
Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond




Abstract
1. Introduction
2. Class-by-Class Overview of Chelation Relevance

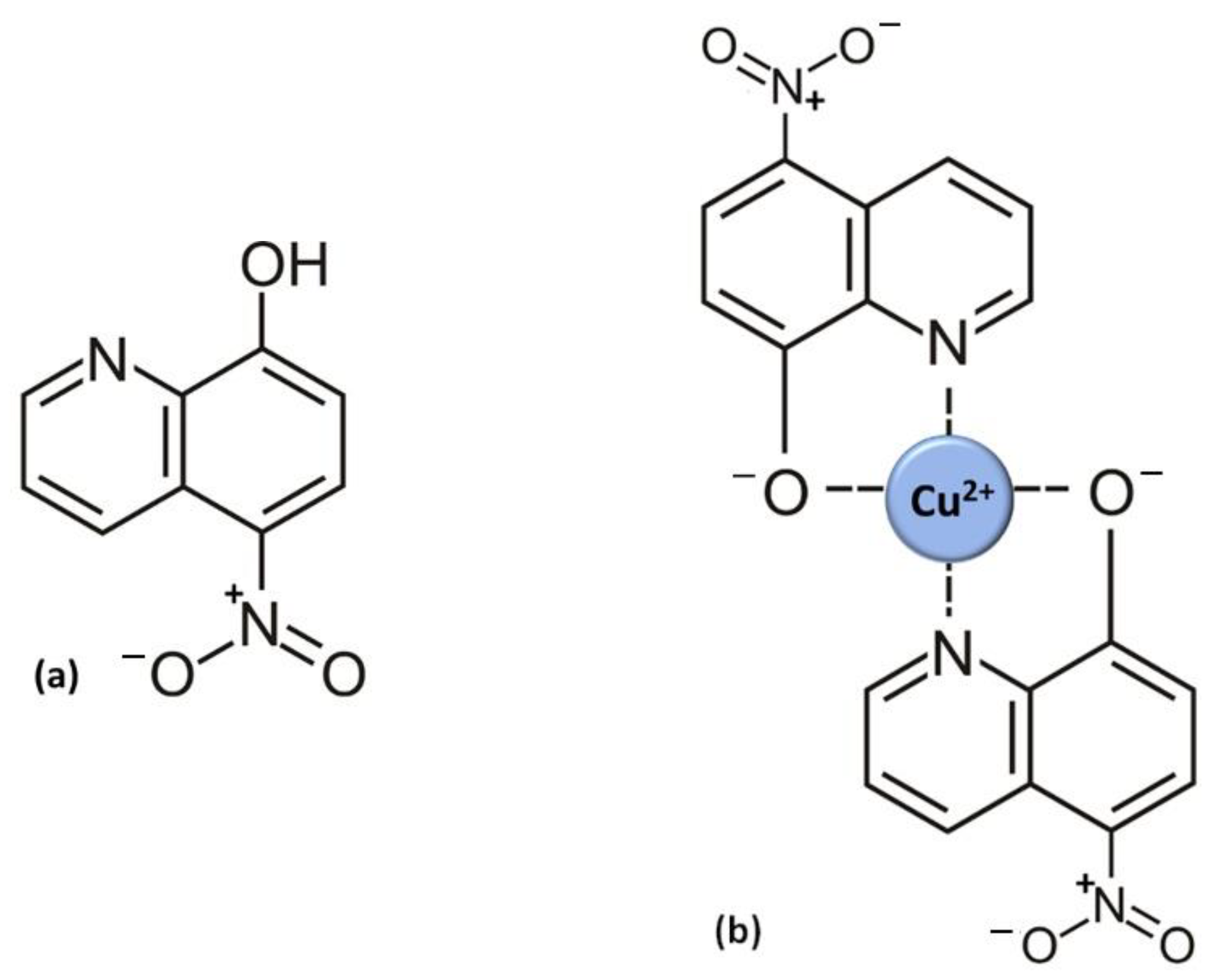{kind=link}
