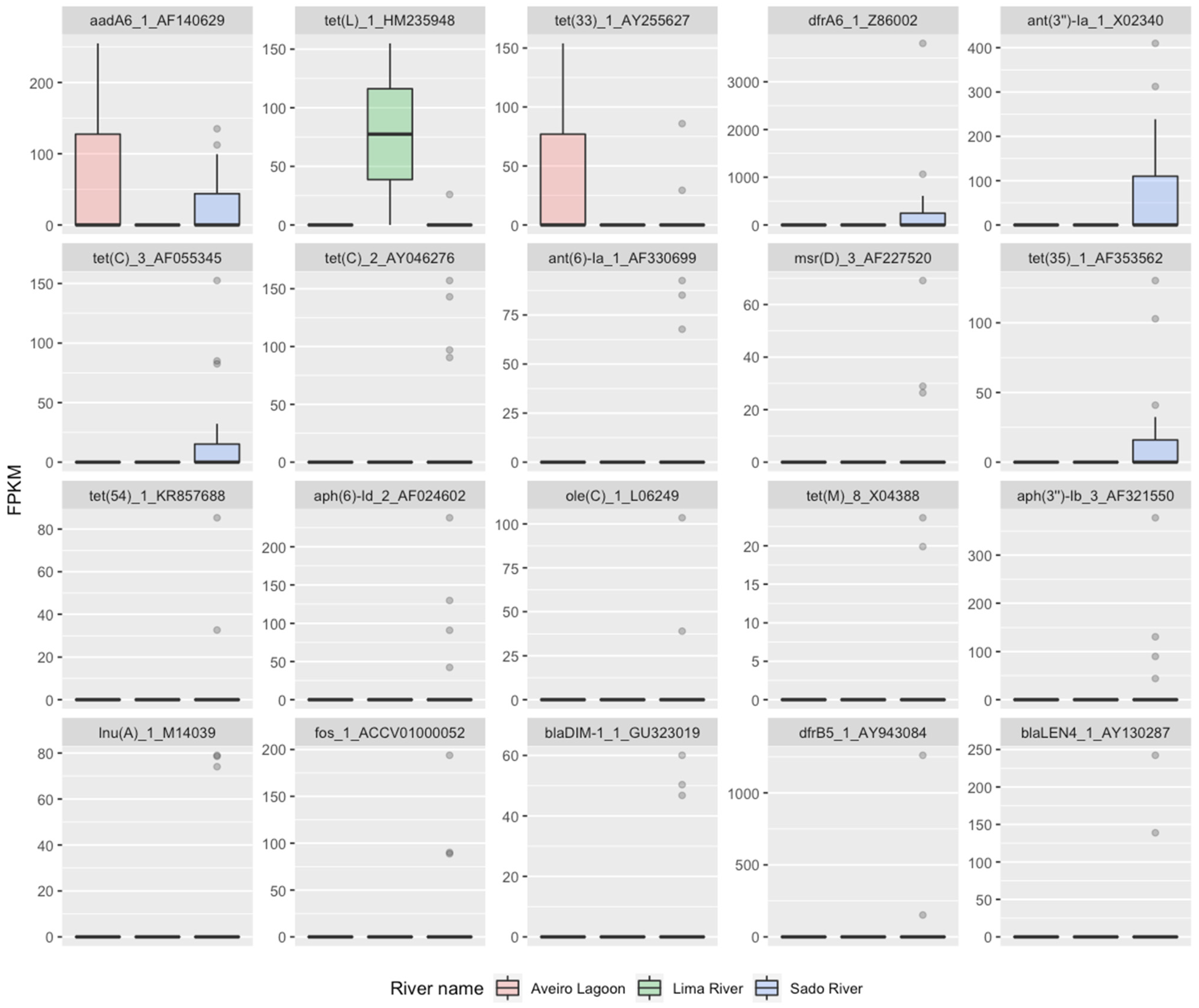
2.1. RF1—Source Attribution to Aquatic Environment Using Resistomes of Mussel, Gilt-Head Sea Bream and Oyster Aquaculture Sediments
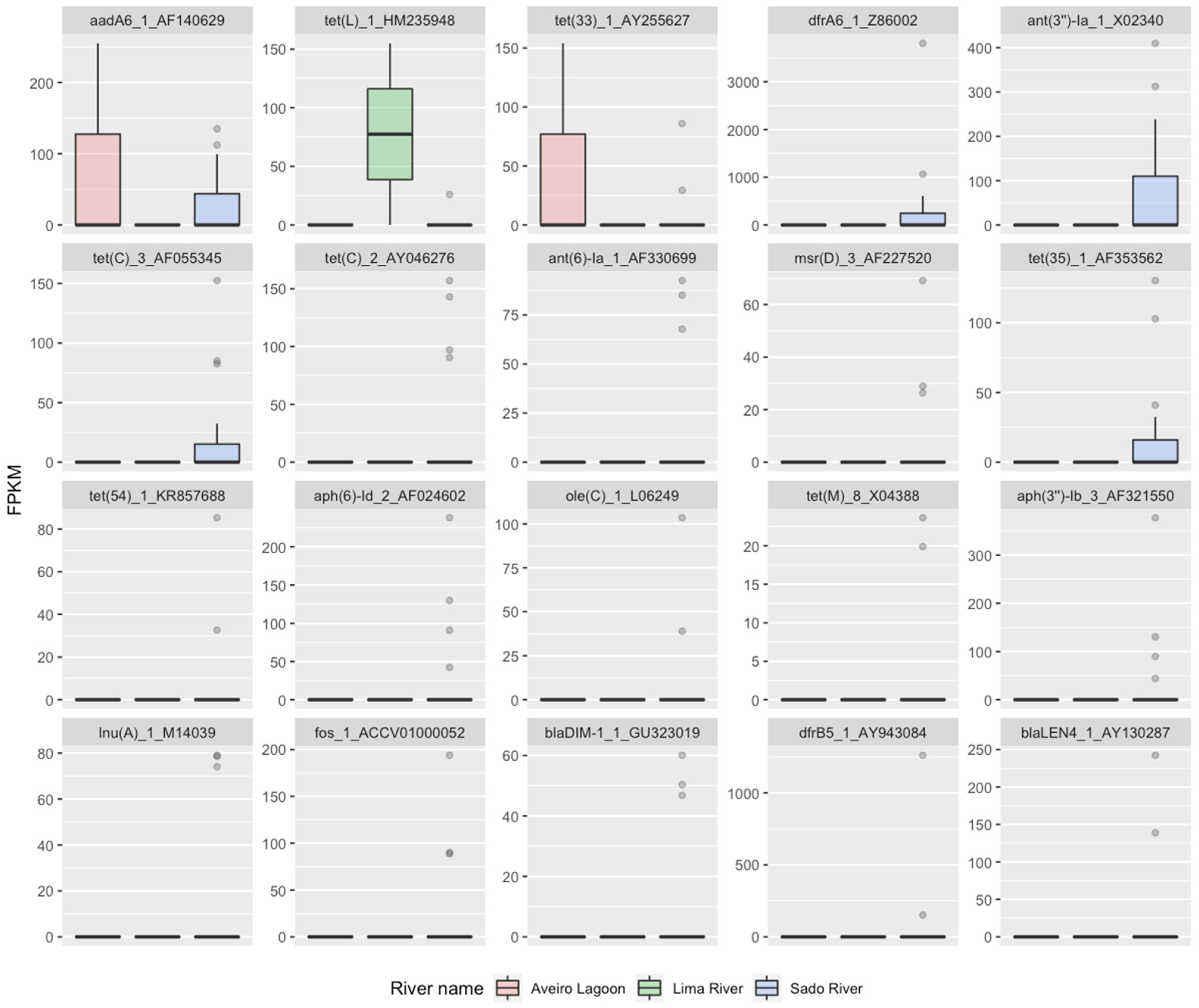
2.2. RF2—Source Attribution to Aquatic Environment Using Resistomes of Oyster Aquaculture Sediments
3. Discussion
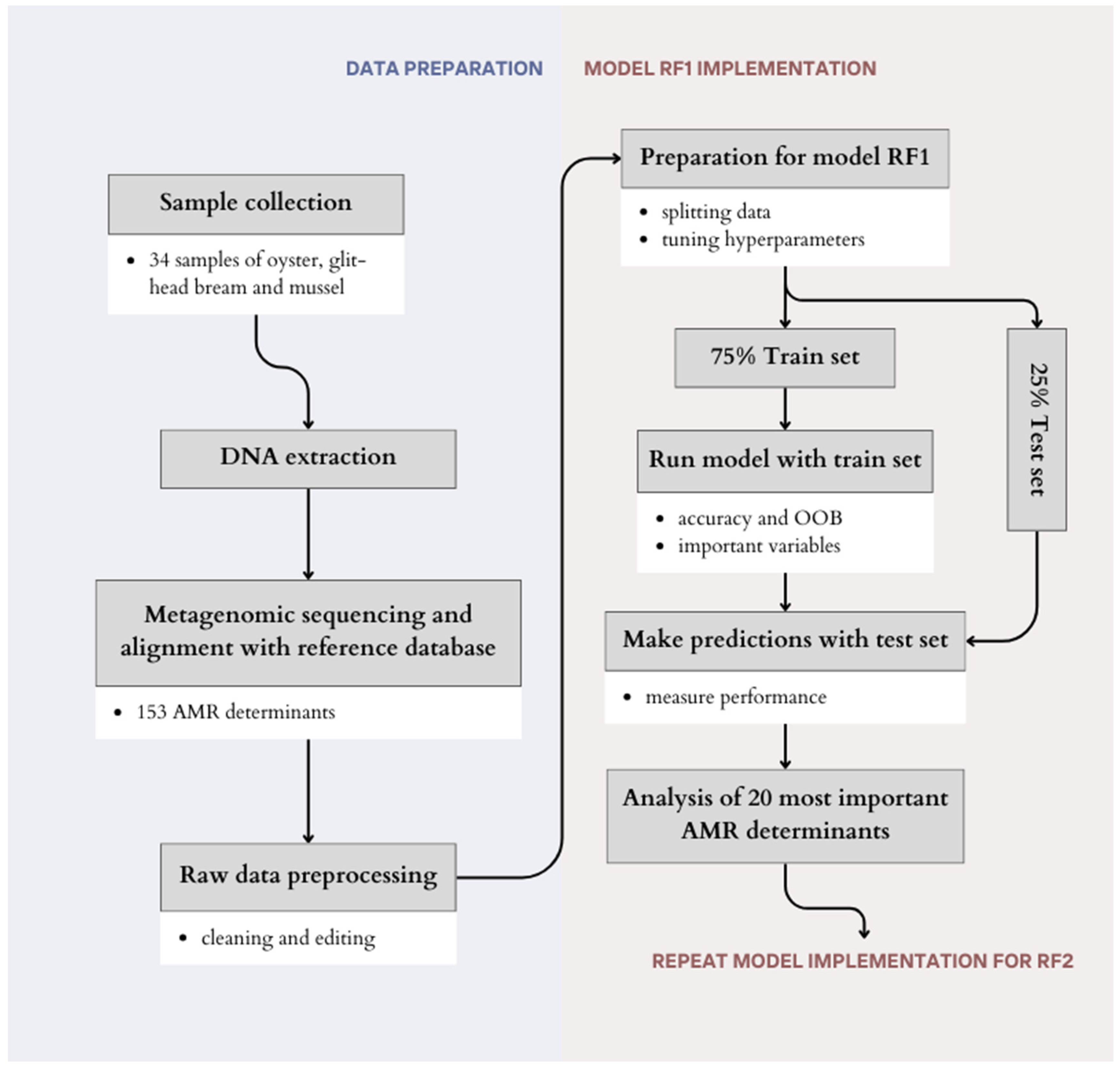
4. Materials and Methods
4.1. Sample Collection and DNA Extraction
4.2. Metagenomic Sequencing, Reference Databases and Bioinformatic Analysis
4.3. Machine-Learning Based Source-Attribution
