

Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa


 ,
, 
Abstract
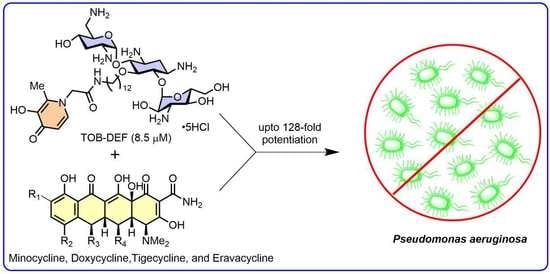
1. Introduction
2. Results
2.1. Synthesis of TOB-DEF Conjugates 1a–c and Control Compounds 2 and 3
2.2. Antibacterial Activity of TOB-DEF Conjugates 1a–c and Compounds 2–3
2.3. TOB-DEF Conjugates 1a–c Potentiate Multiple Classes of Antibiotics against P. aeruginosa PAO1
2.4. Conjugate 1c Synergizes with a Panel of Antibiotics against MDR Isolates of P. aeruginosa
2.5. Conjugate 1c Exhibited Superior Potentiation of Tetracyclines When Compared with Control Compounds 2 and 3
2.6. Tetracycline Potentiation of Conjugate 1c Is Reduced under Iron-Depleted (ID) Conditions
2.7. Compound 1c Disrupts the Outer Membrane of P. aeruginosa Isolates
2.8. Cytotoxicity Study of Conjugates 1a–c
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Compounds 1a–c, and 2–9
4.2.1. Synthesis of 1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-tobramycin (4)
4.2.2. General Procedure A: Preparation of 5-O-(N-Bromoalkane)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (5a–c)
4.2.3. 5-O-(4-Bromobutyl)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (5a)
4.2.4. 5-O-(4-Bromooctane)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (5b)
4.2.5. 5-O-(4-Bromododecane)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (5c)
4.2.6. Procedure B: Preparation of 5-O-(N-Amino-Alkylated)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (6a–c)
4.2.7. 5-O-(4-Aminobutyl)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (6a)
4.2.8. 5-O-(8-Aminooctyl)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (6b)
4.2.9. 5-O-(12-Aminododecyl)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (6c)
4.2.10. Synthesis of 2-(3-Hydroxy-4-Oxo-1,4-Dihydropyridin-1-Yl) Acetic Acid (7)
4.2.11. General Procedure C: Amide Coupling Reaction for the Preparation of Compounds (8a–c)
4.2.12. 5-O-(Butyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (8a)
4.2.13. 5-O-(Octyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (8b)
4.2.14. 5-O-(Dodecyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-1,3,2′,6′,3″-Penta-N-(Tert-Butoxycarbonyl)-4′,2″,4″,6″-Tetra-O-TBDMS-Tobramycin (8c)
4.2.15. 5-O-(Dodecyl)-1,3,2′,6′,3″-Penta-N-Boc-4′,2″,4′′,6′′-Tetra-O-TBDMS-Tobramycin (9)
4.2.16. General Procedure D: Removal of All the Protecting Groups for the Preparation of Compounds (1a–c)
4.2.17. 5-O-(Butyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-Tobramycin∙5HCl (1a)
4.2.18. 5-O-(Octyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-Tobramycin∙5HCl (1b)
4.2.19. 5-O-(Dodecyl-2-(3-Hydroxy-2-Methyl-4-Oxopyridin-1(4H)-Yl)Acetamide)-Tobramycin∙5HCl (1c)
4.2.20. 5-O-(Dodecyl)-Tobramycin (2)
4.2.21. N-Dodecyl-2-(3-Hydroxy-4-Oxo-1,4-Dihydropyridin-1-Yl) Acetamide (3)
4.3. Microbiology
4.3.1. Antibacterial Susceptibility Assay
4.3.2. Checkerboard Assay
4.4. Cell Viability Assay
Toxicity against HEK293 and HepG2 Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CDC. Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Atlanta, GA, USA, 2019.
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017.