{kind=link}
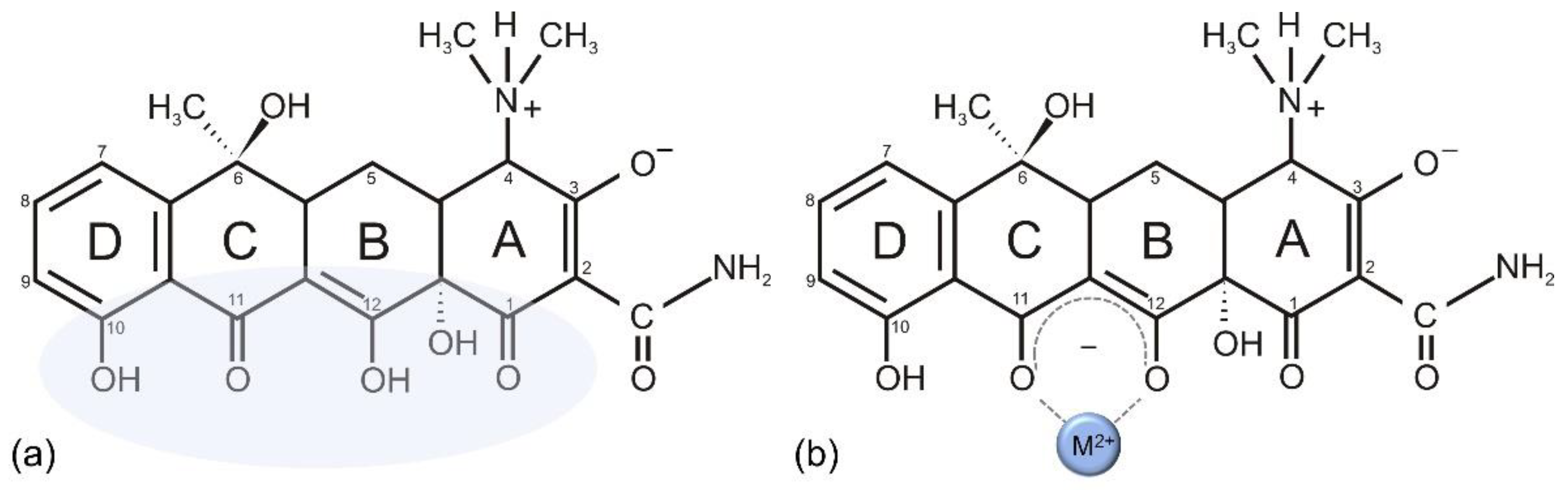{kind=link}
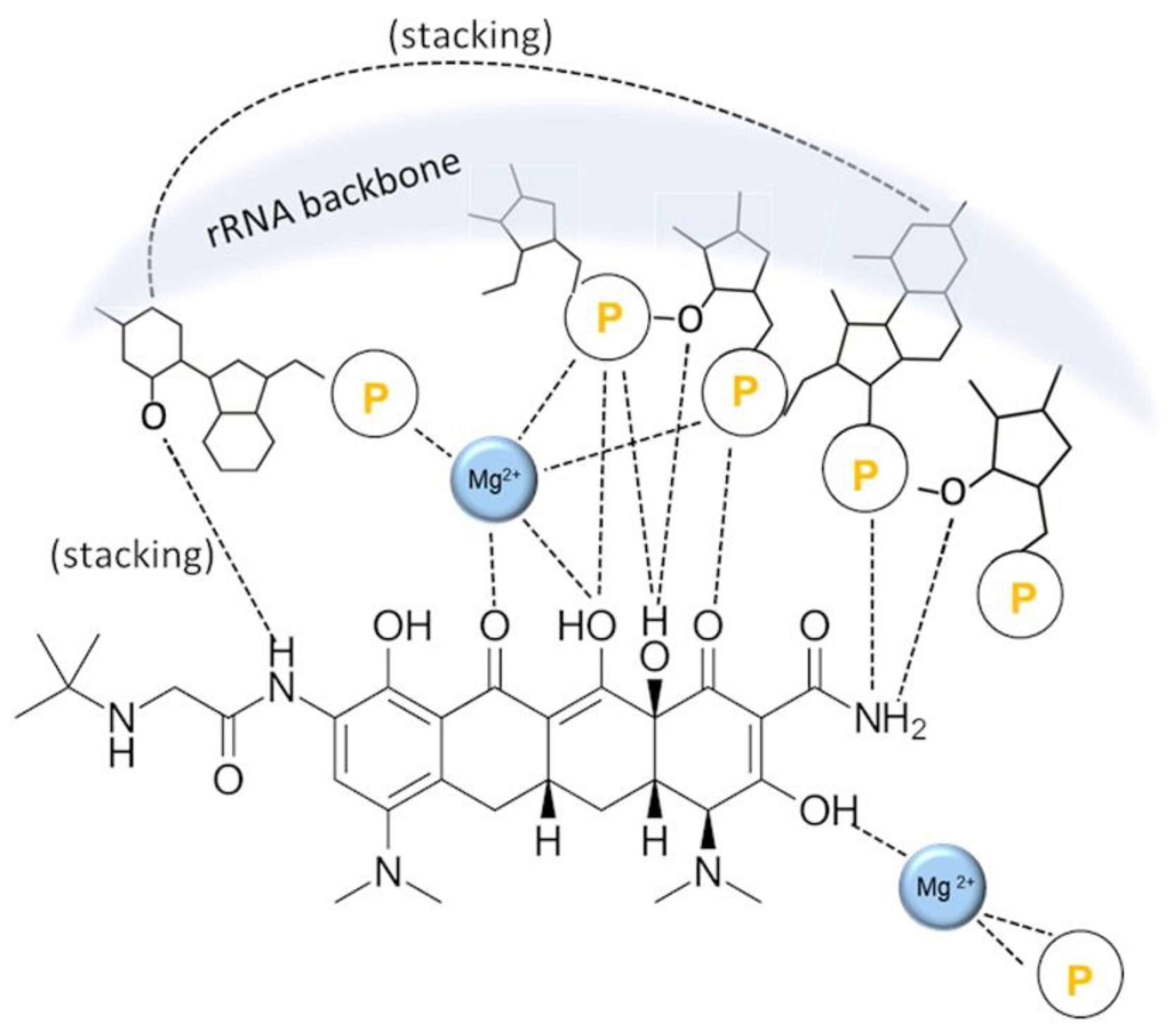{kind=link}
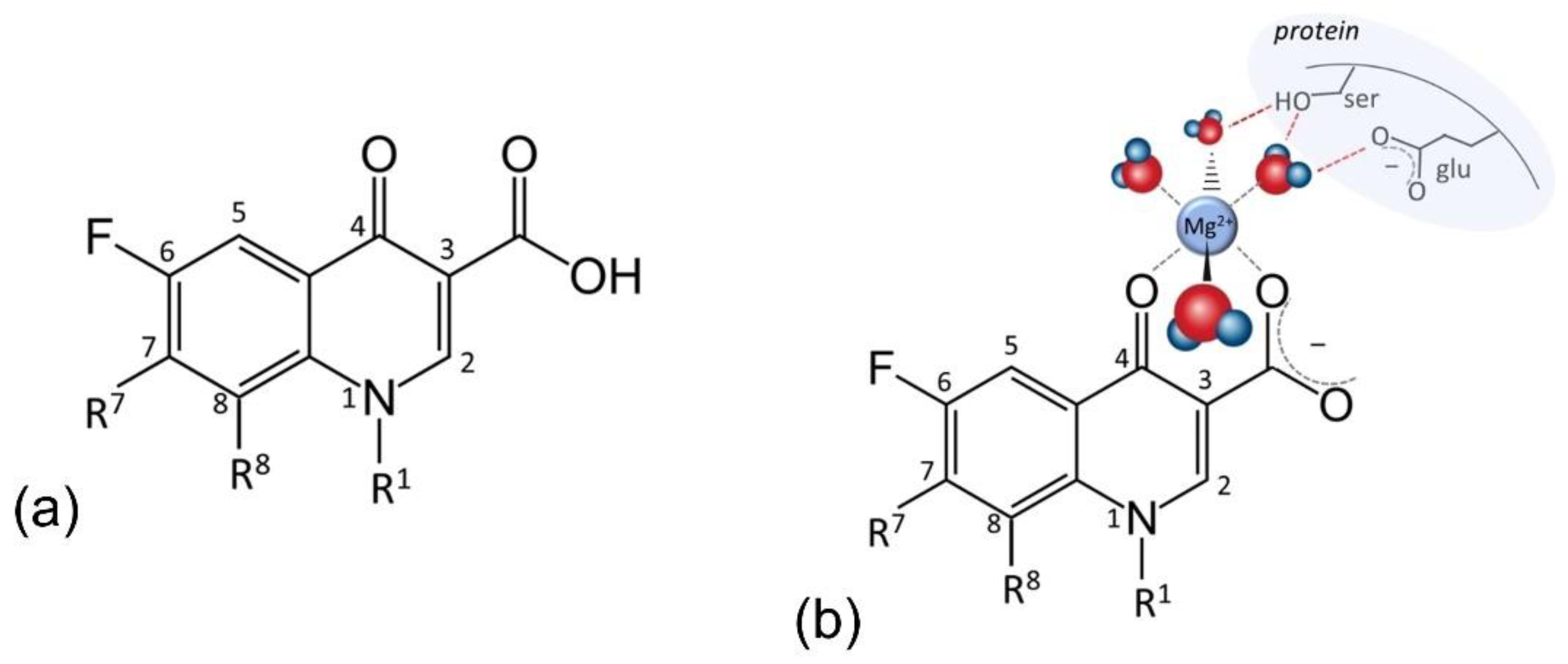{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibacterial CLASS/DRUG | References |
|---|---|
| Nitroxoline | |
| [11,18,19,21,31,32,33,34,35] |
| Tetracyclines | |
| [36,37,38,39,40,41,42,43] |
| Fluoroquinolones | |
| [44,45,46] |
| Sulfonamides | |
| [47,48,49] |
| Polypeptide Antibacterials | |
| [50,51,52,53,54,55,56,57,58,59,60,61,62,63] |
| Macrolides and Lincosamides | |
| |
| Cefiderocol and Other Beta-Lactam Antibacterials | |
| [24,25,27,64,65,66,67,68,69,70,71] |
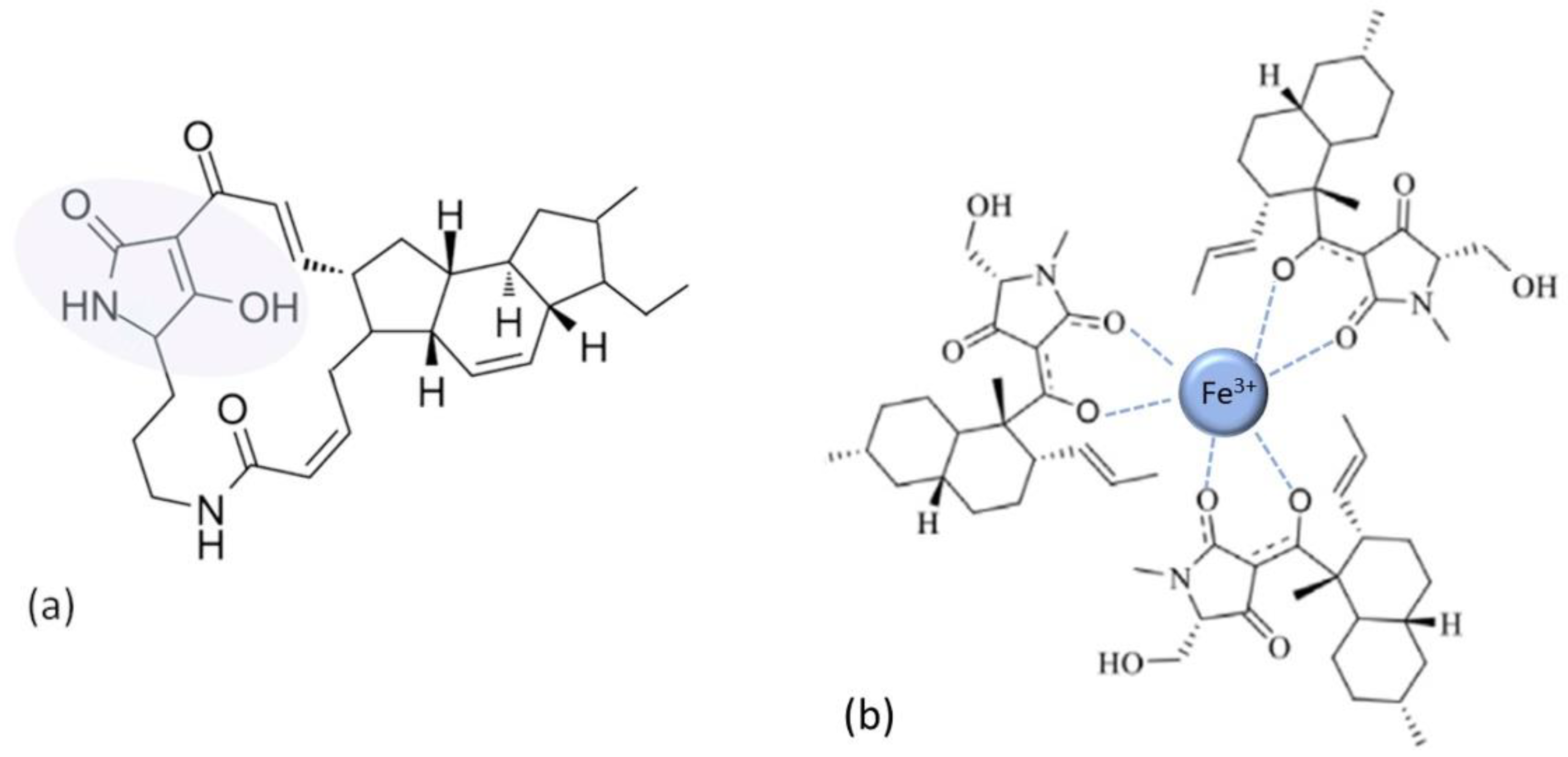
| Tetramates | |
| [72,73,74] |
3. Nitroxoline
4. Tetracyclines
5. Fluoroquinolones
6. Sulfonamides
