
ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets

Abstract
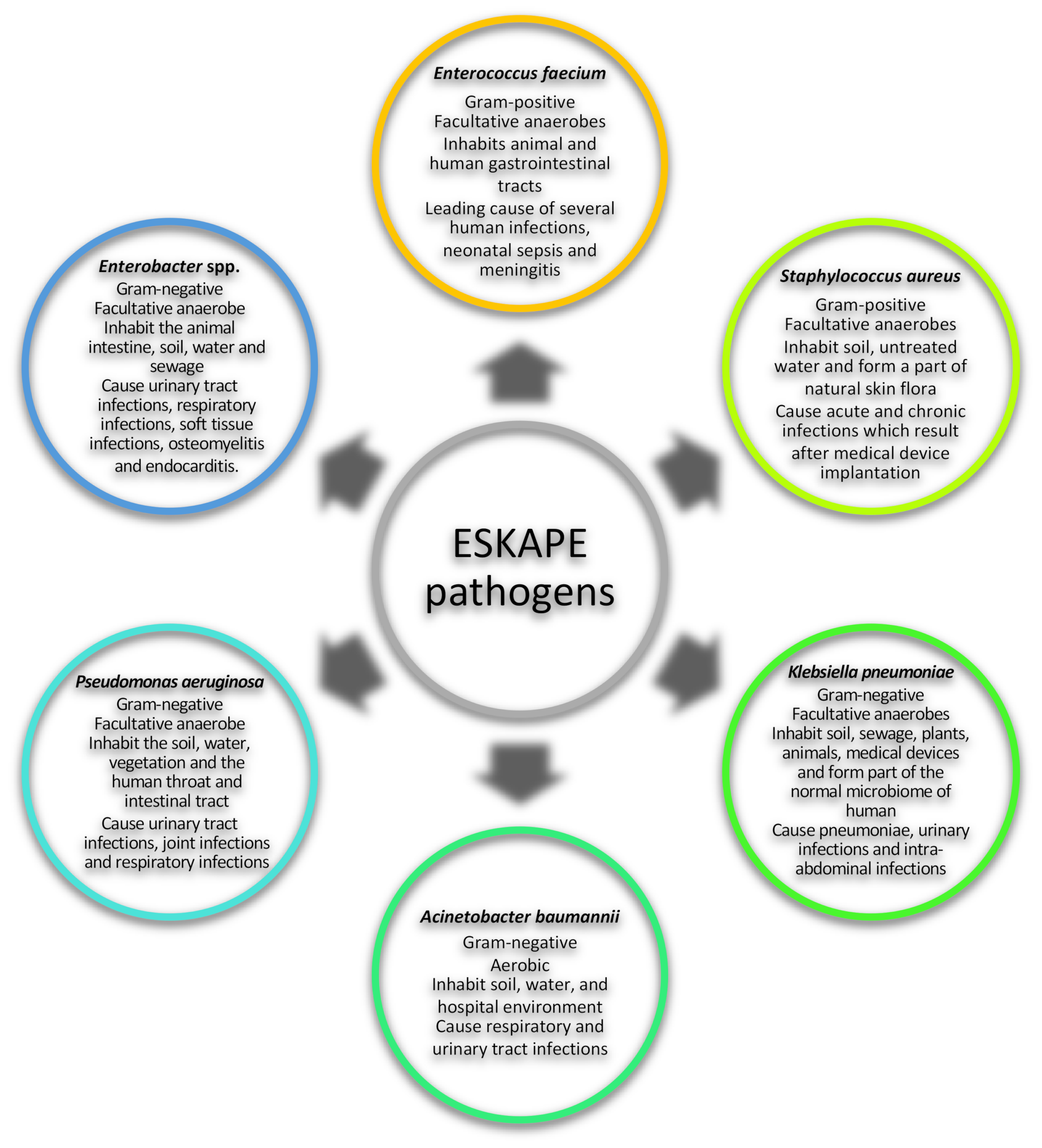
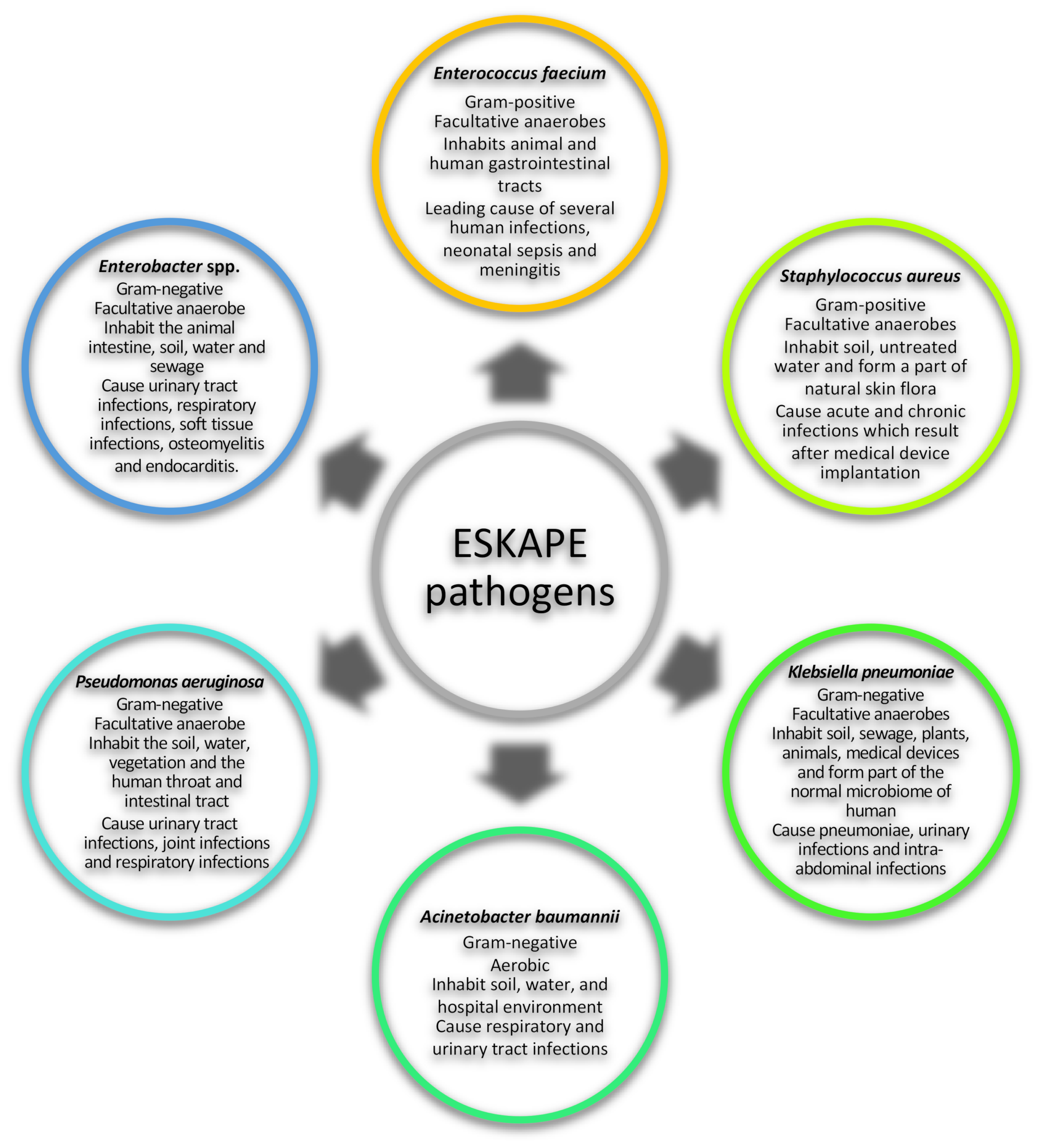
:1. Introduction
2. Caseinolytic Proteins: Classification, Function and Structure
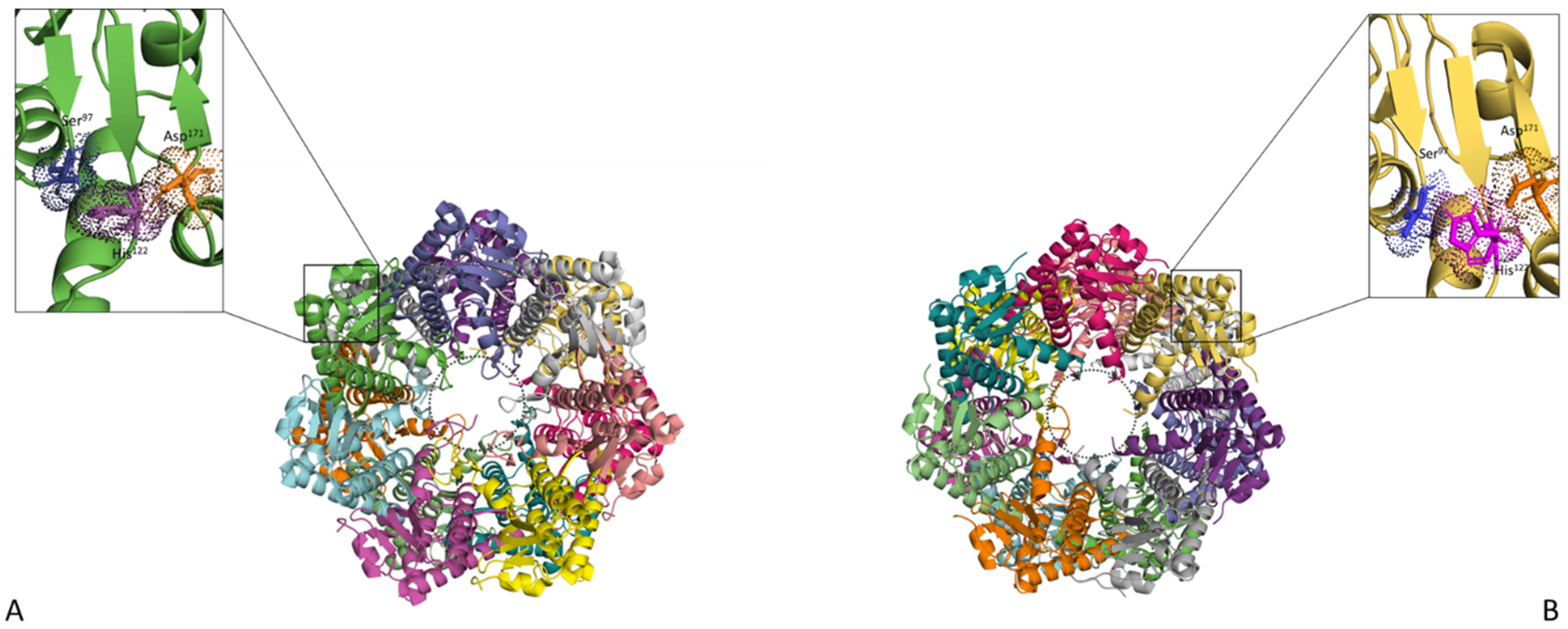
2.1. Catalytic Subunit—ClpP
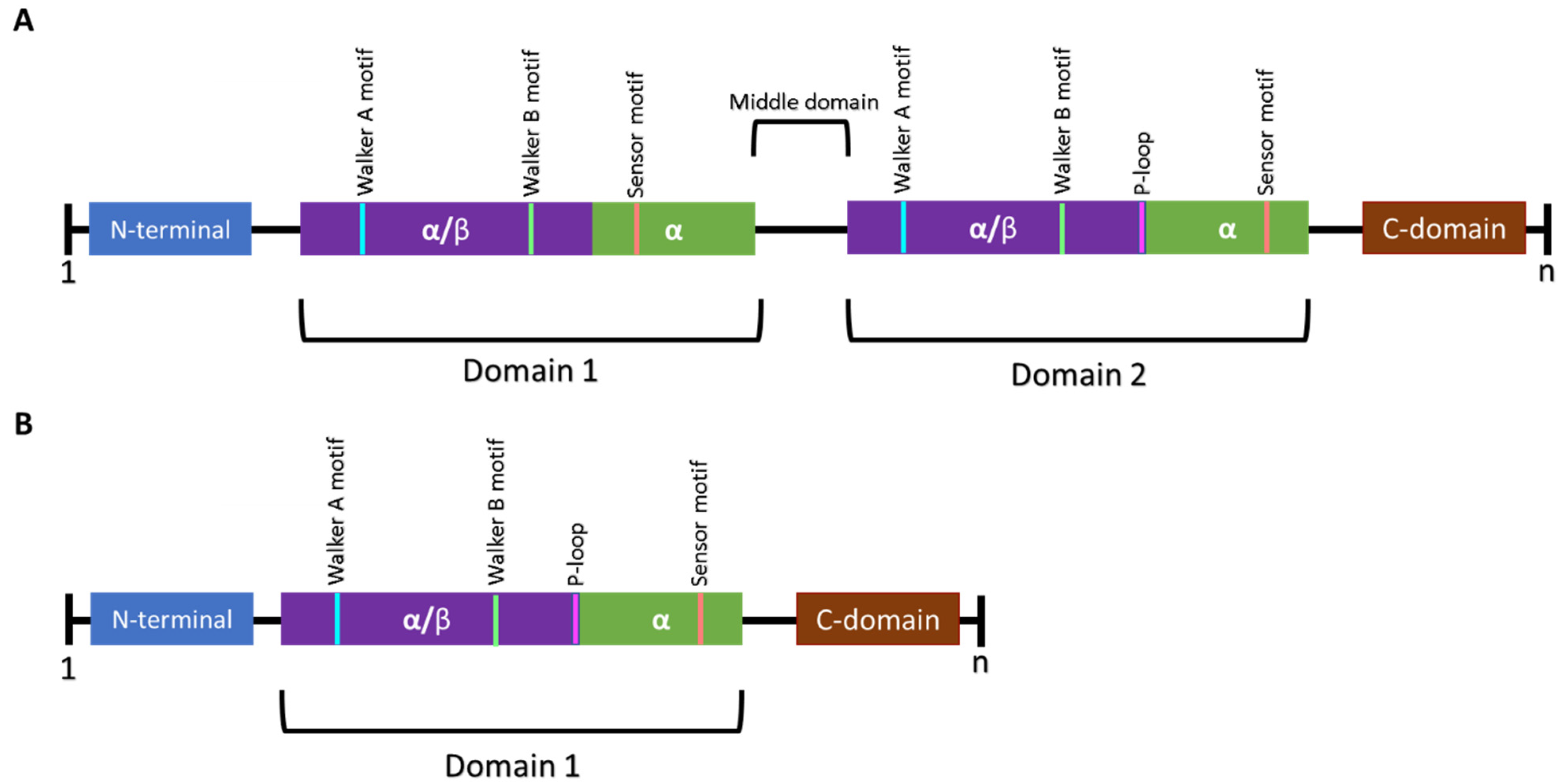
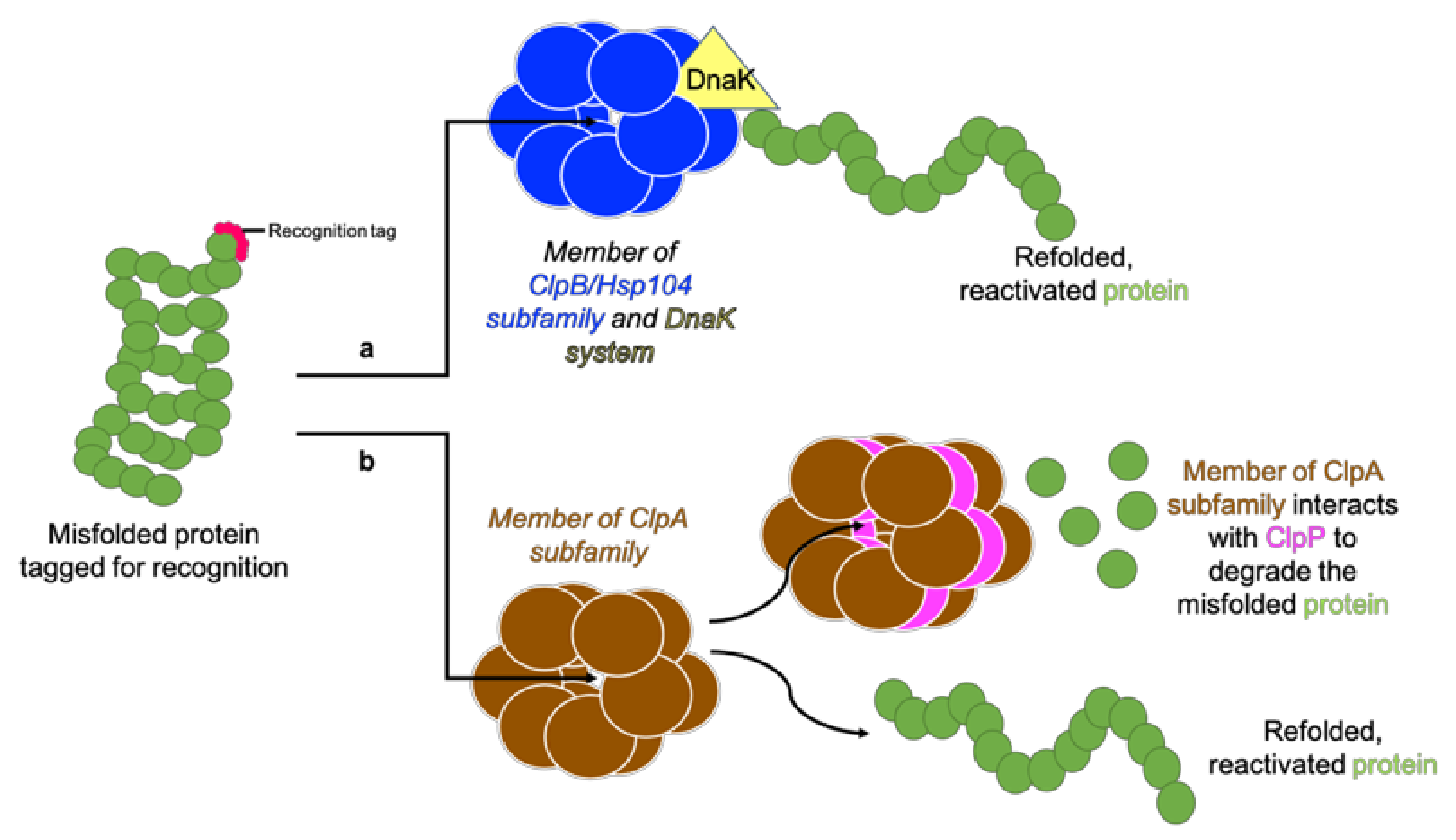
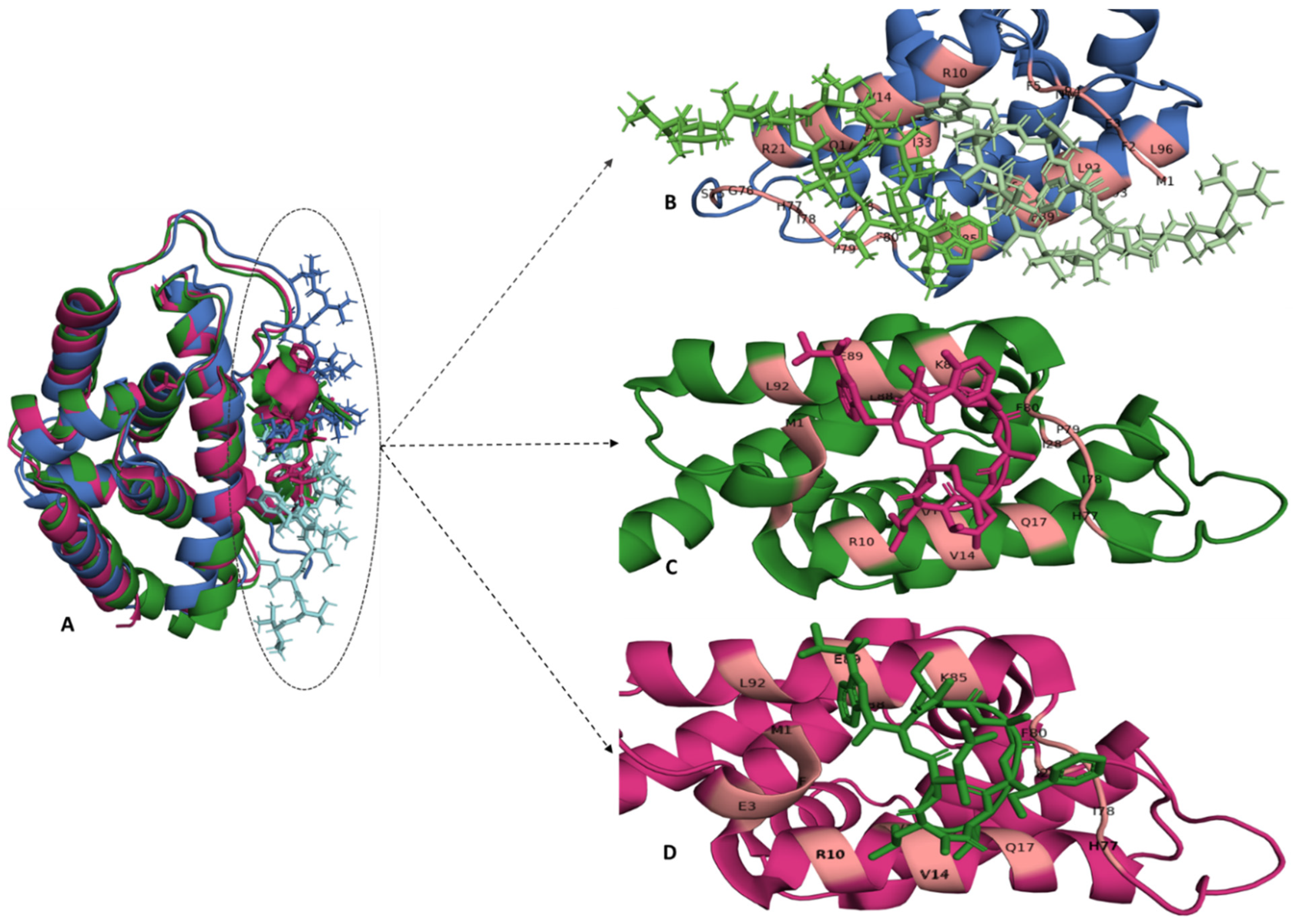
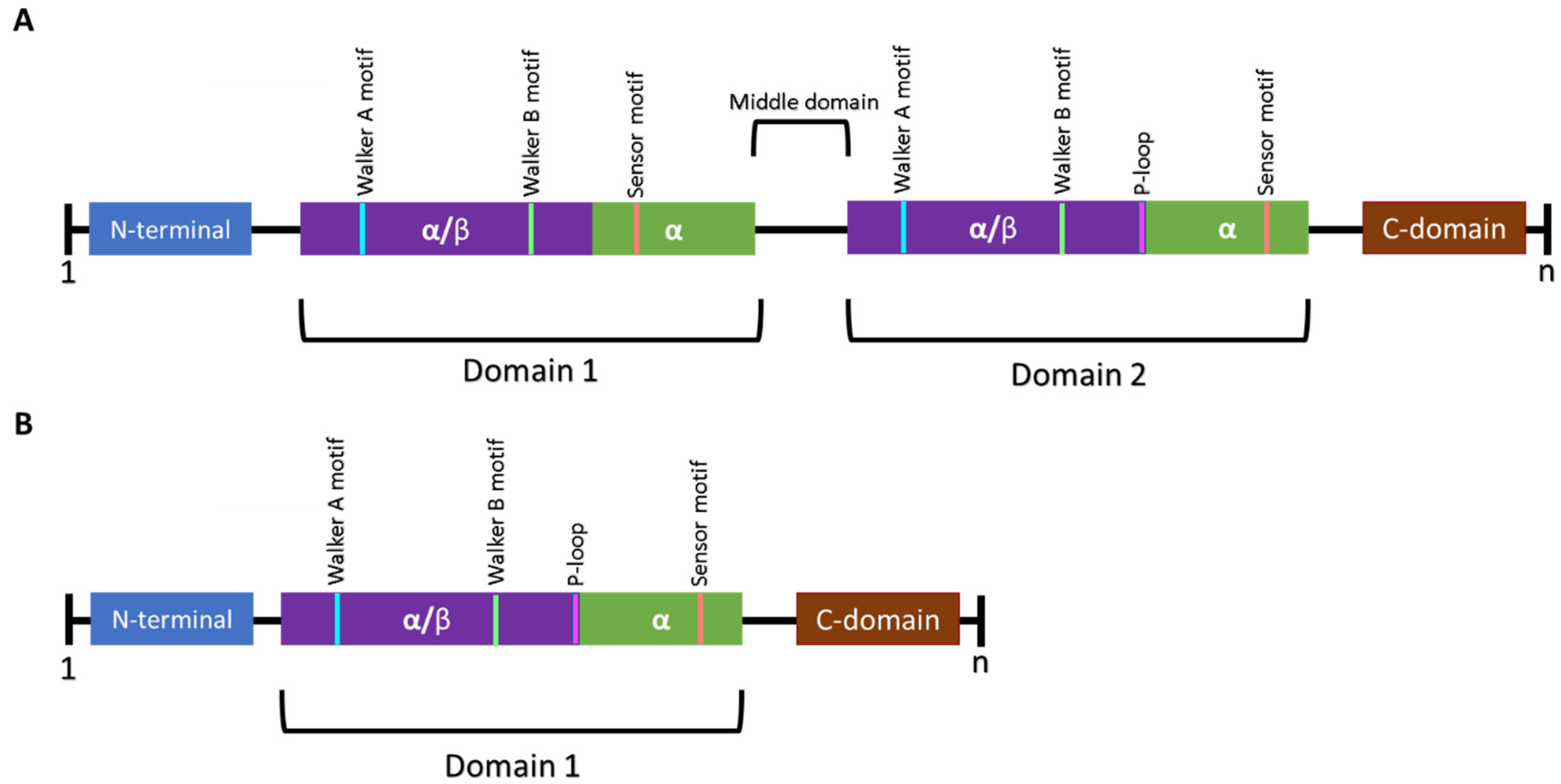
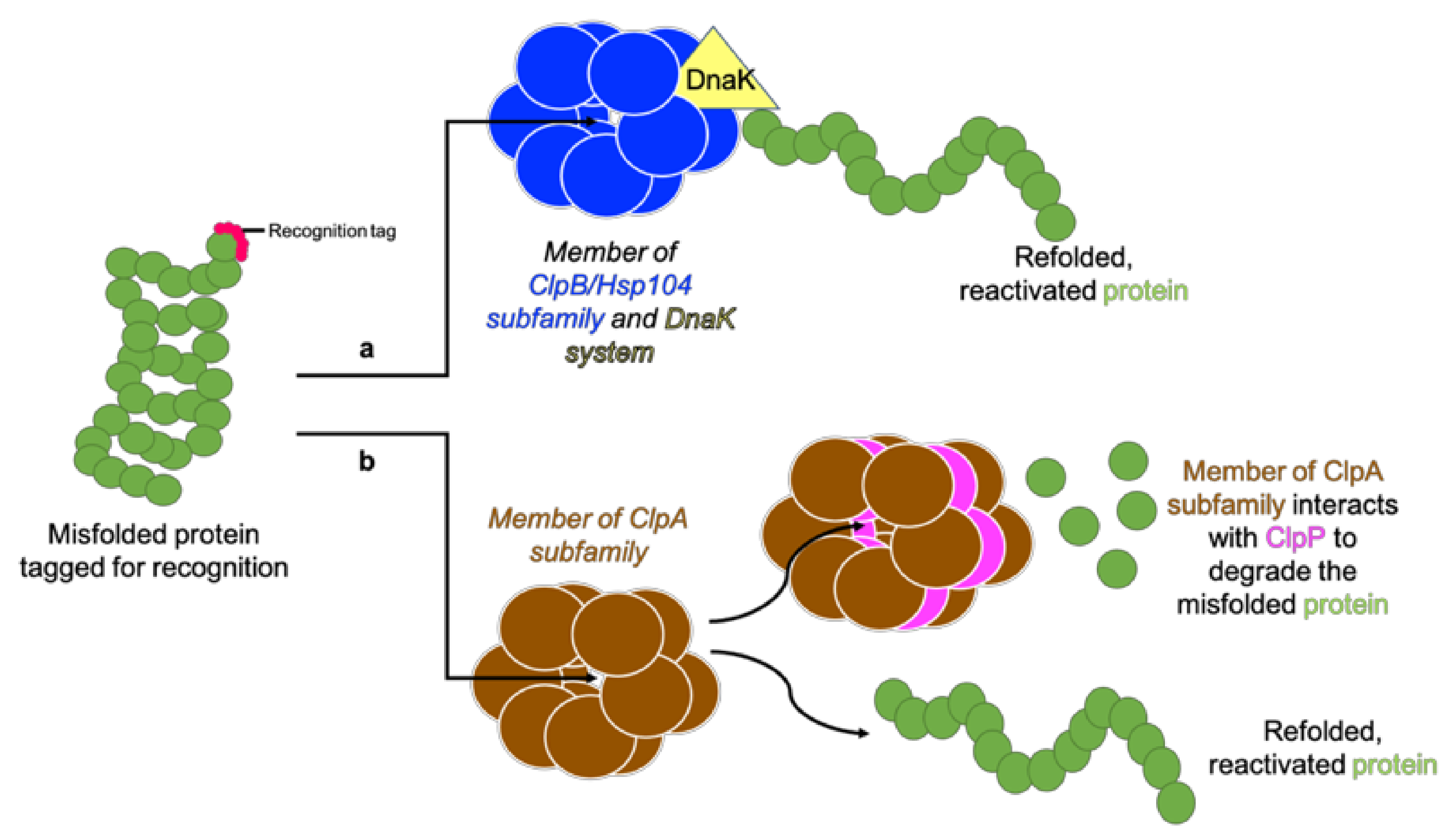
2.2. Regulatory Subunits—Clp ATPases
3. Caseinolytic Proteins Targeted in ESKAPE Pathogens
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Ramsamy, Y.; Essack, S.Y.; Sartorius, B.; Patel, M.; Mlisana, K.P. Antibiotic resistance trends of ESKAPE pathogens in Kwazulu-Natal, South Africa: A five-year retrospective analysis. Afr. J. Lab. Med. 2018, 7, 887. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed]
- Inweregbu, K.; Dave, J.; Pittard, A. Nosocomial infections. Contin. Educ. Anaesth. Crit. Care Pain 2005, 5, 14–17. [Google Scholar] [CrossRef]
- Haque, M.; Sartelli, M.; McKimm, J.; Bin Abu Bakar, M. Health care-associated infections—An overview. Infect. Drug Resist. 2018, 11, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Progress and Challenges in Implementing the Research on ESKAPE Pathogens. Infect. Control Hosp. Epidemiol. 2010, 31 (Suppl. 1), S7–S10. [Google Scholar] [CrossRef]
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; Bardanca, M.G.; Ambroa, A.; López, M.; Bou, G.; Tomás, M. Strategies to Combat Multidrug-Resistant and Persistent Infectious Diseases. Antibiotics 2020, 9, 65. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- AhYoung, A.P.; Koehl, A.; Cascio, D.; Egea, P.F. Structural mapping of the C lp B ATP ases of Plasmodium falciparum: Targeting protein folding and secretion for antimalarial drug design. Protein Sci. 2015, 24, 1508–1520. [Google Scholar] [CrossRef]
- Higaki, S.; Morohashi, M.; Yamagishi, T. Isolation of Enterococcus species from infectious skin lesions. Drugs Under Exp. Clin. Res. 2002, 28, 91–93. [Google Scholar]
- Kafil, H.S.; Asgharzadeh, M. Vancomycin-resistant enteroccus faecium and enterococcus faecalis isolated from education hospital of iran. Maedica 2014, 9, 323–327. [Google Scholar]
- Giacometti, A.; Cirioni, O.; Schimizzi, A.M.; Del Prete, M.S.; Barchiesi, F.; D’Errico, M.M.; Petrelli, E.; Scalise, G. Epidemiology and Microbiology of Surgical Wound Infections. J. Clin. Microbiol. 2000, 38, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Calfee, D.P. Recent advances in the understanding and management of Klebsiella pneumoniae. F1000Research 2017, 6, 1760. [Google Scholar] [CrossRef]
- Martin, R.M.; Bachman, M.A. Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. [Google Scholar] [CrossRef]
- Podschun, R.; Ullmann, U. Klebsiella spp. as Nosocomial Pathogens: Epidemiology, Taxonomy, Typing Methods, and Pathogenicity Factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef]
- Bojer, M.S.; Struve, C.; Ingmer, H.; Hansen, D.S.; Krogfelt, K.A. Heat Resistance Mediated by a New Plasmid Encoded Clp ATPase, ClpK, as a Possible Novel Mechanism for Nosocomial Persistence of Klebsiella pneumoniae. PLoS ONE 2010, 5, e15467. [Google Scholar] [CrossRef]