4.3.1. RF1—Source Attribution to Aquatic Environment Using Mussel, Gilt-Head Bream and Oyster Resistomes
4.3.2. RF2—Source Attribution to Aquatic Environment Using Oyster Resistomes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reverter, M.; Sarter, S.; Caruso, D.; Avarre, J.-C.; Combe, M.; Pepey, E.; Pouyaud, L.; Vega-Heredía, S.; de Verdal, H.; Gozlan, R.E. Aquaculture at the crossroads of global warming and antimicrobial resistance. Nat. Commun. 2020, 11, 1870. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.; Day, C.; Krzanowski, J.; Nutt, D.; Carhart-Harris, R. Patients’ Accounts of Increased “Connectedness” and “Acceptance” After Psilocybin for Treatment-Resistant Depression. J. Humanist. Psychol. 2017, 57, 520–564. [Google Scholar] [CrossRef]
- Koch, N.; Islam, N.F.; Sonowal, S.; Prasad, R.; Sarma, H. Environmental antibiotics and resistance genes as emerging contaminants: Methods of detection and bioremediation. Curr. Res. Microb. Sci. 2021, 2, 100027. [Google Scholar] [CrossRef] [PubMed]
- European Comission. Proposal for a Directive of the European Parliament and of the Council Concerning Urban Wastewater Treatment (Recast); COM/2022/0541; European Commission: Brussels, Belgium, 2022.
