Abstract
Pseudomonas aeruginosa has the genetic potential to acquire colistin resistance through the modification of lipopolysaccharide by the addition of 4-amino-4-deoxy-L-arabinose (L-Ara4N) or phosphoethanolamine (PEtN), mediated by the arn operon or the eptA gene, respectively. However, in vitro evolution experiments and genetic analysis of clinical isolates indicate that lipopolysaccharide modification with L-Ara4N is invariably preferred over PEtN addition as the colistin resistance mechanism in this bacterium. Since little is known about eptA regulation in P. aeruginosa, we generated luminescent derivatives of the reference strain P. aeruginosa PAO1 to monitor arn and eptA promoter activity. We performed transposon mutagenesis assays to compare the likelihood of acquiring mutations leading to arn or eptA induction and to identify eptA regulators. The analysis revealed that eptA was slightly induced under certain stress conditions, such as arginine or biotin depletion and accumulation of the signal molecule diadenosine tetraphosphate, but the induction did not confer colistin resistance. Moreover, we demonstrated that spontaneous mutations leading to colistin resistance invariably triggered arn rather than eptA expression, and that eptA was not induced in resistant mutants upon colistin exposure. Overall, these results suggest that the contribution of eptA to colistin resistance in P. aeruginosa may be limited by regulatory restraints.
1. Introduction
The increase in antibiotic resistance among bacterial pathogens is becoming a critical global crisis, mainly due to the emergence of multidrug-resistant (MDR) strains, especially among Gram-negative bacterial pathogens [1]. One of the few remaining options for treating life-threatening infections caused by MDR Gram-negative bacteria is colistin, a cationic antibiotic belonging to the polymyxin family that is now considered a “last-resort” antibacterial drug [2,3]. Unfortunately, the use of colistin in human and veterinary medicine has led to the emergence and spread of colistin-resistant isolates, often leaving clinicians without suitable resources to treat patients [4,5].
Colistin is a cyclic heptapeptide with a tripeptide side chain acylated at the N terminus, whose positive charge is due to the presence of multiple α,γ-diaminobutyric acid residues [6]. According to the “self-promoted uptake pathway” model, the initial binding of colistin with the outer membrane (OM) of Gram-negative bacteria occurs via electrostatic interactions with the negatively charged phosphate groups of the lipid A moiety of lipopolysaccharide (LPS). Then, colistin competitively displaces the divalent cations Mg2+ and Ca2+ that stabilize the LPS layer, thus weakening the OM and facilitating its uptake [5,7]. Recently, it was demonstrated that colistin can also interact with LPS molecules that are transiently present in the cytoplasmic membrane before transport to the OM, accounting for membrane destabilization, loss of cytoplasmatic content, and finally cell lysis [8].
In many Gram-negative bacteria, polymyxin resistance relies on the remodelling of lipid A by the covalent addition of positively charged molecules, such as phosphoethanolamine (PEtN) and 4-amino-4-deoxy-L-arabinose (L-Ara4N), which decrease the negative charge of lipid A and reduce the affinity of polymyxins for LPS [9]. Other polymyxin-resistance mechanisms have been proposed, such as lipid A glycosylation or acylation, capsule overproduction, expression of efflux pumps, and overexpression of basic outer membrane proteins that can bind and mask the divalent cation-binding sites of LPS [9,10]. However, their contribution to polymyxin resistance remains unclear.
Pseudomonas aeruginosa is an opportunistic pathogen that causes both acute and chronic infections with limited therapeutic options [11]. In this bacterium, colistin resistance is always associated with the induction of the arn operon, which is responsible for L-Ara4N-dependent modification of lipid A and is controlled by a complex regulatory network involving several two-component systems (TCSs) [4,12,13]. Indeed, mutations in TCSs that control the expression of arn genes have frequently been identified in colistin-resistant strains [12,13,14,15,16]. By combining reverse genetics with experimental evolution assays, it has been confirmed that a functional lipid A aminoarabinosylation pathway is essential for the acquisition of colistin resistance in vitro, in clinical P. aeruginosa isolates and reference samples [15,16]. On the other hand, analysis of recombinant P. aeruginosa strains constitutively expressing arn genes revealed that lipid A aminoarabinosylation is sufficient to confer colistin resistance in some but not all P. aeruginosa strains [17]. This is in line with the observation that the evolution towards high levels of colistin resistance also involves mutations in other genetic loci that are not directly related to lipid A modification, and are probably required to improve and/or support the effect of L-Ara4N-modified lipid A on colistin resistance [15]. Nonetheless, these studies overall confirm lipid A aminoarabinosylation as the main colistin-resistance mechanism in P. aeruginosa. Accordingly, inhibitors of the Ara-4N transferase ArnT, which catalyses the last step of lipid A aminoarabinosylation, were found to restore susceptibility in colistin-resistant P. aeruginosa strains [18,19].
Conversely, the role of lipid A phosphoethanolamination in P. aeruginosa is less clear. P. aeruginosa has an endogenous gene, named eptA, which encodes a functional PEtN transferase capable of adding PEtN to lipid A [20,21]. It was reported that eptA gene expression in P. aeruginosa is induced by extracellular zinc, but not other metals, through the TCS ColRS, and that zinc-induced PEtN addition to lipid A does not confer polymyxin resistance [20]. Furthermore, Liu and colleagues observed that the introduction into P. aeruginosa of plasmids carrying the mobile gene mcr-1, which encodes the PEtN transferase MCR-1, led to colistin resistance, although the increase in colistin MIC varied between isolates [22,23]. More recently, by cloning the eptA and mcr-1 genes in a plasmid for IPTG-inducible expression, it was demonstrated that (i) exogenous and endogenous PEtN transferases (MCR-1 and EptA) in P. aeruginosa have comparable lipid A PEtN transferase activity, (ii) both are effective in promoting colistin resistance, and (iii) the modification of lipid A with PEtN confers levels of colistin resistance comparable to those conferred by L-Ara4N-modified lipid A [21].
Given the efficacy of PEtN transferases in conferring colistin resistance in P. aeruginosa when ectopically expressed from plasmids [21,22,23], it is unclear why lipid A aminoarabinosylation is always preferred over lipid A phosphoethanolamination as a colistin-resistance mechanism [12,13,14,15,16]. Since little is known about the regulatory landscape of P. aeruginosa eptA, in this study we generated reporter strains and performed random mutagenesis screenings to evaluate the propensity of P. aeruginosa to acquire mutations that induce eptA, and to identify genes that influence eptA gene expression and/or colistin resistance.