- Nakonieczna, J.; Woźniak, A.; Pierański, M.K.; Rapacka-Zdonczyk, A.; Ogonowska, P.; Grinholc, M. Photoinactivation of ESKAPE pathogens: Overview of novel therapeutic strategy. Future Med. Chem. 2019, 11, 443–461. [Google Scholar] [CrossRef]
- Llaca-Díaz, J.M.; Mendoza-Olazarán, S.; Camacho-Ortiz, A.; Flores, S.; Garza-González, E. One-Year Surveillance of ESKAPE Pathogens in an Intensive Care Unit of Monterrey, Mexico. Chemotherapy 2012, 58, 475–481. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The Epidemiology, Pathogenesis and Treatment of Pseudomonas aeruginosa Infections. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Xie, G.; Traglia, G.M.; Johnson, S.L.; Davenport, K.W.; van Duin, D.; Ramazani, A.; Perez, F.; Jacobs, M.R.; Sherratt, D.J.; et al. Whole-Genome Comparative Analysis of Two Carbapenem-Resistant ST-258 Klebsiella pneumoniae Strains Isolated during a North-Eastern Ohio Outbreak: Differences within the High Heterogeneity Zones. Genome Biol. Evol. 2016, 8, 2036–2043. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Obi, L.; Morobe, I.; Bisi-Johnson, M. Molecular Characteristics and Antibiotic Resistance Profiles of Klebsiella Isolates in Mthatha, Eastern Cape Province, South Africa. Int. J. Microbiol. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pons, M.J.; Ruiz, J. Current trends in epidemiology and antimicrobial resistance in intensive care units. J. Emerg. Crit. Care Med. 2019, 3, 5. [Google Scholar] [CrossRef]
- Kidd, T.J.; Mills, G.; Sá-Pessoa, J.; Dumigan, A.; Frank, C.G.; Insua, J.L.; Ingram, R.; Hobley, L.; Bengoechea, J.A. A Klebsiella pneumoniae antibiotic resistance mechanism that subdues host defences and promotes virulence. EMBO Mol. Med. 2017, 9, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 464–473. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-Virulence Strategies To Combat Bacteria-Mediated Disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Mühlen, S.; Dersch, P. Anti-Virulence Strategies to Target Bacterial Infections. In How to Overcome the Antibiotic Crisis; Springer: Cham, Switzerland, 2015; pp. 147–183. [Google Scholar]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different Drugs for Bad Bugs: Antivirulence Strategies in the Age of Antibiotic Resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Glover, J.R.; Singer, M.A.; Lindquist, S. HSP100/Clp proteins: A common mechanism explains diverse functions. Trends Biochem. Sci. 1996, 21, 289–296. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van Der Veken, P.; De Winter, H.; Augustyns, K. ClpP Protease, a Promising Antimicrobial Target. Int. J. Mol. Sci. 2019, 20, 2232. [Google Scholar] [CrossRef]
- Ingmer, H.; Vogensen, F.K.; Hammer, K.; Kilstrup, M. Disruption and Analysis of the clpB, clpC, and clpE Genes in Lactococcus lactis: ClpE, a New Clp Family in Gram-Positive Bacteria. J. Bacteriol. 1999, 181, 2075–2083. [Google Scholar] [CrossRef]
- Lee, S.; Sowa, M.E.; Watanabe, Y.-H.; Sigler, P.B.; Chiu, W.; Yoshida, M.; Tsai, F.T. The Structure of ClpB: A Molecular Chaperone that Rescues Proteins from an Aggregated State. Cell 2003, 115, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Pietrosiuk, A.; Lenherr, E.D.; Falk, S.; Bönemann, G.; Kopp, J.; Zentgraf, H.; Sinning, I.; Mogk, A. Molecular Basis for the Unique Role of the AAA+ Chaperone ClpV in Type VI Protein Secretion. J. Biol. Chem. 2011, 286, 30010–30021. [Google Scholar] [CrossRef]