- Helsens, N.; Calvez, S.; Prevost, H.; Bouju-Albert, A.; Maillet, A.; Rossero, A.; Hurtaud-Pessel, D.; Zagorec, M.; Magras, C. Antibiotic Resistance Genes and Bacterial Communities of Farmed Rainbow Trout Fillets (Oncorhynchus mykiss). Front Microbiol. 2020, 11, 590902. [Google Scholar] [CrossRef]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards); Koutsoumanis, K.; Allende, A.; Álvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; et al. Role played by the environment in the emergence and spread of antimicrobial resistance (AMR) through the food chain. EFSA J. 2021, 19, 188. [Google Scholar] [CrossRef]
- Carvalho, I.T.; Santos, L. Antibiotics in the aquatic environments: A review of the European scenario. Environ. Int. 2016, 94, 736–757. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Jia, W.-L.; Song, C.; He, L.-Y.; Wang, B.; Gao, F.-Z.; Zhang, M.; Ying, G.-G. Antibiotics in soil and water: Occurrence, fate, and risk. Curr. Opin. Environ. Sci. Health 2023, 32, 100437. [Google Scholar] [CrossRef]
- Bottery, M.J.; Pitchford, J.W.; Friman, V.-P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 2020, 15, 939–948. [Google Scholar] [CrossRef]
- Li, L.-G.; Yin, X.; Zhang, T. Tracking antibiotic resistance gene pollution from different sources using machine-learning classification. Microbiome 2018, 6, 93. [Google Scholar] [CrossRef]
- de Abreu, V.A.C.; Perdigão, J.; Almeida, S. Metagenomic Approaches to Analyze Antimicrobial Resistance: An Overview. Front. Genet. 2021, 11, 575592. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Arango-Argoty, G.; Zhang, L.; Pruden, A.; Vikesland, P. Identification of discriminatory antibiotic resistance genes among environmental resistomes using extremely randomized tree algorithm. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.S.R.; Röder, T.; Van Gompel, L.; Petersen, T.N.; Hansen, R.B.; Hansen, I.M.; Bossers, A.; Aarestrup, F.M.; Wagenaar, J.A.; Hald, T. Metagenomics-Based Approach to Source-Attribution of Antimicrobial Resistance Determinants—Identification of Reservoir Resistome Signatures. Front. Microbiol. 2021, 11, 601407. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.G.; Domingues, C.P.F.; Figueiredo, J.F.; Dionisio, F.; Botelho, A.; Nogueira, T. Estuarine Aquacultures at the Crossroads of Animal Production and Antibacterial Resistance: A Metagenomic Approach to the Resistome. Biology 2022, 11, 1681. [Google Scholar] [CrossRef] [PubMed]
- Muziasari, W.I.; Pitkänen, L.K.; Sørum, H.; Stedtfeld, R.D.; Tiedje, J.M.; Virta, M. The Resistome of Farmed Fish Feces Contributes to the Enrichment of Antibiotic Resistance Genes in Sediments below Baltic Sea Fish Farms. Front. Microbiol. 2017, 7, 2137. [Google Scholar] [CrossRef] [PubMed]
- Cabello, F.C.; Godfrey, H.P.; Tomova, A.; Ivanova, L.; Dölz, H.; Millanao, A.; Buschmann, A.H. Antimicrobial use in aquaculture re-examined: Its relevance to antimicrobial resistance and to animal and human health. Environ. Microbiol. 2013, 15, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Salgueiro, V.; Manageiro, V.; Bandarra, N.M.; Reis, L.; Ferreira, E.; Caniça, M. Bacterial Diversity and Antibiotic Susceptibility of Sparus aurata from Aquaculture. Microorganisms 2020, 8, 1343. [Google Scholar] [CrossRef]
- Watts, J.E.M.; Schreier, H.J.; Lanska, L.; Hale, M.S. The Rising Tide of Antimicrobial Resistance in Aquaculture: Sources, Sinks and Solutions. Mar. Drugs 2017, 15, 158. [Google Scholar] [CrossRef]
- Rocha, C.P.; Cabral, H.N.; Marques, J.C.; Gonçalves, A.M.M. A Global Overview of Aquaculture Food Production with a Focus on the Activity’s Development in Transitional Systems—The Case Study of a South European Country (Portugal). J. Mar. Sci. Eng. 2022, 10, 417. [Google Scholar] [CrossRef]