2. Results
2.1. PEtN Transferases Support the Evolution of P. aeruginosa towards High Levels of Colistin Resistance
Previous studies demonstrated that ectopic expression of endogenous or exogenous PEtN transferases (EptA and MCR-1, respectively) is able to confer colistin resistance in P. aeruginosa [21,22,23]. However, evolution experiments and genomic analyses revealed that the acquisition of high levels of colistin resistance in P. aeruginosa requires a functional L-Ara4N lipid A modification pathway and mutations in several independent loci that synergistically produce the resistance phenotype [15,16]. Thus, to further verify the potential of PEtN to contribute to colistin resistance acquisition in P. aeruginosa, we performed in vitro evolution assays for L-Ara4N-defective mutants (ΔarnBCA) of two different reference strains (PAO1 and PA14) transformed with plasmids for expression of either EptA or MCR-1, using an empty plasmid as the control. With this aim, five independent cultures for each strain were subsequently refreshed in the presence of increasing concentrations of colistin (up to 64 µg/mL), and cultures were considered extinct when they showed no visible growth after five days. In line with previous evidence [16], the ΔarnBCA mutants containing the empty plasmid were unable to evolve the ability to grow at colistin concentrations higher than 2 or 4 µg/mL (Figure 1 and Supplementary Materials Figure S1), confirming the necessity of L-Ara4N-modified lipid A for the acquisition of high levels of colistin-resistance in wild-type backgrounds. In contrast, strains ectopically expressing PEtN transferases successfully evolved high levels of colistin resistance, although the evolutionary process appeared to be slightly faster in MCR-1-expressing cells compared with EptA-expressing cells (Figure 1 and Supplementary Materials Figure S1). This could be explained by the previously observed detrimental effect of ectopic EptA expression on P. aeruginosa growth [21]. Nevertheless, the experimental results demonstrate that PEtN transferases, including the endogenous enzyme EptA, when ectopically expressed from plasmids can support the evolution of P. aeruginosa towards high-level colistin resistance. This led us to hypothesize that in P. aeruginosa the role of the chromosomal eptA gene in the acquisition of colistin resistance could be limited by regulatory restraints.
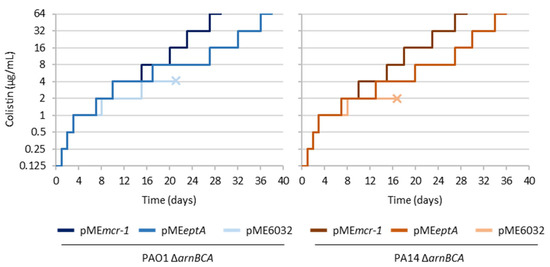
Figure 1.
In vitro evolution assays for L-Ara4N deficient mutants of P. aeruginosa PAO1 and PA14 (ΔarnBCA) carrying plasmids for the ectopic expression of EptA (pMEeptA) or MCR-1 (pMEmcr-1), or the empty plasmid pME6032 as the control. Strains were evolved through serial passages in the presence of increasing concentrations of colistin (up to 64 μg/mL). Five biological replicates (independent cultures) were analyzed for each strain. The graphs show a representative curve for each strain, while the curves of the other replicates are provided in Figure S1. The “×” symbol highlights cultures that went extinct during the evolution experiment.
2.2. Generation and Validation of Reporter Strains for PeptA and Parn Activity
To investigate the regulatory network(s) involved in the control of eptA gene expression, we generated a luminescent reporter in the reference strain PAO1 (PAO1 PeptA::lux) harboring the luxCDABE operon under the control of the eptA promoter inserted into a neutral site of the chromosome. Similarly, we generated another reporter strain to monitor the activity of the arn promoter (PAO1 Parn::lux), and a control strain carrying only the lux operon inserted into the same chromosomal site (PAO1 lux). The PAO1 PeptA::lux reporter was validated by assessing luminescence emission in response to different concentrations of extracellular zinc (ZnCl2) during both planktonic and colony growth. As expected, luminescence was induced by zinc in a dose-dependent manner, and such induction was not observed upon deletion of the colRS genes (PAO1 PeptA::lux ΔcolRS) (Figure 2a), confirming that eptA regulation by zinc occurs through the TCS ColRS [20]. Interestingly, we also confirmed that zinc-mediated induction of eptA gene expression does not increase colistin resistance in P. aeruginosa [20], and observed that extracellular zinc had a negative impact on bacterial growth at eptA-inducing concentrations (Figure S2).
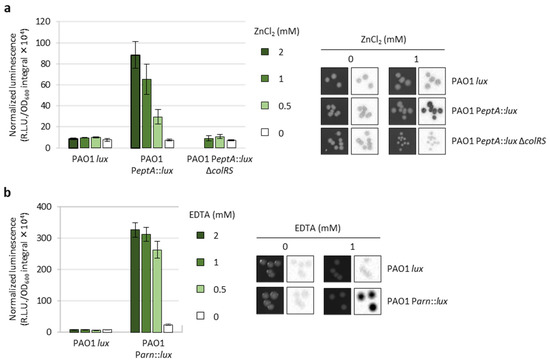
Figure 2.
Validation of the PAO1 PeptA::lux and PAO1 Parn::lux reporter strains. (a) Area under the normalized luminescence curve for PAO1 lux, PAO1 PeptA::lux, and PAO1 PeptA::lux ΔcolRS cultured in lysogeny broth (LB) supplemented with different ZnCl2 concentrations, and colony growth (left images) and luminescence (right images) of the same strains on LB agar plates in the presence or absence of 1 mM ZnCl2. PAO1 PeptA::lux ΔcolRS did not grow at 2 mM ZnCl2 (data not shown and [20]). (b) Area under the normalized luminescence curve for PAO1 lux and PAO1 Parn::lux cultured in LB supplemented with different EDTA concentrations, and colony growth (left images) and luminescence (right images) of the same strains on LB agar plates in the presence or absence of 1 mM EDTA. Values are the mean (±SD) of three independent assays. Images are representative of three independent experiments providing similar results.
The PAO1 Parn::lux strain was validated by demonstrating that the luminescence emitted by this reporter was proportional to the concentration of the divalent cation chelator EDTA in the growth medium (Figure 2b), which removes Ca2+ and Mg2+ that stabilize the OM and induces the arn operon [24]. As expected, neither zinc nor EDTA had any effect on luminescence emission in the control strain PAO1 lux (Figure 2).