- Zheng, B.; Halperin, T.; Hruskova-Heidingsfeldova, O.; Adam, Z.; Clarke, A.K. Characterization of chloroplast Clp proteins in Arabidopsis: Localization, tissue specificity and stress responses. Physiol. Plant. 2002, 114, 92–101. [Google Scholar] [CrossRef]
- Capestany, C.A.; Tribble, G.D.; Maeda, K.; Demuth, D.R.; Lamont, R.J. Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J. Bacteriol. 2008, 190, 1436–1446. [Google Scholar] [CrossRef]
- Nair, S.; Milohanic, E.; Berche, P. ClpC ATPase Is Required for Cell Adhesion and Invasion of Listeria monocytogenes. Infect. Immun. 2000, 68, 7061–7068. [Google Scholar] [CrossRef]
- Bojer, M.S.; Struve, C.; Ingmer, H.; Krogfelt, K.A. ClpP-dependent and-independent activities encoded by the polycistronic clpK-encoding locus contribute to heat shock survival in Klebsiella pneumoniae. Res. Microbiol. 2013, 164, 205–210. [Google Scholar] [CrossRef]
- Park, S.-S.; Kwon, H.-Y.; Tran, T.D.-H.; Choi, M.-H.; Jung, S.-H.; Lee, S.; Briles, D.E.; Rhee, D.-K. ClpL is a chaperone without auxiliary factors. FEBS J. 2015, 282, 1352–1367. [Google Scholar] [CrossRef]
- Thibault, G.; Tsitrin, Y.; Davidson, T.; Gribun, A.; Houry, W.A. Large nucleotide-dependent movement of the N-terminal domain of the ClpX chaperone. EMBO J. 2006, 25, 3367–3376. [Google Scholar] [CrossRef]
- Ali, M.S.; Baek, K.-H. Protective Roles of Cytosolic and Plastidal Proteasomes on Abiotic Stress and Pathogen Invasion. Plants 2020, 9, 832. [Google Scholar] [CrossRef]
- Frees, D.; Qazi, S.N.A.; Hill, P.; Ingmer, H. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol. Microbiol. 2003, 48, 1565–1578. [Google Scholar] [CrossRef]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Silbergleit, M.; Vasquez, A.A.; Miller, C.J.; Sun, J.; Kato, I. Chapter Five-Oral and intestinal bacterial exotoxins: Potential linked to carcinogenesis. In Progress in Molecular Biology and Translational Science; Sun, J., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 171, pp. 131–193. [Google Scholar]
- Farrand, A.J.; Reniere, M.L.; Ingmer, H.; Frees, D.; Skaar, E.P. Regulation of Host Hemoglobin Binding by the Staphylococcus aureus Clp Proteolytic System. J. Bacteriol. 2013, 195, 5041–5050. [Google Scholar] [CrossRef]
- Fsihi, H.; Steffen, P.; Cossart, P. CHAPTER 16-Listeria monocytogenes. In Principles of Bacterial Pathogenesis; Groisman, E.A., Ed.; Academic Press: San Diego, CA, USA, 2001; pp. 751–803. [Google Scholar]
- Gaillot, O.; Pellegrini, E.; Bregenholt, S.; Nair, S.; Berche, P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol. 2002, 35, 1286–1294. [Google Scholar] [CrossRef]
- Qiu, D.; Eisinger, V.M.; Head, N.E.; Pier, G.B.; Yu, H.D. ClpXP proteases positively regulate alginate overexpression and mucoid conversion in Pseudomonas aeruginosa. Microbiology 2008, 154, 2119–2130. [Google Scholar] [CrossRef]
- Kwon, H.-Y.; Ogunniyi, A.D.; Choi, M.-H.; Pyo, S.-N.; Rhee, D.-K.; Paton, J.C. The ClpP Protease of Streptococcus pneumoniae Modulates Virulence Gene Expression and Protects against Fatal Pneumococcal Challenge. Infect. Immun. 2004, 72, 5646–5653. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Carney, D.W.; Sello, J.K.; Sauer, R.T. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc. Natl. Acad. Sci. USA 2014, 111, E4587–E4595. [Google Scholar] [CrossRef]