- Silva, I.; Tacão, M.; Henriques, I. Selection of antibiotic resistance by metals in a riverine bacterial community. Chemosphere 2021, 263, 127936. [Google Scholar] [CrossRef]
- Phakhounthong, K.; Chaovalit, P.; Jittamala, P.; Blacksell, S.D.; Carter, M.J.; Turner, P.; Chheng, K.; Sona, S.; Kumar, V.; Day, N.P.J.; et al. Predicting the severity of dengue fever in children on admission based on clinical features and laboratory indicators: Application of classification tree analysis. BMC Pediatr. 2018, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.G.; Hoffmann, T.; Greenbury, S.F.; Cominetti, O.; Jallow, M.; Kwiatkowski, D.; Barahona, M.; Jones, N.S.; Casals-Pascual, C. Precision identification of high-risk phenotypes and progression pathways in severe malaria without requiring longitudinal data. NPJ Digit. Med. 2019, 2, 63. [Google Scholar] [CrossRef] [PubMed]
- Kabaria, C.W.; Molteni, F.; Mandike, R.; Chacky, F.; Noor, A.M.; Snow, R.W.; Linard, C. Mapping intra-urban malaria risk using high resolution satellite imagery: A case study of Dar es Salaam. Int. J. Health Geogr. 2016, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haddawy, P.; Hasan, A.I.; Kasantikul, R.; Lawpoolsri, S.; Sa-Angchai, P.; Kaewkungwal, J.; Singhasivanon, P. Spatiotemporal Bayesian networks for malaria prediction. Artif. Intell. Med. 2018, 84, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Clemente, L.; Lu, F.; Santillana, M. Improved Real-Time Influenza Surveillance: Using Internet Search Data in Eight Latin American Countries. JMIR Public Health Surveill. 2019, 5, e12214-58. [Google Scholar] [CrossRef] [PubMed]
- Rosas, M.A.; Bezerra, A.F.B.; Duarte-Neto, P.J. Use of artificial neural networks in applying methodology for allocating health resources. Public Health Prac. 2013, 47, 128–136. [Google Scholar] [CrossRef]
- Yousefi, M.; Yousefi, M.; Ferreira, R.P.M.; Kim, J.H.; Fogliatto, F.S. Chaotic genetic algorithm and Adaboost ensemble metamodeling approach for optimum resource planning in emergency departments. Artif. Intell. Med. 2018, 84, 23–33. [Google Scholar] [CrossRef]
- Elyan, E.; Hussain, A.; Sheikh, A.; Elmanama, A.A.; Vuttipittayamongkol, P.; Hijazi, K. Antimicrobial Resistance and Machine Learning: Challenges and Opportunities. IEEE Access 2022, 10, 31561–31577. [Google Scholar] [CrossRef]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef]
- Luan, J.; Zhang, C.; Xu, B.; Xue, Y.; Ren, Y. The predictive performances of random forest models with limited sample size and different species traits. Fish. Res. 2020, 227, 105534. [Google Scholar] [CrossRef]
- Ghojogh, B.; Crowley, M. The Theory Behind Overfitting, Cross Validation, Regularization, Bagging, and Boosting: Tutorial. Cornell University. arXiv 2019, arXiv:1905.12787. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M.C. Tetracycline antibiotics: Mode of action, applications, molecular biology and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, A.J.; Markley, J.L.; Kumar, H.; Wang, B.; Fang, L.; Irum, S.; Symister, C.T.; Wallace, M.; Burnham, C.-A.D.; Andleeb, S.; et al. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun. Biol. 2020, 3, 241. [Google Scholar] [CrossRef] [PubMed]
- Ungemach, F.R.; Müller-Bahrdt, D.; Abraham, G. Guidelines for prudent use of antimicrobials and their implications on antibiotic usage in veterinary medicine. Int. J. Med. Microbiol. 2006, 296 (Suppl. S41), 33–38. [Google Scholar] [CrossRef]