2.3. Identification of Genes Influencing eptA Promoter Activity and Evaluation of Their Impact on Colistin Resistance
To identify novel regulators of eptA gene expression, we carried out transposon-mediated random mutagenesis in PAO1 PeptA::lux. For comparison, transposon mutagenesis was also performed in PAO1 Parn::lux. Transconjugants were picked from conjugation plates, spotted onto new agar plates, and screened for luminescence emission using PAO1 PeptA::lux (or PAO1 Parn::lux), PAO1 lux, and the PAO1 wild type as controls representing non-induced or basal luminescence levels. Luminescence was quantified for each clone (see Section 4) and compared among clones in order to identify possible outliers producing higher or lower levels of luminescence. An example of a screening plate is provided in Figure S3.
The screening of 12,000 transposon mutants for each reporter led to identification of only clones with increased luminescence levels, probably because the eptA and arn promoters were poorly active under the culture conditions used for the screening (LB agar plates without any supplement) (Figure 2). The outliers identified in the screening plates were verified for luminescence emission in liquid cultures and tested by MIC assays for colistin sensitivity. Overall, we identified 30 transposon mutants for PAO1 Parn::lux and five for PAO1 PeptA::lux that emitted significantly higher levels of luminescence with respect to the corresponding controls or the other mutant clones (Figure 3). Half of the selected PAO1 Parn::lux transposon mutants also showed increased colistin resistance (≥4-fold increase in MIC), while colistin MIC was identical to the parental strain for all PAO1 PeptA::lux transposon mutants (Figure 3). Notably, we did not observe a clear correlation between Parn activity and colistin MIC (Figure 3b), suggesting that arn gene expression is not always sufficient to confer colistin resistance. This is in line with previous results obtained with evolution experiments or genetically engineered strains [15,16].
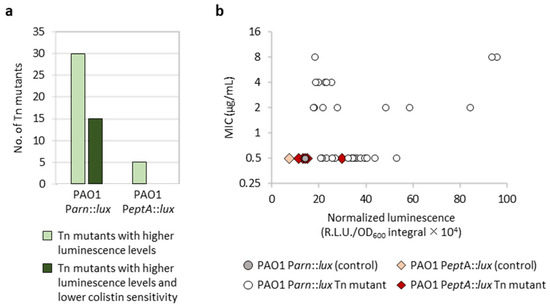
Figure 3.
Overview of the results of the transposon mutagenesis screening. (a) Number of transposon mutants of PAO1 Parn::lux or PAO1 PeptA::lux showing higher luminescence emission, both on agar plates and during planktonic growth, and/or higher colistin MIC compared with the corresponding parental strain. (b) Correlation between PeptA or Parn activity (x axis) and colistin MIC (y axis) for the selected transposon mutants of PAO1 Parn::lux (circles) or PAO1 PeptA::lux (diamonds) and the parental strains for comparison. Luminescence levels were quantified from liquid cultures and correspond to the mean of three independent experiments.
Overall, these results suggest that transposon insertions leading to increased promoter activity are more likely to occur for Parn than PeptA, leading to the proposal that the arn operon might be more connected to the P. aeruginosa regulatory network with respect to the eptA gene. Moreover, even if the number of mutants (only five) with enhanced PeptA activity identified in our screening is poorly indicative, the observation that none of these mutants showed any increase in colistin resistance is consistent with previous evidence that eptA expression and lipid A phosphoethanolamination induced by extracellular zinc are not sufficient to confer colistin resistance in P. aeruginosa (Figure 2a and Figure S2) [20].
Since our study was aimed at identifying novel regulators of eptA gene expression, we identified the transposon insertion site for the five PeptA::lux mutants showing increased luminescence emission. Transposon insertions were found in anabolic genes involved in biotin (bioA and bioB) or arginine biosynthesis (argF), and in the apaH and rmcA genes involved in the degradation of the intracellular signaling molecules diadenosine tetraphosphate (Ap4A) and cyclic diguanylate (c-di-GMP), respectively (Table 1) [25,26]. While the function of c-di-GMP in promoting cell aggregation and the biofilm lifestyle has been well documented in P. aeruginosa [27,28], less is known about the role of Ap4A in bacteria. Ap4A production is generally increased under stress, but it is still debated whether it represents a bona fide secondary messenger or a damage metabolite [29,30]. Notably, none of these genes have previously been linked to regulation of lipid A modification genes and/or colistin resistance.

Table 1.
Genes disrupted in transposon mutants with increased PeptA activity.
2.4. Further Characterization of Genes Influencing eptA Promoter Activity
Given that transposon insertions within coding sequences are expected to cause loss of gene function, to confirm the transposon mutagenesis results we generated clean deletion mutants of the PAO1 PeptA::lux reporter in all the identified genes except bioA (Table S1), as the impact of the loss of biotin biosynthesis could be inferred from the ΔbioB mutant. The deletion mutants in apaH, bioB, and argF showed increased luminescence emission compared with the parental strain, in line with what observed for the corresponding transposon mutants (Figure 4). In contrast, the ΔrmcA mutant had luminescence levels comparable to the parental reporter strain, during both planktonic and colony growth. This mutant was therefore not included in the following analyses. Interestingly, the ΔapaH, ΔbioB, and ΔargF mutants also showed impaired and/or delayed growth (Figure 4a), indicating that the absence of biotin or arginine biosynthesis has a negative impact on growth even in a rich medium such as LB. The growth defects of the ΔapaH mutant could be explained by the intracellular accumulation of Ap4A, probably caused by the lack of Ap4A tetraphosphatase activity, and indeed it has been reported that high levels of Ap4A can have deleterious effects on bacterial fitness [29]. Growth and luminescence emission in deletion mutants were restored to parental levels by complementation with plasmids expressing the cognate gene (Figure S4), confirming that the observed phenotypes were caused by the lack of the genes under investigation.
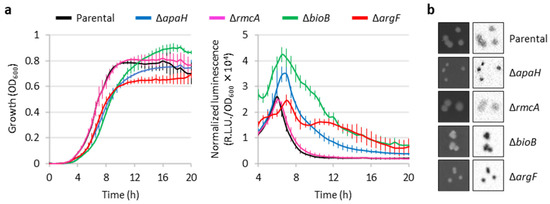
Figure 4.