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.J. Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol. Rev. 2018, 42, fux053. [Google Scholar] [CrossRef]
- Planet, P.J. Pseudomonas aeruginosa. In Principles and Practice of Pediatric Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2018; pp. 866–870. [Google Scholar]
- Rhodes, N.J.; Cruce, C.E.; O’Donnell, J.N.; Wunderink, R.G.; Hauser, A.R. Resistance trends and treatment options in gram-negative ventilator-associated pneumonia. Curr. Infect. Dis. Rep. 2018, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Finnan, S.; Morrissey, J.P.; O’gara, F.; Boyd, E.F. Genome diversity of Pseudomonas aeruginosa isolates from cystic fibrosis patients and the hospital environment. J. Clin. Microbiol. 2004, 42, 5783–5792. [Google Scholar] [CrossRef] [PubMed]
- LiPuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Jain, M.; Bar-Meir, M.; McColley, S. Clinical significance of microbial infection and adaptation in cystic fibrosis. Clin. Microbiol. Rev. 2011, 24, 29–70. [Google Scholar] [CrossRef]
- Abdul Wahab, A.; Zahraldin, K.; Ahmed, M.A.S.; Jarir, S.A.; Muneer, M.; Mohamed, S.F.; Hamid, J.M.; Hassan, A.A.; Ibrahim, E.B. The emergence of multidrug-resistant Pseudomonas aeruginosa in cystic fibrosis patients on inhaled antibiotics. Lung India Off. Organ Indian Chest Soc. 2017, 34, 527. [Google Scholar] [CrossRef]
- Kunz Coyne, A.J.; El Ghali, A.; Holger, D.; Rebold, N.; Rybak, M. Therapeutic strategies for emerging multidrug-resistant Pseudomonas aeruginosa. Infect. Dis. Ther. 2022, 11, 661–682. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.; Lin, T.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin. Infect. Dis. 2019, 69, 538–543. [Google Scholar] [CrossRef]
- Cornelis, P.; Dingemans, J. Pseudomonas aeruginosa adapts its iron uptake strategies in function of the type of infections. Front Cell Infect. Microbiol. 2013, 3, 75. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Cheng, W.; Wei, H.; Yuan, Y.; Yang, Z.; Zhang, X. An overview of recent progress in siderophore-antibiotic conjugates. Eur. J. Med. Chem. 2019, 182, 111615. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Srinivas, P.; Pogue, J. Cefiderocol: A novel agent for the management of multidrug-resistant gram-negative organisms. Infect. Dis. Ther. 2020, 9, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Nishikawa, T.; Matsumoto, S.; Yoshizawa, H.; Sato, T.; Nakamura, R.; Tsuji, M.; Yamano, Y. Siderophore cephalosporin cefiderocol utilizes ferric iron transporter systems for antibacterial activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 7396–7401. [Google Scholar] [CrossRef]
- Zhanel, G.; Golden, A.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.; Adam, H.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.; et al. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline Antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Tabcheh, J.; Vergalli, J.; Davin-Régli, A.; Ghanem, N.; Pages, J.-M.; Al-Bayssari, C.; Brunel, J.M. Rejuvenating the Activity of Usual Antibiotics on Resistant Gram-Negative Bacteria: Recent Issues and Perspectives. Int. J. Mol. Sci. 2023, 24, 1515. [Google Scholar] [CrossRef]
- Si, Z.; Pethe, K.; Chan-Park, M.B. Chemical Basis of Combination Therapy to Combat Antibiotic Resistance. JACS Au 2023, 3, 276–292. [Google Scholar] [CrossRef]
- Rusu, A.; Buta, E.L. The Development of Third-Generation Tetracycline Antibiotics and New Perspectives. Pharmaceutics 2021, 13, 2085. [Google Scholar] [CrossRef]
- Eliopoulos, G.; Roberts, M. Tetracycline therapy: Update. Clin. Infect. Dis. 2003, 36, 462–467. [Google Scholar]