- Raju, R.M.; Unnikrishnan, M.; Rubin, D.H.F.; Krishnamoorthy, V.; Kandror, O.; Akopian, T.N.; Goldberg, A.L.; Rubin, E.J. Mycobacterium tuberculosis ClpP1 and ClpP2 Function Together in Protein Degradation and Are Required for Viability in vitro and During Infection. PLoS Pathog. 2012, 8, e1002511. [Google Scholar] [CrossRef]
- Dahmen, M.; Vielberg, M.-T.; Groll, M.; Sieber, S.A. Structure and Mechanism of the Caseinolytic Protease ClpP1/2 Heterocomplex from Listeria monocytogenes. Angew. Chem. Int. Ed. 2015, 54, 3598–3602. [Google Scholar] [CrossRef]
- Nagpal, J.; Paxman, J.; Zammit, J.E.; Alhuwaider, A.; Truscott, K.N.; Heras, B.; Dougan, D.A. Molecular and structural insights into an asymmetric proteolytic complex (ClpP1P2) from Mycobacterium smegmatis. Sci. Rep. 2019, 9, 18019. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Malik, I.T.; Thomy, D.; Henrichfreise, B.; Sass, P. The functional ClpXP protease of Chlamydia trachomatis requires distinct clpP genes from separate genetic loci. Sci. Rep. 2019, 9, 14129. [Google Scholar] [CrossRef] [PubMed]
- Mawla, G.D.; Hall, B.M.; Cárcamo-Oyarce, G.; Grant, R.A.; Zhang, J.J.; Kardon, J.R.; Ribbeck, K.; Sauer, R.T.; Baker, T.A. ClpP1P2 peptidase activity promotes biofilm formation in Pseudomonas aeruginosa. Mol. Microbiol. 2020, 115, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Chaudhary, H.; Marsee, J.D. Phylogenetic analysis predicts structural divergence for proteobacterial ClpC proteins. J. Struct. Biol. 2018, 201, 52–62. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. 2020. Available online: https://www.schrodinger.com/products/pymol (accessed on 18 August 2022).
- Hanson, P.I.; Whiteheart, S.W. AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 2005, 6, 519–529. [Google Scholar] [CrossRef]
- Baker, T.A.; Sauer, R.T. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim. Biophys. Acta 2012, 1823, 15–28. [Google Scholar] [CrossRef]
- Maurizi, M.R.; Xia, D. Protein binding and disruption by Clp/Hsp100 chaperones. Structure 2004, 12, 175–183. [Google Scholar] [CrossRef]
- Sauer, R.T.; Baker, T.A. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Frees, D.; Gerth, U.; Ingmer, H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 142–149. [Google Scholar] [CrossRef]
- Leigh, R.J.; McKenna, C.; McWade, R.; Lynch, B.; Walsh, F. Comparative genomics and pangenomics of vancomycin resistant and susceptible Enterococcus faecium from Irish hospitals across 20 years. BioRxiv 2021. [Google Scholar] [CrossRef]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef]
- Chatterjee, I.; Becker, P.; Grundmeier, M.; Bischoff, M.; Somerville, G.A.; Peters, G.; Sinha, B.; Harraghy, N.; Proctor, R.A.; Herrmann, M. Staphylococcus aureus ClpC Is Required for Stress Resistance, Aconitase Activity, Growth Recovery, and Death. J. Bacteriol. 2005, 187, 4488–4496. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, S.B.; Bojer, M.S.; Boll, E.J.; Martin, Y.; Helmersen, K.; Skogstad, M.; Struve, C. Heat-resistant, extended-spectrum β-lactamase-producing Klebsiella pneumoniae in endoscope-mediated outbreak. J. Hosp. Infect. 2016, 93, 57–62. [Google Scholar] [CrossRef]
- Belisario, J.; Lee, H.; Luknauth, H.; Rigel, N.; Martinez, L. Acinetobacter baumannii Strains Deficient in the Clp Chaperone-Protease Genes Have Reduced Virulence in a Murine Model of Pneumonia. Pathogens 2021, 10, 204. [Google Scholar] [CrossRef]