Validation of the putative eptA regulators by deletion mutagenesis. (a) Growth (left panel) and normalized luminescence curves (right panel) of PAO1 PeptA::lux and its deleted apaH, rmcA, bioB, or argF derivatives, cultured in LB. Values are the mean (±SD) of three independent assays. (b) Colony growth (left images) and luminescence (right images) of the above-mentioned strains on LB agar plates. Images are representative of three independent experiments giving similar results.
Based on the function of ApaH, BioB, and ArgF (Table 1), it is plausible to predict that these proteins have an indirect effect on eptA promoter activity. Since the only regulatory system demonstrated to control eptA transcription is the TCS ColRS, which is activated by extracellular zinc [20], we verified whether the lack of the apaH, bioB, or argF genes could influence zinc-mediated induction of eptA expression and/or whether the deletion of colRS genes could reduce the effect of ΔapaH, ΔbioB, or ΔargF mutations on eptA promoter activity. As shown in Figure 5a, the PAO1 PeptA::lux reporter was induced by extracellular zinc in a dose-dependent manner even in the absence of apaH, bioB, or argF. Moreover, the levels of luminescence emitted by PAO1 PeptA::lux ΔapaH, ΔbioB, and ΔargF mutants, respectively, were basically identical in the presence or absence of a functional ColRS system (Figure 5b). Overall, these results clearly indicate that ApaH, BioB, and ArgF affect PeptA activity in a ColRS-independent manner.
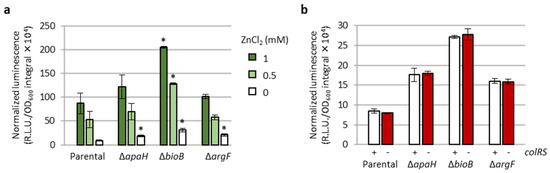
Figure 5.
ApaH, BioB, and ArgF affect eptA promoter activity in a ColRS-independent manner. (a) PeptA promoter activity (area under the normalized luminescence curves) in PAO1 PeptA::lux and its deleted apaH, bioB, or argF derivatives, cultured in LB supplemented with different concentrations of ZnCl2. (b) PeptA promoter activity (area under the normalized luminescence curves) in the above-mentioned strains and in isogenic deletion mutants lacking the colRS genes, cultured in LB. Values are the mean (±SD) of three independent assays. Asterisks (*) indicate a statistically significant increase in promoter activity with respect to the parental strain cultured in the presence of the same ZnCl2 concentration, according to the unpaired t test (p < 0.01).
In the attempt to further characterize the impact of impaired anabolic metabolism on eptA gene expression, the ΔbioB and ΔargF mutants were also assayed in minimal medium. As expected, both mutants did not grow unless biotin or arginine was added to the medium, and their growth was promoted by exogenously supplied biotin or arginine in a dose-dependent manner (Figure S5), confirming the auxotrophy of the ΔbioB and ΔargF mutants for biotin and arginine, respectively. Interestingly, PeptA activity in these strains was found to be inversely proportional to growth rates and yields, with maximum PeptA induction observed at biotin or arginine concentrations that sustained growth at levels far below those of the parental prototrophic strain (Figure S5). These data indicate that depletion of arginine or biotin, rather than the lack of the specific ArgF or BioB enzymes, is the signal that triggers eptA gene expression. However, the mechanism of induction was not further investigated in this study.
2.5. The eptA Gene Is Not Induced in Colistin-Resistant Spontaneous Mutants
The results described above suggest that P. aeruginosa is more prone to acquire mutations leading to the induction of the arn operon rather than the eptA gene, leading to postulation that eptA could be poorly connected to P. aeruginosa regulatory networks. However, random mutagenesis by transposon insertion is strongly biased toward loss-of-function mutations, as it often results in gene inactivation. While this can be useful to identify negative regulators (e.g., transcriptional repressors), it makes the identification of positive regulators much less probable. This is particularly true in our case, as the promoters under investigation (PeptA and Parn) were poorly active in the culture conditions used for the screening of transposon mutants (Figure 2, Figure 3 and Figure S3).
Thus, we decided to use a complementary approach by verifying whether PeptA induction occurs in spontaneous mutants that acquire resistance to colistin. To this aim, we selected colistin-resistant mutants of the reporter strains PAO1 PeptA::lux and PAO1 Parn::lux, and of the strain PAO1 lux as control, on agar plates containing colistin at 20 × MIC (10 μg/mL). The frequency of resistant mutants, calculated from 12 independent cultures, was overall comparable between the three strains (Table 2) and similar to values previously obtained for wild-type strains [16], indicating that the reporter constructs do not significantly affect the mutation rate nor probably the mutation patterns that can confer resistance.
Table 2.
Frequency of colistin-resistant spontaneous mutants.
For each strain, 160 colistin-resistant mutants were taken from independent replicate cultures and spotted onto LB agar plates, in order to quantify luminescence emission for each mutant. As shown in Figure 6, all the colistin-resistant mutants of the reporter strain PAO1 Parn::lux showed huge increases in luminescence emission, demonstrating that all these mutants had acquired mutations that (directly or indirectly) induce arn gene expression. In contrast, colistin-resistant mutants of PAO1 PeptA::lux had luminescence levels comparable to the controls, indicating that none of the mutations that conferred resistance in this group led to the induction of the eptA gene. Identical results were obtained when spontaneous resistant mutants were spotted on agar plates containing 10 μg/mL colistin (Figure S6), implying that eptA is not induced in P. aeruginosa even as a response to colistin exposure.
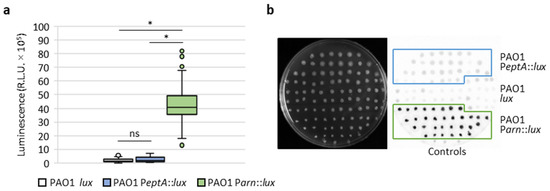
Figure 6.
Induction of eptA and arn genes in colistin-resistant spontaneous mutants. (a) Box-and-whisker plots showing the luminescence emitted by 160 colistin-resistant spontaneous mutants obtained for each of the PAO1 lux, PAO1 PeptA::lux, and PAO1 Parn::lux strains, on LB agar plates. (b) Example of a screening plate, showing the growth (left image) and luminescence emission (right panel) for a subset (n = 32) of the colistin-resistant mutants of the above-mentioned strains. The parental (colistin-sensitive) strains PAO1 lux, PAO1 PeptA::lux, and PAO1 Parn::lux were included as controls. Asterisks (*) indicate a statistically significant difference (p < 0.001), according to the Kruskal–Wallis test. Abbreviation: ns, not significant.