- Grossman, T. Tetracycline antibiotics and resistance. Cold Spring Harb Perspect Med. 2016, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Hancock, R. Adaptive and mutational resistance: Role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.; Lee, C. Distinct roles of outer membrane porins in antibiotic resistance and membrane integrity in Escherichia coli. Front. Microbiol. 2019, 953, 1–9. [Google Scholar] [CrossRef]
- Gasparrini, A.J.; Markley, J.L.; Kumar, H.; Wang, B.; Fang, L.; Irum, S.; Symister, C.T.; Wallace, M.; Burnham, C.-A.D.; Andleeb, S.; et al. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun. Biol. 2020, 3, 241. [Google Scholar] [CrossRef]
- White, J.; Cantor, C. Role of magnesium in the binding of tetracycline to Escherichia coli ribosomes. J. Mol. Biol. 1971, 58, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Cilibrizzi, A.; Abbate, V.; Bruce, K.; Hider, R. Effect of iron chelation on anti-pseudomonal activity of doxycycline. Int. J. Antimicrob. Agents. 2021, 58, 106438. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Domalaon, R.; Lyu, Y.; Zhanel, G.G.; Schweizer, F. Tobramycin-linked efflux pump inhibitor conjugates synergize fluoroquinolones, rifampicin and fosfomycin against multidrug-resistant Pseudomonas aeruginosa. J. Clin. Med. 2018, 7, 158. [Google Scholar] [CrossRef]
- Dhiman, S.; Ramirez, D.; Li, Y.; Kumar, A.; Arthur, G.; Schweizer, F. Chimeric Tobramycin-Based Adjuvant TOB-TOB-CIP Potentiates Fluoroquinolone and β-Lactam Antibiotics against Multidrug-Resistant Pseudomonas aeruginosa. ACS Infect. Dis. 2023, 9, 864–885. [Google Scholar] [CrossRef]
- Gorityala, B.; Guchhait, G.; Fernando, D.; Deo, S.; McKenna, S.; Zhanel, G.; Kumar, A.; Schweizer, F. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem. Int. Ed. 2016, 55, 555–559. [Google Scholar] [CrossRef]
- Domalaon, R.; Idowu, T.; Zhanel, G.; Schweizer, F. Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 2018, 31, e00077-17. [Google Scholar] [CrossRef]
- Barman Balfour, J.A.; Foster, R.H. Deferiprone: A review of its clinical potentitial in iron overload in beta-thalassaemia and other transfusion-dependent diseases. Drugs 1999, 58, 553–578. [Google Scholar] [CrossRef] [PubMed]
- Mawani, Y.; Cawthray, J.; Chang, S.; Sachs-Barrable, K.; Weekes, D.; Wasan, K.; Orvig, C. In vitro studies of lanthanide complexes for the treatment of osteoporosis. Dalton Trans. 2013, 42, 5999–6011. [Google Scholar] [CrossRef]
- Odds, F. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 31st ed.; CLSI Supplement M100; Clinical and Laboratory Standards Institute: Wayne, NJ, USA, 2021. [Google Scholar]
- Clifton, L.A.; Skoda, M.W.; Le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of divalent cation removal on the structure of gram-negative bacterial outer membrane models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef]
- Klobucar, K.; Côté, J.-P.; French, S.; Borrillo, L.; Guo, A.B.Y.; Serrano-Wu, M.H.; Lee, K.K.; Hubbard, B.; Johnson, J.W.; Gaulin, J.L.; et al. Chemical Screen for Vancomycin Antagonism Uncovers Probes of the Gram-Negative Outer Membrane. ACS Chem. Biol. 2021, 16, 929–942. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Bell, A. Antibiotic uptake into Gram-negative bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 713–720. [Google Scholar] [CrossRef]
- Hancock, R.E.W. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef]
- Silver, L.L. A gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 2016, 24, 6379–6389. [Google Scholar] [CrossRef]