- Wang, N.; Ozer, E.; Mandel, M.; Hauser, A.R. Genome-Wide Identification of Acinetobacter baumannii Genes Necessary for Persistence in the Lung. mBio 2014, 5, e01163-14. [Google Scholar] [CrossRef]
- Boyd, A.; Chakrabarty, A.M. Pseudomonas aeruginosa biofilms: Role of the alginate exopolysaccharide. J. Ind. Microbiol. Biotechnol. 1995, 15, 162–168. [Google Scholar] [CrossRef]
- Lee, C.; Franke, K.B.; Kamal, S.M.; Kim, H.; Lünsdorf, H.; Jäger, J.; Nimtz, M.; Trček, J.; Jänsch, L.; Bukau, B.; et al. Stand-alone ClpG disaggregase confers superior heat tolerance to bacteria. Proc. Natl. Acad. Sci. USA 2017, 115, E273–E282. [Google Scholar] [CrossRef]
- Kirstein, J.; Hoffmann, A.; Lilie, H.; Schmidt, R.; Rübsamen-Waigmann, H.; Brötz-Oesterhelt, H.; Mogk, A.; Turgay, K. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med. 2009, 1, 37–49. [Google Scholar] [CrossRef]
- Lee, B.-G.; Park, E.Y.; Lee, K.-E.; Jeon, H.; Sung, K.H.; Paulsen, H.; Rübsamen-Schaeff, H.; Brötz-Oesterhelt, H.; Song, H.K. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol. 2010, 17, 471–478. [Google Scholar] [CrossRef]
- Fetzer, C.; Korotkov, V.S.; Thänert, R.; Lee, K.M.; Neuenschwander, M.; Von Kries, J.P.; Medina, E.; Sieber, S.A. A Chemical Disruptor of the ClpX Chaperone Complex Attenuates the Virulence of Multidrug-Resistant Staphylococcus aureus. Angew. Chem. Int. Ed. 2017, 56, 15746–15750. [Google Scholar] [CrossRef]
- Gao, W.; Kim, J.-Y.; Anderson, J.R.; Akopian, T.; Hong, S.; Jin, Y.-Y.; Kandror, O.; Kim, J.-W.; Lee, I.-A.; Lee, S.-Y.; et al. The Cyclic Peptide Ecumicin Targeting ClpC1 Is Active against Mycobacterium tuberculosis In Vivo. Antimicrob. Agents Chemother. 2015, 59, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Gavrish, E.; Sit, C.S.; Cao, S.; Kandror, O.; Spoering, A.; Peoples, A.; Ling, L.; Fetterman, A.; Hughes, D.; Bissell, A.; et al. Lassomycin, a Ribosomally Synthesized Cyclic Peptide, Kills Mycobacterium tuberculosis by Targeting the ATP-Dependent Protease ClpC1P1P2. Chem. Biol. 2014, 21, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, N.M.; Lee, H.; Choules, M.P.; Pauli, G.F.; Phansalkar, R.; Anderson, J.R.; Gao, W.; Ren, J.; Santarsiero, B.D.; Lee, H.; et al. High-Resolution Structure of ClpC1-Rufomycin and Ligand Binding Studies Provide a Framework to Design and Optimize Anti-Tuberculosis Leads. ACS Infect. Dis. 2019, 5, 829–840. [Google Scholar] [CrossRef]
- Labana, P.; Dornan, M.H.; Lafrenière, M.; Czarny, T.L.; Brown, E.D.; Pezacki, J.P.; Boddy, C.N. Armeniaspirols inhibit the AAA+ proteases ClpXP and ClpYQ leading to cell division arrest in Gram-positive bacteria. Cell Chem. Biol. 2021, 28, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Fetzer, C.; Korotkov, V.S.; Sieber, S.A. Hydantoin analogs inhibit the fully assembled ClpXP protease without affecting the individual peptidase and chaperone domains. Org. Biomol. Chem. 2019, 17, 7124–7127. [Google Scholar] [CrossRef]
- Cherinka, B.; Andrews, B.H.; Sánchez-Gallego, J.; Brownstein, J.; Argudo-Fernández, M.D.C.; Blanton, M.; Bundy, K.; Jones, A.; Masters, K.; Law, D.R.; et al. Marvin: A Tool Kit for Streamlined Access and Visualization of the SDSS-IV MaNGA Data Set. Astron. J. 2019, 158, 74. [Google Scholar] [CrossRef]