3. Discussion
Although the P. aeruginosa EptA enzyme is proficient in lipid A phosphoethanolamination, confers colistin resistance, and supports the evolution towards high-level colistin resistance when ectopically expressed from plasmids [20,21] (Figure 1), P. aeruginosa generally acquires colistin resistance by mutations that trigger the expression of the alternative lipid A modification system Arn, responsible for the addition of L-Ara4N to lipid A [15,16,31]. We therefore hypothesized that there could be some genetic and/or regulatory barriers to the exploitation of EptA and thus of PEtN-modified lipid A as a resistance mechanism during the natural evolution of this bacterium towards colistin resistance. To test this hypothesis, in this study we generated luminescent reporter strains to monitor eptA and arn promoter activities, and used transposon mutagenesis and a selection of spontaneous mutants resistant to colistin to compare the likelihood of mutations that induce eptA or arn gene expression and/or colistin resistance.
Transposon mutagenesis revealed that transposon insertions leading to arn gene induction are more probable than those activating the eptA promoter. Moreover, arn-inducing transposon insertions overall resulted in higher luminescence levels than eptA-inducing insertions, and increased levels of colistin resistance were observed for transposon mutants with induced arn gene expression but not for mutants showing PeptA induction (Figure 3). The genetic and phenotypic characterization of knock-out mutants showing eptA induction revealed that eptA transcription is influenced by certain metabolic stresses, such as arginine and biotin depletion (ΔargF and ΔbioB, respectively) or intracellular accumulation of the stress signal Ap4A (ΔapaH), and that this regulation is independent from the TCS ColRS (Figure 4, Figure 5 and Figure S5). Notably, the lack of ApaH, BioB, or ArgF had a much lower impact on eptA promoter activity than the exogenous signal Zn2+ (Figure 5), which, however, is unable to promote colistin resistance [20] (Figure S2). Since PEtN addition to lipid A in response to extracellular Zn2+ was confirmed by mass spectrometry [20], it is likely that the inability of Zn2+ to confer colistin resistance is at least partly due to the highly deleterious effect of this metal on bacterial growth under eptA inducing conditions (Figure S2). Interestingly, the genetic conditions identified in this study to promote eptA expression (bioB, argF, or apaH inactivation) also caused relevant growth defects (Figure 4, Figures S4 and S5), suggesting that eptA gene expression might mainly occur under stress conditions that do not facilitate the acquisition of an antibiotic-resistant phenotype. As an example, we can consider the case of biotin depletion (ΔbioB mutant), which led to the highest increase in PeptA activity among our knockout mutants (Figure 4 and Figure S5). Biotin can be produced by de novo synthesis or environmentally acquired; however, as biotin synthesis is expensive, bacteria generally shut down synthesis when an exogenous source of this cofactor is available [32]. Recently, biotin biosynthesis was shown to be essential during P. aeruginosa infection in mice in which the low biotin concentrations of human plasma were artificially mimicked [33]. This implies that mutations leading to inactivation of the biotin de novo pathway might be counter-selected in vivo, irrespective of their impact on antibiotic resistance. Another example of likely counter-selectable mutation is apaH inactivation, which causes accumulation of the stress signal molecule Ap4A. Additionally to impairing growth [29], Ap4A accumulation has recently been reported to increase sensitivity to killing by aminoglycosides in different bacterial pathogens, including P. aeruginosa [34].
Two main drawbacks of the transposon mutagenesis screening process should be noted. First, we analyzed a limited number of transposon mutants (12,000) in a strain carrying about 5500 genes [35]. According to genome-wide transposon mutagenesis projects, such a number of transposon mutants should be enough to cover about 50–60% of nonessential genes [36,37], so it is plausible that additional (direct or indirect) regulators of eptA may exist that were not identified in our study. Second, transposon mutagenesis generates mainly loss-of-function mutations, thus hindering the identification of genes encoding for (transcriptional) activators, especially in the case of the eptA promoter which is basically inactive under the culture conditions used for transposon mutant screening (Figure 2). To overcome these limitations, we also analyzed PeptA and Parn activity in hundreds of colistin-resistant spontaneous mutants selected from several independent bacterial cultures. This analysis revealed that Parn was induced in all colistin-resistant mutants included in our analysis, while PeptA was induced in none of them (Figure 6), strongly suggesting that gain-of-function mutations leading to relevant levels of eptA gene expression may be very rare and/or difficult to select for in P. aeruginosa.
In conclusion, although the mutagenesis screenings were conducted on a single strain (P. aeruginosa PAO1) and we cannot exclude the possibility that other strains might behave differently, our results confirm the evidence from in vitro evolution experiments and clinical isolates that lipid A aminoarabinosylation is preferred over phosphoethanolamination as a colistin-resistance mechanism in P. aeruginosa. Moreover, our results suggest the proposal that this could be due to the fitness costs related to the exogenous and/or endogenous signals that trigger eptA gene expression, and/or to the ineffectiveness of the P. aeruginosa regulatory networks in providing sufficient levels of eptA gene expression.
4. Materials and Methods
4.1. Bacterial Strains, Plasmids and Growth Media
Bacterial strains and plasmids used in this study are listed in Supplementary Materials Tables S1 and S2, respectively. Unless otherwise stated, bacteria were cultured in LB (Lennox formulation) for genetic manipulation, transposon mutagenesis, growth, and luminescence assays, or in cation-adjusted Mueller–Hinton broth CAMHB for in vitro evolution experiments and MIC assay. When required, antibiotics were added at the following concentrations for E. coli (the concentration used for P. aeruginosa is shown in brackets): ampicillin, 100 μg/mL; carbenicillin (500 μg/mL); nalidixic acid, 15 μg/mL; chloramphenicol, 30 μg/mL (375 μg/mL); tetracycline, 12.5 μg/mL (100 μg/mL); gentamicin, 10 μg/mL (50 μg/mL).
4.2. In Vitro Evolution Assays
To select for mutants with high-level colistin resistance, P. aeruginosa strains were sequentially cultured in the presence of increasing concentrations of colistin as previously described [16]. Briefly, strains were precultured in 2 mL of CAMHB at 37 °C and 250 rpm to late-exponential phase and then sequentially refreshed 1:100 in 1 mL of fresh medium in the presence of two-fold higher concentrations of colistin (from 0.25 to 64 µg/mL) as soon as cultures reached an OD600 > 0.5 (starting inoculum ≥ 107 cells/mL). For each colistin concentration, three serial passages were performed before moving to the two-fold higher concentration. Five independent cultures were performed for each strain. Cultures showing no visible growth after five days were considered extinct [16].