- Gao, P.; Mao, D.; Luo, Y.; Wang, L.; Xu, B.; Xu, L. Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res. 2012, 46, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Sun, Y.; Zhang, T.; Ding, X.; Li, Y.; Wang, M.; Zeng, Z. Antibiotics, Antibiotic Resistance Genes, and Bacterial Community Composition in Fresh Water Aquaculture Environment in China. Microb. Ecol. 2015, 70, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mao, C.; Lei, L.; Yan, B.; Yuan, J.; Guo, Y.; Li, T.; Xiong, X.; Cao, X.; Huang, J.; et al. Antibiotic resistance genes and their links with bacteria and environmental factors in three predominant freshwater aquaculture modes. Ecotoxicol. Environ. Saf. 2022, 241, 113832. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.; Barton, G.; Kietzmann, M.; Meißner, J. Treatment of pigs with enrofloxacin via different oral dosage forms—environmental contaminations and resistance development of Escherichia coli. J. Vet Sci. 2022, 23, e23. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cutler, A.; Cutler, D.R.; Stevens, J.R. Random forests. In Ensemble Machine Learning; Zhang, C., Ma, Y.Q., Eds.; Springer: Boston, MA, USA, 2012. [Google Scholar]
- Biau, G.; Devroye, L.; Lugosi, G. Consistency of Random Forests and Other Averaging Classifiers. J. Mach. Learn. Res. 2008, 9, 2015–2033. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Ling, C.X.; Li, C. Data mining for direct marketing: Problems and solutions. In Proceedings of the Fourth International Conference on Knowledge Discovery and Data Mining (KDD), New York, NY, USA, 27–31 August 1998; pp. 73–79. [Google Scholar]
- Ljumovic, M.; Klar, M. Estimating expected error rates of random forest classifiers: A comparison of cross-validation and bootstrap. In Proceedings of the 4th Mediterranean Conference on Embedded Computing (MECO), Budva, Montenegro, 14–18 June 2015; pp. 212–215. [Google Scholar]
- Goldstein, B.A.; Hubbard, A.E.; Cutler, A.; Barcellos, L.F. An application of Random Forests to a genome-wide association dataset: Methodological considerations & new findings. BMC Genet. 2010, 11, 49. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, D.; Grisoni, F.; Todeschini, R. Multivariate comparison of classification performance measures. Chemom. Intell. Lab. Syst. 2018, 174, 33–44. [Google Scholar] [CrossRef]
- Kuhn, M.; Johnson, K. Measuring Predictor Importance. In Applied Predictive Modeling, 1st ed.; Springer: New York, NY, USA, 2013; pp. 463–485. [Google Scholar] [CrossRef]
- Yuan, Y.; Wu, L.; Zhang, X. Gini-Impurity Index Analysis. IEEE Trans. Inf. Forensics Secur. 2021, 16, 3154–3169. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salgueiro, H.S.; Ferreira, A.C.; Duarte, A.S.R.; Botelho, A. Source Attribution of Antibiotic Resistance Genes in Estuarine Aquaculture: A Machine Learning Approach. Antibiotics 2024, 13, 107. https://doi.org/10.3390/antibiotics13010107
Salgueiro HS, Ferreira AC, Duarte ASR, Botelho A. Source Attribution of Antibiotic Resistance Genes in Estuarine Aquaculture: A Machine Learning Approach. Antibiotics. 2024; 13(1):107. https://doi.org/10.3390/antibiotics13010107
Chicago/Turabian StyleSalgueiro, Helena Sofia, Ana Cristina Ferreira, Ana Sofia Ribeiro Duarte, and Ana Botelho. 2024. "Source Attribution of Antibiotic Resistance Genes in Estuarine Aquaculture: A Machine Learning Approach" Antibiotics 13, no. 1: 107. https://doi.org/10.3390/antibiotics13010107
APA StyleSalgueiro, H. S., Ferreira, A. C., Duarte, A. S. R., & Botelho, A. (2024). Source Attribution of Antibiotic Resistance Genes in Estuarine Aquaculture: A Machine Learning Approach. Antibiotics, 13(1), 107. https://doi.org/10.3390/antibiotics13010107