- Vasudevan, D.; Rao, S.P.S.; Noble, C.G. Structural Basis of Mycobacterial Inhibition by Cyclomarin A. J. Biol. Chem. 2013, 288, 30883–30891. [Google Scholar] [CrossRef]
- Wolf, N.M.; Lee, H.; Zagal, D.; Nam, J.-W.; Oh, D.-C.; Lee, H.; Suh, J.-W.; Pauli, G.F.; Cho, S.; Abad-Zapatero, C. Structure of the N-terminal domain of ClpC1 in complex with the antituberculosis natural product ecumicin reveals unique binding interactions. Acta Crystallogr. Sect. D Struct. Biol. 2020, 76, 458–471. [Google Scholar] [CrossRef]
- Choules, M.P.; Wolf, N.M.; Lee, H.; Anderson, J.R.; Grzelak, E.M.; Wang, Y.; Ma, R.; Gao, W.; McAlpine, J.B.; Jin, Y.-Y.; et al. Rufomycin Targets ClpC1 Proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob. Agents Chemother. 2019, 63, e02204-18. [Google Scholar] [CrossRef]
- Kazmaier, U.; Junk, L. Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Mar. Drugs 2021, 19, 446. [Google Scholar] [CrossRef]
- Hall, B.M.; Breidenstein, E.B.M.; de la Fuente-Núñez, C.; Reffuveille, F.; Mawla, G.D.; Hancock, R.E.W.; Baker, T.A. Two Isoforms of Clp Peptidase in Pseudomonas aeruginosa Control Distinct Aspects of Cellular Physiology. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [PubMed]
- Zolkiewski, M.; Zhang, T.; Nagy, M. Aggregate reactivation mediated by the Hsp100 chaperones. Arch. Biochem. Biophys. 2012, 520, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Sharma, A.K.; Vatamaniuk, O.K. N-Terminal Extension and C-Terminal Domains Are Required for ABCB6/HMT-1 Protein Interactions, Function in Cadmium Detoxification, and Localization to the Endosomal-Recycling System in Caenorhabditis elegans. Front. Physiol. 2018, 9, 885. [Google Scholar] [CrossRef]
- Lo, J.H.; Baker, T.A.; Sauer, R.T. Characterization of the N-terminal repeat domain of Escherichia coli ClpA—A class I Clp/HSP100 ATPase. Protein Sci. 2001, 10, 551–559. [Google Scholar] [CrossRef]
- Kedzierska, S.; Akoev, V.; Barnett, A.M.E.; Zolkiewski, M. Structure and Function of the Middle Domain of ClpB from Escherichia coli. Biochemistry 2003, 42, 14242–14248. [Google Scholar] [CrossRef]
- Cashikar, A.G.; Schirmer, E.C.; Hattendorf, D.A.; Glover, J.R.; Ramakrishnan, M.S.; Ware, D.M.; Lindquist, S.L. Defining a Pathway of Communication from the C-Terminal Peptide Binding Domain to the N-Terminal ATPase Domain in a AAA Protein. Mol. Cell 2002, 9, 751–760. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
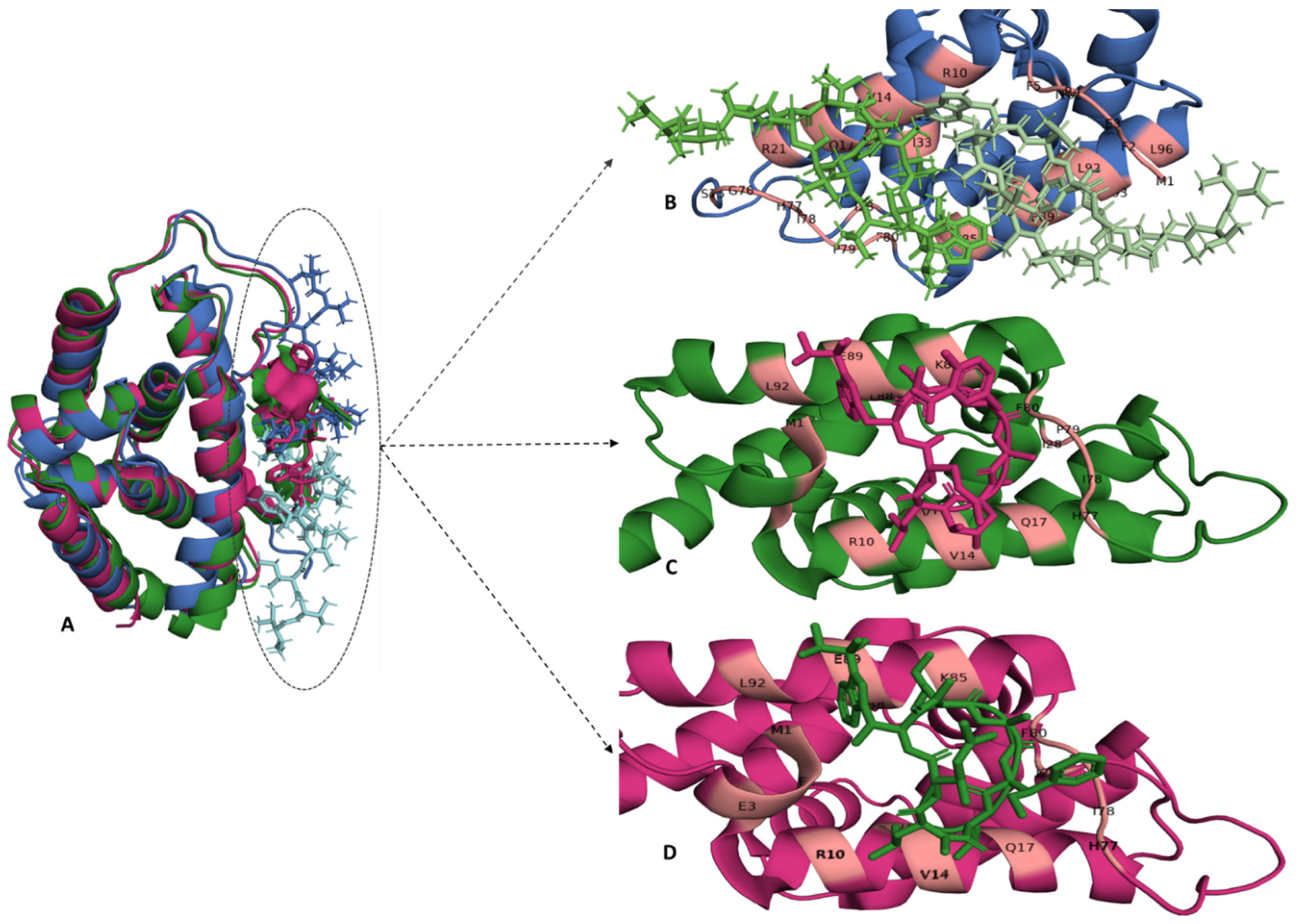{kind=link}
| Resistance Strategy | Resistance Mechanism | Antibiotics | ESKAPE Pathogens | References |
|---|---|---|---|---|
| Drug inactivation | Production of β-lactamase enzyme, which hydrolyses β-lactam rings | β-lactam (penicillin, carbapenems and | K. pneumoniae | [1,3,22] |
| cephalosporins) | P. aeruginosa, Enterobacter | |||
| Contains chromosomally encoded AAC(6′)-Ii, which is responsible for enzymatic inactivation and EfmM ribosomal methylation | Tobramycin | E. faecium | [23] | |
| Carbapenemases, metallo-β-lactamases 2 and oxacillinase serine β-lactamases produced to catalyse antibiotic hydrolysis | Colistin, imipenem | A. baumannii | [3] | |
| Decreased drug influx | Reduce the amount of porin protein OprD, or via loss of an outer membrane protein (Omp) | β-lactam (Imipenem and meropenem) | P. aeruginosa, A. baumannii | [3,23] |