4.3. Generation of Luminescent Reporter Strains, Deletion Mutants and Complementing Plasmids
To obtain the constructs for the generation of PAO1 PeptA::lux and PAO1 Parn::lux reporter strains, a 514-bp and a 416-bp DNA region encompassing PeptA or Parn, respectively, were PCR amplified, individually cloned into the sequencing plasmid pBluescript II KS (pBS; Table S2) and verified by DNA sequencing. Primers and restriction enzymes used for PCR and cloning are listed in Table S3. Then, PeptA and Parn were excised from pBS and directionally sub-cloned into the self-proficient integration plasmid mini-CTX1lux [38], yielding mini-CTX1PeptA::lux and mini-CTX1Parn::lux (Table S2). These plasmids, or the control plasmid mini-CTX1lux, were transferred into P. aeruginosa PAO1 by conjugation, and transconjugants were selected on LB agar plates containing 15 μg/mL nalidixic acid and 100 μg/mL tetracycline. The plasmid backbone was removed using the sacB-based suicide vector pFLP2 [39] as previously described [40], and pFLP2 was cured by plating onto LB agar plates supplemented with 10% sucrose. Carbenicillin-sensitive clones were analyzed by colony PCR to verify the insertion of the PeptA::lux or Parn::lux construct in the P. aeruginosa chromosome.
To obtain the constructs for the generation of P. aeruginosa colRS, apaH, rmcA, bioB, and argF deletion mutants (Tables S1 and S2), two DNA fragments of approximately 500 bp encompassing the upstream (↑) and downstream (↓) region of each gene of interest were PCR amplified, directionally cloned into pBS, and verified by DNA sequencing. Then, the ↑↓ fragments of each gene were excised from pBS and sub-cloned into the sacB-based suicide vector pDM4 [41]. Primers and restriction enzymes used for PCR and cloning are listed in Table S3. The resulting pDM4 derivatives were transferred into P. aeruginosa by conjugation, and transconjugants were selected on LB agar plates containing 15 μg/mL nalidixic acid and 375 μg/mL chloramphenicol. Deletion mutations were obtained by recombination and sucrose-based selection as described [40]. Gene deletions were verified by PCR and DNA sequencing.
The complementing plasmids pMEapaH, pMEbioB, and pMEargF (Table S2) were generated by individually cloning the PCR-amplified coding sequence of apaH, bioB, or argF into the shuttle vector pME6032, under the control of an IPTG-inducible promoter [42]. All constructs were verified by DNA sequencing.
4.4. Growth and Luminescence Assays
For planktonic growth and promoter activity assays, reporter strains were precultured in LB and then refreshed 1:1000 in LB in the presence or absence of different ZnCl2 or EDTA concentrations. Bacterial cultures were incubated in 96-well microtiter plates (200 μL in each well) at 37 °C in a Spark 10M microtiter plate reader (Tecan, Männedorf, Switzerland), in order to measure growth (OD600) and luminescence emission (relative light units, RLU) over time. Luminescence was normalized to growth by dividing the RLU values by OD600 values. Area under the normalized luminescence curves was calculated using the trapezoidal method, employing GraphPad Prism software. When appropriate, growth and luminescence assays were also performed in the minimal medium M9 containing 20 mM succinate as the carbon source [43] supplemented or not with different concentrations of arginine or biotin.
For colony growth assays, reporter strains were precultured in LB, harvested by centrifugation, and resuspended in sterile saline solution at OD600 = 1. Serial ten-fold dilutions were prepared and 5 μL aliquots of selected dilutions were spotted onto LB agar plates containing or not 1 mM ZnCl2 or 1 mM EDTA. After incubation at 37 °C for 16–18 h, white light and luminescence images of plates were acquired with ChemiDoc (Bio-Rad, Segrate, Italy).
4.5. Transposon-Mediated Random Mutagenesis
Transposon mutagenesis was performed as previously described [44], using the pLM1 vector that holds a Tn5 transposon derivative containing an origin of replication that is functional in E. coli, and a gentamycin resistance gene [45]. This vector was transferred into PAO1 PeptA::lux and PAO1 Parn::lux by conjugation using E. coli S17.1λpir as the donor strain. Transposon mutants were selected on LB agar plates supplemented with 15 μg/mL nalidixic acid and 50 μg/mL gentamycin. Colonies were picked with sterile toothpicks and spotted onto new LB agar plates. Plates were then visualized using ChemiDoc and luminescence was measured with the ImageLab software (Bio-Rad) as the adjusted volume of each clone, i.e., the sum of all intensities detected by the software within the boundaries of each colony from which the background value was subtracted. Mutants selected for further analyses were those that appeared as outliers in the box-and-whisker plots and showed a ≥2-fold increase in luminescence with respect to the median luminescence value of mutants from the same screening plate.
To map transposon insertions, genomic DNA was extracted from selected transposon mutants and digested with BamHI, XbaI, or NcoI, whose restriction sites are not present in the transposon. The digested DNA was self-ligated with T4 DNA ligase and introduced by transformation into E. coli S17.1 λpir. Plasmid DNA was isolated from gentamicin-resistant colonies and sequenced using the Tn5 specific primers TnpRL17-1 and TnpRL13-2 (Table S3) [44].
4.6. MIC Assays
MIC was determined using the standard broth microdilution method. Strains were precultured in LB or CAMHB and refreshed at ca. 5 × 105 cells/mL in the same medium containing increasing concentrations of colistin. MIC values were visually assessed after 24 h of growth at 37 °C under static conditions. Each strain was tested in at least three independent experiments.
4.7. Selection of Colistin-Resistant Spontaneous Mutants
Strains were cultured in LB broth at 37 °C until the late exponential or early stationary phase, harvested by centrifugation and resuspended in sterile saline solution at OD600 = 1. Serial dilutions were performed and plated onto LB agar plates to measure total numbers of CFUs. Colistin-resistant mutants were selected by plating 200 µL aliquots of the undiluted samples onto LB agar plates supplemented with 10 µg/mL colistin (20 × MIC). After 48 h of incubation at 37 °C, colistin-resistant CFUs were counted, picked with sterile toothpicks and spotted onto new LB agar plates supplemented or not with 10 µg/mL colistin. In order to evaluate the promoter activity, plates were visualized with ChemiDoc and luminescence quantified as described above (see Section 4.5). The frequency of spontaneous resistant mutants was calculated as the ratio between the number of colistin-resistant CFUs and the total number of CFUs.