- Akhoundsadegh, N.; Belanger, C.R.; Hancock, R.E.W. Outer Membrane Interaction Kinetics of New Polymyxin B Analogs in Gram-Negative Bacilli. Antimicrob. Agents Chemother. 2019, 63, e00935-19. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.; DeCorby, M.; Laing, N.; Weshnoweski, B.; Vashisht, R.; Tailor, F.; Nichol, K.; Wierzbowski, A.; Baudry, P.; Karlowsky, J. Canadian antimicrobial resistance alliance (CARA) Hoban DJ; Antimicrobial resistant pathogens in intensive care units in Canada: Results of the Canadian National Intensive Care Unit (CAN-ICU) study, 2005–2006. Antimicrob. Agents Chemother. 2008, 52, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Hoban, D.J.; Zhanel, G.G. Introduction to the CANWARD study (2007–11). J. Antimicrob. Chemother. 2013, 68, i3–i5. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.; Brizuela, M.; Jackson, G.; Schweizer, F. A niclosamide–tobramycin hybrid adjuvant potentiates cefiderocol against P. aeruginosa. RSC Med. Chem. 2021, 12, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.M.; Ramirez, D.; Arthur, G.; Zhanel, G.; Schweizer, F. Guanidinylated polymyxins as outer membrane permeabilizers capable of potentiating rifampicin, erythromycin, ceftazidime and aztreonam against gram-negative bacteria. Antibiotics 2022, 11, 1277. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Antibiotic | MIC (μg/mL) of Antibiotic | |||
|---|---|---|---|---|---|
| Alone | + Compound 1c | Alone | + Compound 1c | ||
| ID-CAMHB | CAMHB | ||||
| PAO1 | MIN | 32 | 8 | 32 | 1 |
| DOX | 16 | 0.25 | 64 | 2 | |
| TIG | 64 | 2 | 32 | 4 | |
| ERV | 8 | 2 | 8 | 8 | |
| PA259 | MIN | 32 | 16 | 128 | 2 |
| DOX | 64 | 2 | 128 | 4 | |
| TIG | 64 | 2 | 64 | 1 | |
| ERV | 16 | 4 | 16 | 2 | |
| PA262 | MIN | 128 | 16 | 128 | 8 |
| DOX | 256 | 32 | 256 | 16 | |
| TIG | 64 | 16 | 64 | 16 | |
| ERV | 16 | 8 | 16 | 16 | |
| PA264 | MIN | 64 | 4 | 64 | 2 |
| DOX | 64 | 16 | 32 | 0.25 | |
| TIG | 64 | 4 | 64 | 1 | |
| ERV | 16 | 16 | 16 | 4 | |
| Strain | Antibiotic | Concentration of 1c (μg/mL) | MIC (μg/mL) of Antibiotic | |||
|---|---|---|---|---|---|---|
| Alone | + Compound 1c | Alone | + Compound 1c | |||
| CAMHB | Mg2+ Supplemented CAMHB | |||||
| PAO1 | MIN | 8 | 32 | 1 | >512 | >512 |
| 16 | 32 | 1 | >512 | >512 | ||
| 32 | 32 | 1 | >512 | >512 | ||
| 64 | 32 | 0.5 | >512 | 128 | ||
| DOX | 8 | 64 | 2 | 512 | 512 | |
| 16 | 64 | 2 | 512 | 256 | ||
| 32 | 64 | 1 | 512 | 256 | ||
| 64 | 64 | 0.5 | 512 | 128 | ||
| PA259 | MIN | 8 | 128 | 2 | >512 | >512 |
| 16 | 128 | 2 | >512 | >512 | ||
| 32 | 128 | 2 | >512 | >512 | ||
| 64 | 128 | 1 | >512 | 128 | ||
| DOX | 8 | 128 | 4 | 512 | 512 | |
| 16 | 128 | 2 | 512 | 256 | ||
| 32 | 128 | 2 | 512 | 256 | ||
| 64 | 128 | 2 | 512 | 128 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, K.; Dhiman, S.; Arora, R.; Ramirez, D.M.; Ramirez, D.; Arthur, G.; Schweizer, F. Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics 2023, 12, 1261. https://doi.org/10.3390/antibiotics12081261
Gandhi K, Dhiman S, Arora R, Ramirez DM, Ramirez D, Arthur G, Schweizer F. Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics. 2023; 12(8):1261. https://doi.org/10.3390/antibiotics12081261
Chicago/Turabian StyleGandhi, Karan, Shiv Dhiman, Rajat Arora, Danzel Marie Ramirez, Danyel Ramirez, Gilbert Arthur, and Frank Schweizer. 2023. "Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa" Antibiotics 12, no. 8: 1261. https://doi.org/10.3390/antibiotics12081261
APA StyleGandhi, K., Dhiman, S., Arora, R., Ramirez, D. M., Ramirez, D., Arthur, G., & Schweizer, F. (2023). Exploring Antibiotic-Potentiating Effects of Tobramycin–Deferiprone Conjugates in Pseudomonas aeruginosa. Antibiotics, 12(8), 1261. https://doi.org/10.3390/antibiotics12081261