| Expresses enterococcal surface protein (ESP), which results in the formation of thicker biofilms | Vancomycin | E. faecium | [1] | |
| Thick cell wall traps and reduces antibiotic permeation | Vancomycin | S. aureus | [1] | |
| Mutation of mbrB gene | Colistin | K. pneumoniae | [24] | |
| Outer membrane (OmpF) protein with exclusion limit | MDR 1 | P. aeruginosa | [1] | |
| Developing 4 resistant nodulation division (RND) type MDR efflux pump to remove toxic compounds from the periplasm and cytoplasm | MDR 1 | P. aeruginosa | [3] | |
| Efflux pump system | Nor-like efflux pump | Hydrophilic fluoroquinolones | E. faecium | [23] |
| Expression of Penicillin-binding proteins (PBPs) | β-lactam (Penicillin, Cephalosporins, Carbapenems) | S. aureus | [1,3] | |
| Cephalosporins and aminoglycosides | E. faecium | [23] | ||
| Upregulation of MexAB-OprM | Sulfonamides, cephalosporins, β-lactams, fluoroquinolones | P. aeruginosa | [1] | |
| AcrAB-TolC | Tetracyclines (including tigecycline) | K. pneumoniae | [25] | |
| Alteration of terminal sequence of cell wall precursors | VanA | E. faecium | [1] | |
| Drug site modification | Expresses mecA, which encodes a low-affinity penicillin-binding protein | β-lactam (Penicillin, Methicillin) | S. aureus | [1] |
| Expression of Aminoglycoside-modifying enzymes | Aminoglycosides | P. aeruginosa | [26] | |
| Qnr acts as a DNA homologue to compete for the DNA-binding site of DNA gyrase and topoisomerase IV | Quinolone | K. pneumoniae | [25] |
| Clp Catalytic Subunit | |||
|---|---|---|---|
| Species | Functions | References | |
| ClpP | A number of bacteria including Escherichia coli, Bacillus subtilis, S. aureus | Proteolysis of damaged or misfolded proteins | [31] |
| Clp regulatory subunit 1 | |||
| ClpA 2 | Gram-positive Proteobacteria | Protein quality control | [32] |
| ClpB | Prokaryotes, yeast, and plants | Disaggregation of stress-damaged proteins | [33,34,35] |
| Porphyromonas gingivalis | Intracellular replication and survival | [36] | |
| ClpC | Gram-positive bacteria (Firmicutes and Actinobacteria) and Cyanobacteria | Protein quality control, red blood cell lysis, regulate expression of virulence factors | [32,37] |
| ClpD | Chloroplasts of higher plants | Molecular chaperone | [35] |
| ClpE | Firmicutes | Thermotolerance, cell division and virulence | [32] |
| ClpK | K. pneumonia | Thermotolerance | [38] |
| ClpL | Streptococcus pneumoniae | Nucleotide phosphohydrolase activity, stabilises unfolded proteins, prevents protein aggregation | [39] |
| ClpV | Gram-negative bacteria | Component of the type V1 secrection system | [40] |
| ClpM | Mus musculus | Protein quality control | [35,41] |
| ClpN | Pseudomonas aeruginosa | Cell division | [35,41] |
| ClpX | Proteobacteria, Firmicutes and Thermatogae | Protein quality control, cell division, heat tolerance and virulence | [32,36] |
| ClpY | Gram-positive Proteobacteria | Cell division, heat shock response and capsule transcription | [32] |
| ESKAPE Pathogens | Caseinolytic Proteins | References |
|---|---|---|
| E. faecium | ClpP | [64,65] |
| ClpC | [64,66] | |
| S. aureus | ClpP, ClpB, ClpC | [36,63] |
| ClpX | [36,63] | |
| K. pneumoniae | ClpK | [16,38,67] |
| A. baumannii | ClpP | [68,69] |
| P. aeruginosa | ClpXP and ClpP2 | [48,70] |
| ClpP | [38] | |
| ClpG | [71] | |
| Enterobacter | None reported |