4.8. Statistical Analysis
Statistical analysis was performed with the software GraphPad Instat, using the unpaired t test, or the Kruskal–Wallis test followed by uncorrected Dunn’s multiple comparison test for box-and-whisker plot analysis [46].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics12020200/s1, Figure S1: In vitro evolution assays; Figure S2: Effect of zinc on growth and colistin resistance; Figure S3: Example of a transposon mutagenesis screening plate; Figure S4: Complementation assays; Figure S5: PeptA induction is caused by biotin or arginine depletion; Figure S6: Induction of eptA and arn genes in colistin-resistant spontaneous mutants; Table S1: Bacterial strains used in this study; Table S2: Plasmids used in this study; Table S3: Primers used in this study. References [47,48] are cited in the supplementary materials.
Author Contributions
Conceptualization, F.I.; methodology, M.C., D.S. and A.L.S.; validation, all authors; investigation, M.C., D.S. and A.L.S.; resources, F.I.; data curation, all authors; writing—original draft preparation, M.C., D.S. and F.I.; writing—review and editing, all authors; visualization, all authors; supervision, F.I.; project administration, F.I.; funding acquisition, F.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by PRIN 2017 (protocol 20177J5Y3P), PRIN 2020 (protocol 20208LLXEJ), and Excellence Departments (Art. 1, commi 314-337 Legge 232/2016) grants from the Italian Ministry of University and Research, by the Regione Lazio (Gruppi di Ricerca 2020, POR A0375E0026), and by the Italian Cystic Fibrosis Foundation (project FFC#12/2021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank G. Rampioni and L. Leoni (University Roma Tre) for providing the pDMΔrmcA construct.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Ledger, E.V.K.; Sabnis, A.; Edwards, A.M. Polymyxin and lipopeptide antibiotics: Membrane-targeting drugs of last resort. Microbiology 2022, 168, 001136. [Google Scholar] [CrossRef] [PubMed]
- Binsker, U.; Käsbohrer, A.; Hammerl, J.A. Global colistin use: A review of the emergence of resistant Enterobacterales and the impact on their genetic basis. FEMS Microbiol. Rev. 2022, 46, fuab049. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, K.; Bolard, A.; Plésiat, P. Resistance to polymyxins in Gram-negative organisms. Int. J. Antimicrob. Agents 2017, 49, 526–535. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed Ahmed, M.A.E.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef]
- Sabnis, A.; Hagart, K.L.; Klöckner, A.; Becce, M.; Evans, L.E.; Furniss, R.C.D.; Mavridou, D.A.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef]
- Baron, S.; Hadjadj, L.; Rolain, J.M.; Olaitan, A.O. Molecular mechanisms of polymyxin resistance: Knowns and unknowns. Int. J. Antimicrob. Agents 2016, 48, 583–591. [Google Scholar] [CrossRef]
- Lichtenberg, M.; Jakobsen, T.H.; Kühl, M.; Kolpen, M.; Jensen, P.Ø.; Bjarnsholt, T. The structure-function relationship of Pseudomonas aeruginosa in infections and its influence on the microenvironment. FEMS Microbiol. Rev. 2022, 46, fuac018. [Google Scholar] [CrossRef]
- Schurek, K.N.; Sampaio, J.L.; Kiffer, C.R.; Sinto, S.; Mendes, C.M.; Hancock, R.E. Involvement of pmrAB and phoPQ in polymyxin B adaptation and inducible resistance in non-cystic fibrosis clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 4345–4351. [Google Scholar] [CrossRef]
- Moskowitz, S.M.; Brannon, M.K.; Dasgupta, N.; Pier, M.; Sgambati, N.; Miller, A.K.; Selgrade, S.E.; Miller, S.I.; Denton, M.; Conway, S.P.; et al. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 2012, 56, 1019–1030. [Google Scholar] [CrossRef]
- Barrow, K.; Kwon, D.H. Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 5150–5154. [Google Scholar] [CrossRef]
- Jochumsen, N.; Marvig, R.L.; Damkiær, S.; Jensen, R.L.; Paulander, W.; Molin, S.; Jelsbak, L.; Folkesson, A. The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat. Commun. 2016, 7, 13002. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Imperi, F. Aminoarabinosylation of Lipid A Is Critical for the Development of Colistin Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e01820-17. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Cervoni, M.; Stefanelli, R.; Mancone, C.; Imperi, F. Effect of lipid A aminoarabinosylation on Pseudomonas aeruginosa colistin resistance and fitness. Int. J. Antimicrob. Agents 2020, 55, 105957. [Google Scholar] [CrossRef]
- Ghirga, F.; Stefanelli, R.; Cavinato, L.; Lo Sciuto, A.; Corradi, S.; Quaglio, D.; Calcaterra, A.; Casciaro, B.; Loffredo, M.R.; Cappiello, F.; et al. A novel colistin adjuvant identified by virtual screening for ArnT inhibitors. J. Antimicrob. Chemother. 2020, 75, 2564–2572. [Google Scholar] [CrossRef]
- Quaglio, D.; Mangoni, M.L.; Stefanelli, R.; Corradi, S.; Casciaro, B.; Vergine, V.; Lucantoni, F.; Cavinato, L.; Cammarone, S.; Loffredo, M.R.; et al. ent-Beyerane Diterpenes as a Key Platform for the Development of ArnT-Mediated Colistin Resistance Inhibitors. J. Org. Chem. 2020, 85, 10891–10901. [Google Scholar] [CrossRef]
- Nowicki, E.M.; O’Brien, J.P.; Brodbelt, J.S.; Trent, M.S. Extracellular zinc induces phosphoethanolamine addition to Pseudomonas aeruginosa lipid A via the ColRS two-component system. Mol. Microbiol. 2015, 97, 166–178. [Google Scholar] [CrossRef]
- Cervoni, M.; Lo Sciuto, A.; Bianchini, C.; Mancone, C.; Imperi, F. Exogenous and Endogenous Phosphoethanolamine Transferases Differently Affect Colistin Resistance and Fitness in Pseudomonas aeruginosa. Front. Microbiol. 2021, 12, 778968. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Chandler, C.E.; Leung, L.M.; McElheny, C.L.; Mettus, R.T.; Shanks, R.M.Q.; Liu, J.H.; Goodlett, D.R.; Ernst, R.K.; Doi, Y. Structural Modification of Lipopolysaccharide Conferred by mcr-1 in Gram-Negative ESKAPE Pathogens. Antimicrob. Agents Chemother. 2017, 61, e00580-17. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.B.; Arnosti, D.N.; Chamberlin, M.J.; Ames, B.N. An apaH mutation causes AppppA to accumulate and affects motility and catabolite repression in Escherichia coli. Proc. Natl. Acad. Sci. USA 1989, 86, 5010–5014. [Google Scholar] [CrossRef]
- Paiardini, A.; Mantoni, F.; Giardina, G.; Paone, A.; Janson, G.; Leoni, L.; Rampioni, G.; Cutruzzolà, F.; Rinaldo, S. A novel bacterial l-arginine sensor controlling c-di-GMP levels in Pseudomonas aeruginosa. Proteins 2018, 86, 1088–1096. [Google Scholar] [CrossRef]
- Visaggio, D.; Pasqua, M.; Bonchi, C.; Kaever, V.; Visca, P.; Imperi, F. Cell aggregation promotes pyoverdine-dependent iron uptake and virulence in Pseudomonas aeruginosa. Front. Microbiol. 2015, 6, 902. [Google Scholar] [CrossRef]
- Banerjee, P.; Sahoo, P.K.; Sheenu; Adhikary, A.; Ruhal, R.; Jain, D. Molecular and structural facets of c-di-GMP signalling associated with biofilm formation in Pseudomonas aeruginosa. Mol. Asp. Med. 2021, 81, 101001. [Google Scholar] [CrossRef]
- Despotović, D.; Brandis, A.; Savidor, A.; Levin, Y.; Fumagalli, L.; Tawfik, D.S. Diadenosine tetraphosphate (Ap4A)—An E. coli alarmone or a damage metabolite? FEBS J. 2017, 284, 2194–2215. [Google Scholar] [CrossRef]
- Ferguson, F.; McLennan, A.G.; Urbaniak, M.D.; Jones, N.J.; Copeland, N.A. Re-evaluation of Diadenosine Tetraphosphate (Ap4A) From a Stress Metabolite to Bona Fide Secondary Messenger. Front. Mol. Biosci. 2020, 7, 606807. [Google Scholar] [CrossRef]
- Chung, E.S.; Lee, J.Y.; Rhee, J.Y.; Ko, K.S. Colistin resistance in Pseudomonas aeruginosa that is not linked to arnB. J. Med. Microbiol. 2017, 66, 833–841. [Google Scholar] [CrossRef]
- Sirithanakorn, C.; Cronan, J.E. Biotin, a universal and essential cofactor: Synthesis, ligation and regulation. FEMS Microbiol. Rev. 2021, 45, fuab003. [Google Scholar] [CrossRef]
- Carfrae, L.A.; MacNair, C.R.; Brown, C.M.; Tsai, C.N.; Weber, B.S.; Zlitni, S.; Rao, V.N.; Chun, J.; Junop, M.S.; Coombes, B.K.; et al. Mimicking the human environment in mice reveals that inhibiting biotin biosynthesis is effective against antibiotic-resistant pathogens. Nat. Microbiol. 2020, 5, 93–101. [Google Scholar] [CrossRef]
- Ji, X.; Zou, J.; Peng, H.; Stolle, A.S.; Xie, R.; Zhang, H.; Peng, B.; Mekalanos, J.J.; Zheng, J. Alarmone Ap4A is elevated by aminoglycoside antibiotics and enhances their bactericidal activity. Proc. Natl. Acad. Sci. USA 2019, 116, 9578–9585. [Google Scholar] [CrossRef]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef]
- Liberati, N.T.; Urbach, J.M.; Miyata, S.; Lee, D.G.; Drenkard, E.; Wu, G.; Villanueva, J.; Wei, T.; Ausubel, F.M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 2833–2838. [Google Scholar] [CrossRef]
- Becher, A.; Schweizer, H.P. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques 2000, 29, 948–950, 952. [Google Scholar] [CrossRef]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Spinnato, M.C.; Pasqua, M.; Imperi, F. Generation of Stable and Unmarked Conditional Mutants in Pseudomonas aeruginosa. Methods Mol. Biol. 2022, 2548, 21–35. [Google Scholar] [CrossRef]
- Milton, D.L.; O’Toole, R.; Horstedt, P.; Wolf-Watz, H. Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 1996, 178, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Heeb, S.; Blumer, C.; Haas, D. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J. Bacteriol. 2002, 184, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Rocchio, S.; Santorelli, D.; Rinaldo, S.; Franceschini, M.; Malatesta, F.; Imperi, F.; Federici, L.; Travaglini-Allocatelli, C.; Di Matteo, A. Structural and functional investigation of the Small Ribosomal Subunit Biogenesis GTPase A (RsgA) from Pseudomonas aeruginosa. FEBS J. 2019, 286, 4245–4260. [Google Scholar] [CrossRef]
- Pasqua, M.; Visaggio, D.; Lo Sciuto, A.; Genah, S.; Banin, E.; Visca, P.; Imperi, F. Ferric Uptake Regulator Fur Is Conditionally Essential in Pseudomonas aeruginosa. J. Bacteriol. 2017, 199, e00472-17. [Google Scholar] [CrossRef] [PubMed]
- Dubern, J.F.; Cigana, C.; De Simone, M.; Lazenby, J.; Juhas, M.; Schwager, S.; Bianconi, I.; Döring, G.; Eberl, L.; Williams, P.; et al. Integrated whole-genome screening for Pseudomonas aeruginosa virulence genes using multiple disease models reveals that pathogenicity is host specific. Environ. Microbiol. 2015, 17, 4379–4393. [Google Scholar] [CrossRef]
- Imperi, F.; Fiscarelli, E.V.; Visaggio, D.; Leoni, L.; Visca, P. Activity and Impact on Resistance Development of Two Antivirulence Fluoropyrimidine Drugs in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2019, 9, 49. [Google Scholar] [CrossRef]
- Simon, R.; Priefer, U.; Pühler, A. A broad host range mobilization system for in vivo genetic engineering: Transposon mutagenesis in Gram negative bacteria. Bio/Technology 1983, 1, 784–790. [Google Scholar] [CrossRef]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common virulence factors for bacterial pathogenicity in plants and animals. Science 1995, 268, 1899–1902. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).