| Compound | Structure 1 | Mechanism of Action | References |
|---|---|---|---|
| 334 | 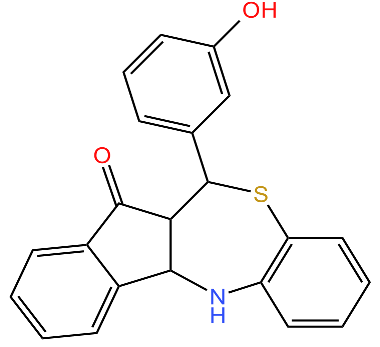 | Deoligomerization of S. aureus ClpX, disrupts the ClpXP complex and blocks ClpX ATPase activity. S. aureus produces lower levels of toxins, such as hemolysins in the presence of the compound. | [43,74] |
| D3 | 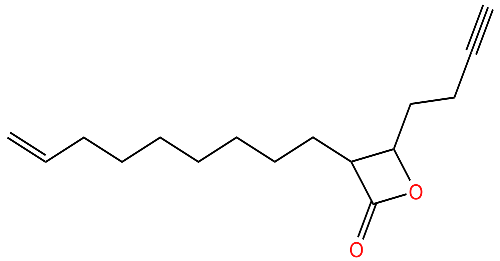 | Irreversibly inhibits ClpP in methicillin-resistant S. aureus. Most potent inhibitor. | [43] |
| E2 | 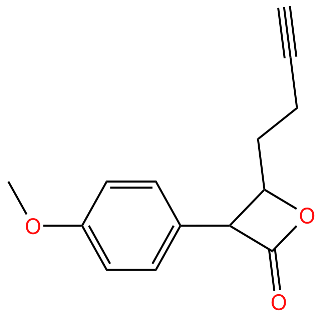 | Irreversibly inhibits ClpP in methicillin-resistant S. aureus | [43] |
| G2 | 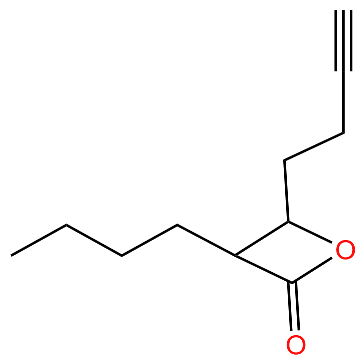 | Irreversibly inhibits ClpP in methicillin-resistant S. aureus | [43] |
| Acyldepsipeptides (ADEPs) | 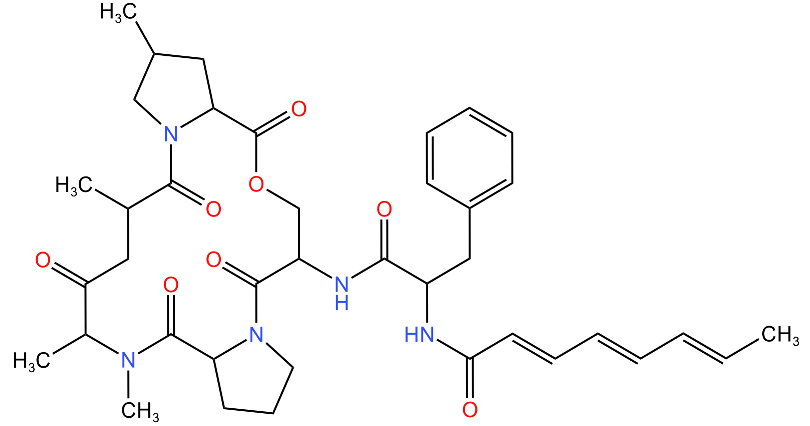 | Prevents complex formation between ClpP and Clp ATPases in Gram-positive bacteria, such as Enterococci and S. aureus | [43,72,73] |
| Ecumicin | 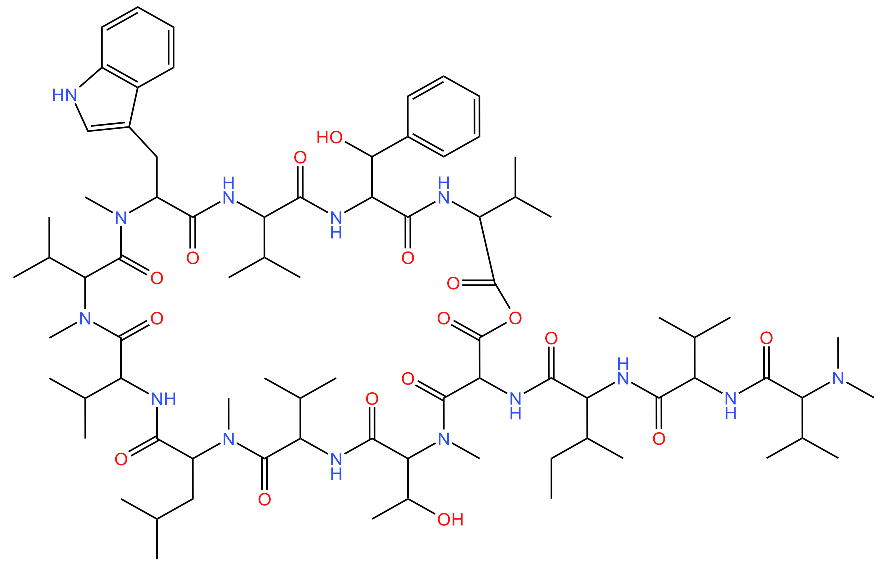 | Binds to the N-terminal domain of ClpC1 of M. tuberculosis. Stimulates the ATPase hydrolysis activity of M. tuberculosis ClpC1 and at the same time decouples ClpC1 and ClpP, therefore inhibiting proteolytic activity and resulting in cell death. | [43,75] |
| Cyclomarin A | 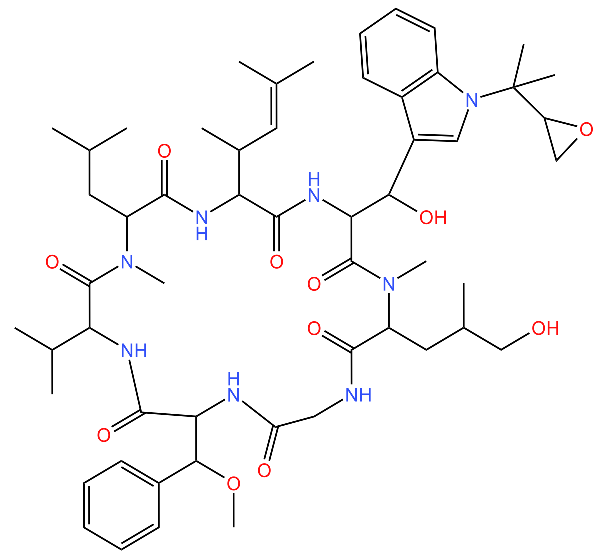 | Binds to the N-terminal domain of ClpC1 of M. tuberculosis and prevents the movement of the N-terminal domain. Causes excessive proteolysis. | [43,76,77] |
| Lassomycin | 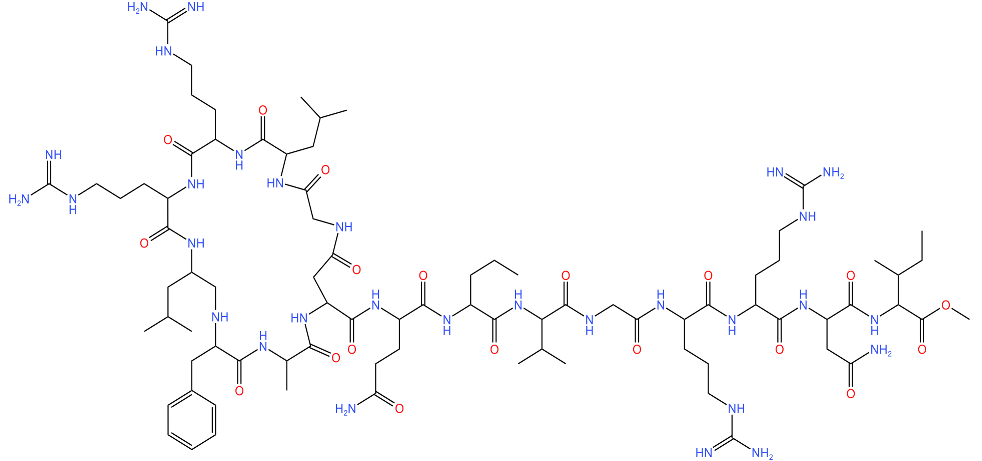 | Binds to an acidic N-terminal pocket on ClpC1. Stimulates ATPase activity of ClpC1 from M. tuberculosis, however it also inhibits ATP-dependent degradation of proteins. Uncouples ClpC1 from ClpP1 and ClpP2, resulting in the death of the cell as unnecessary proteins build up. | [16,76] |
| Rufomycin | 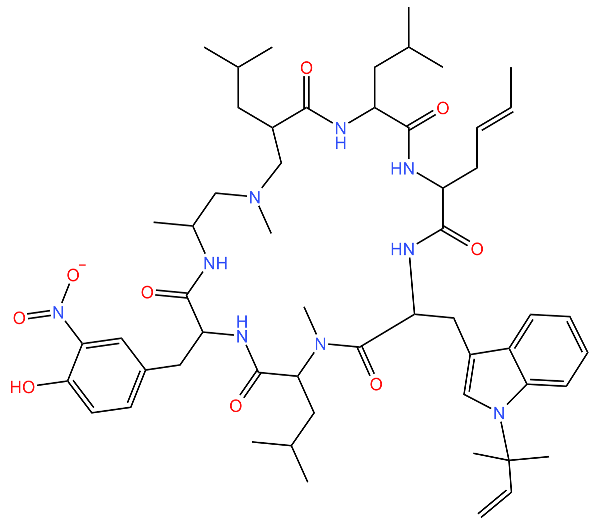 | Interacts with the N-terminal domain of ClpC1 of M. tuberculosis. Decreases the proteolytic activity of the ClpC1 and ClpP complex, therefore resulting in the build-up of proteins in the cell. | [77] |
| Armeiaspirols | 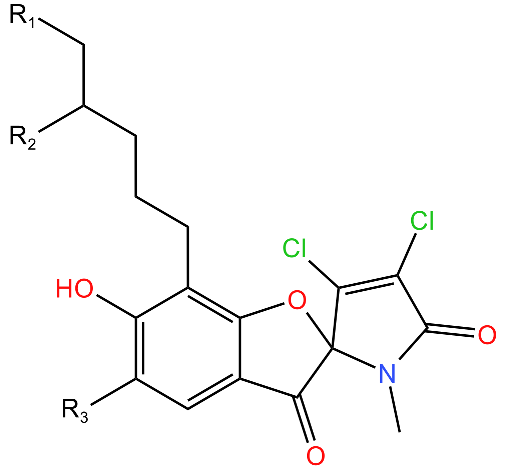 | Inhibits ClpXP and ClpYQ in Bacillus subtilis by binding to the ATPase domains and therefore inhibits the function of the complexes. Inhibits ATP hydrolysis and proteolysis. | [78] |
| Hydantoin analog | 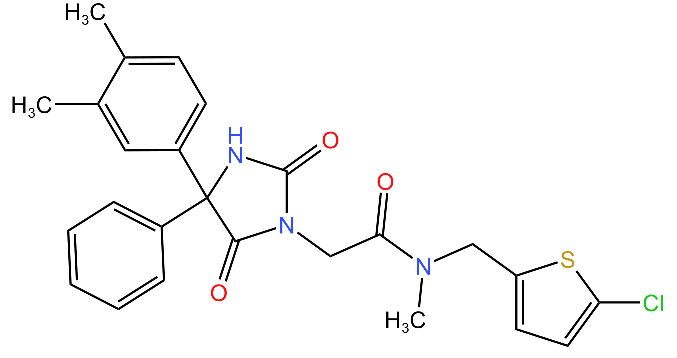 | Inhibits the ClpXP complex. Binds to a binding pocket on ClpP and impairs complex substrate turnover. | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motiwala, T.; Mthethwa, Q.; Achilonu, I.; Khoza, T. ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets. Antibiotics 2022, 11, 1218. https://doi.org/10.3390/antibiotics11091218
Motiwala T, Mthethwa Q, Achilonu I, Khoza T. ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets. Antibiotics. 2022; 11(9):1218. https://doi.org/10.3390/antibiotics11091218
Chicago/Turabian StyleMotiwala, Tehrim, Qiniso Mthethwa, Ikechukwu Achilonu, and Thandeka Khoza. 2022. "ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets" Antibiotics 11, no. 9: 1218. https://doi.org/10.3390/antibiotics11091218
APA StyleMotiwala, T., Mthethwa, Q., Achilonu, I., & Khoza, T. (2022). ESKAPE Pathogens: Looking at Clp ATPases as Potential Drug Targets. Antibiotics, 11(9), 1218. https://doi.org/10.3390/antibiotics11091218