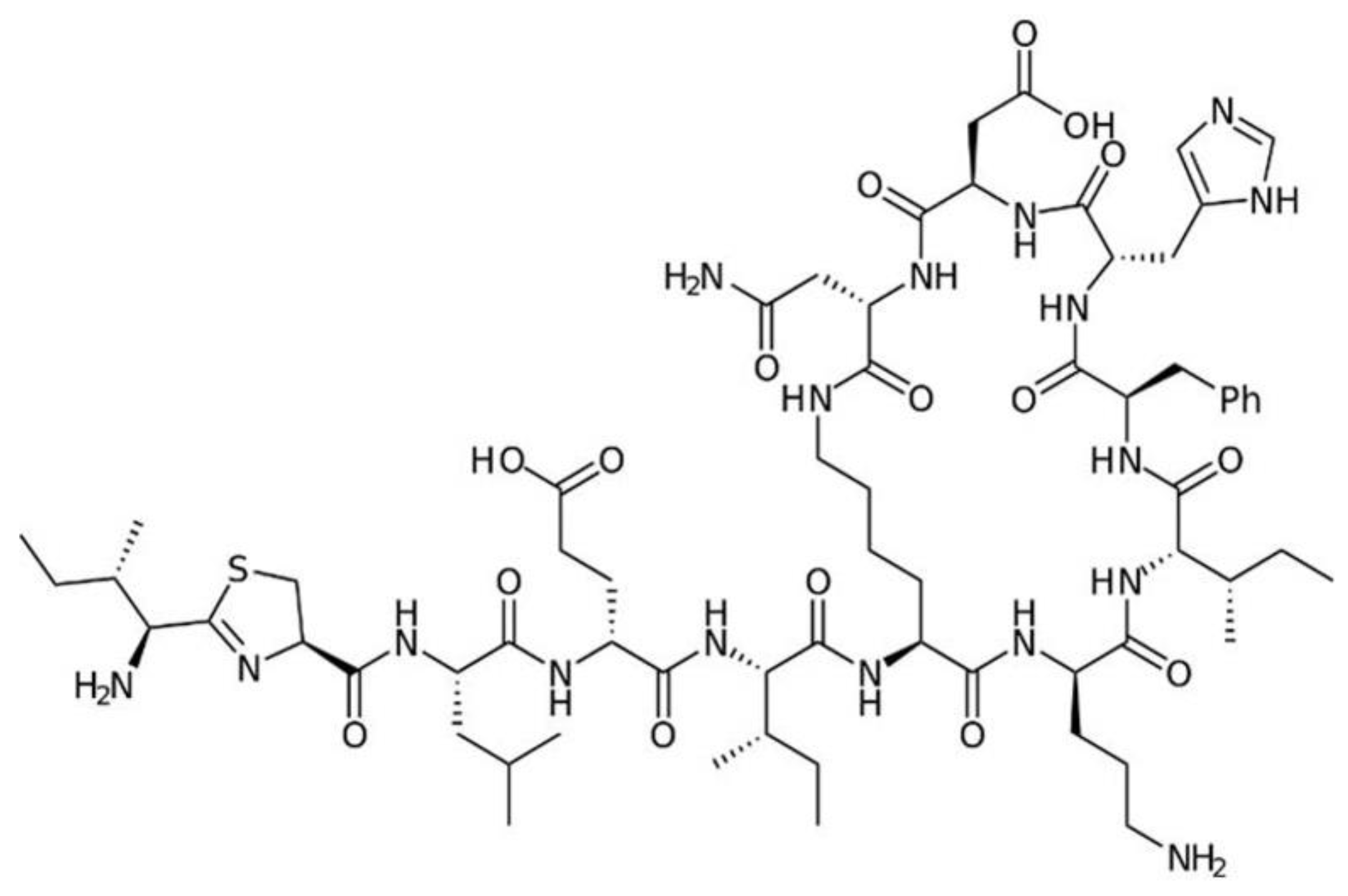
7. Polypeptide Antibacterials
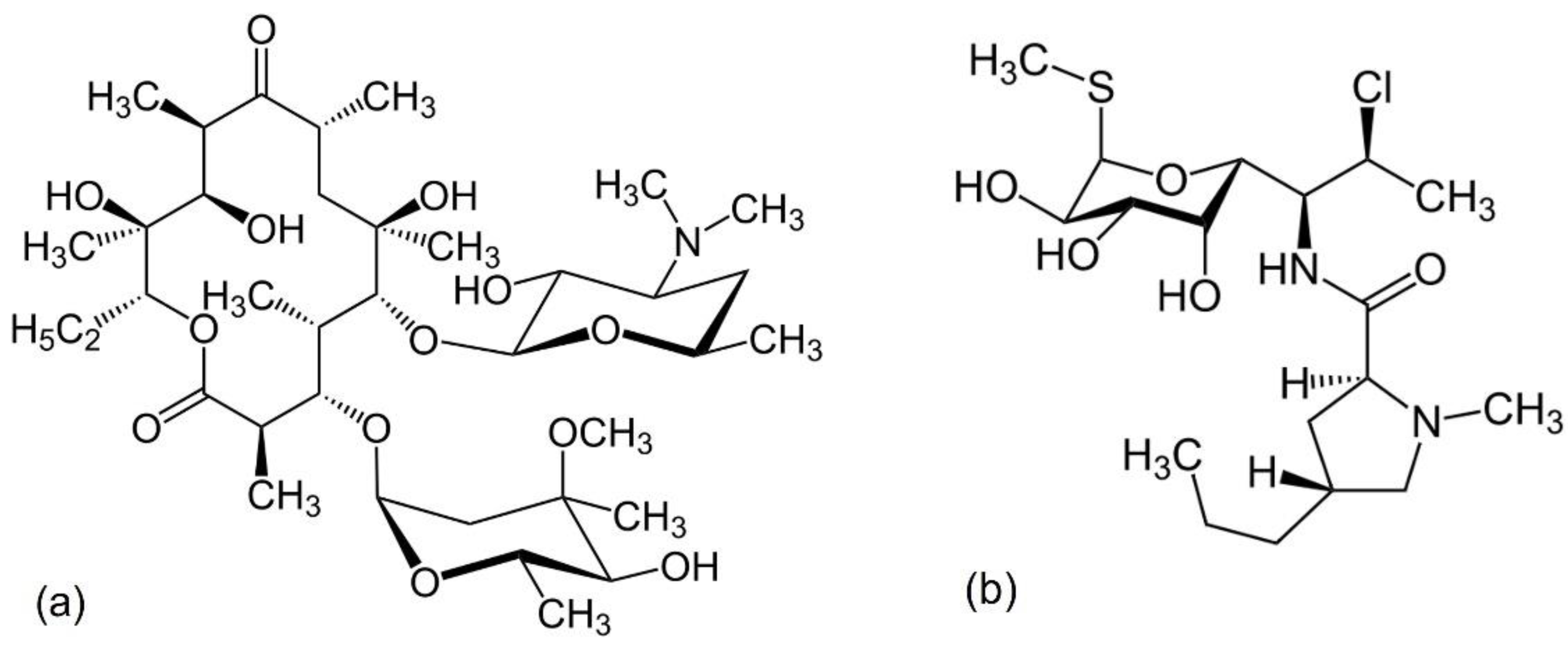
7.1. Vancomycin
7.2. Polymyxins
7.3. Bacitracin
8. Macrolides and Lincosamides
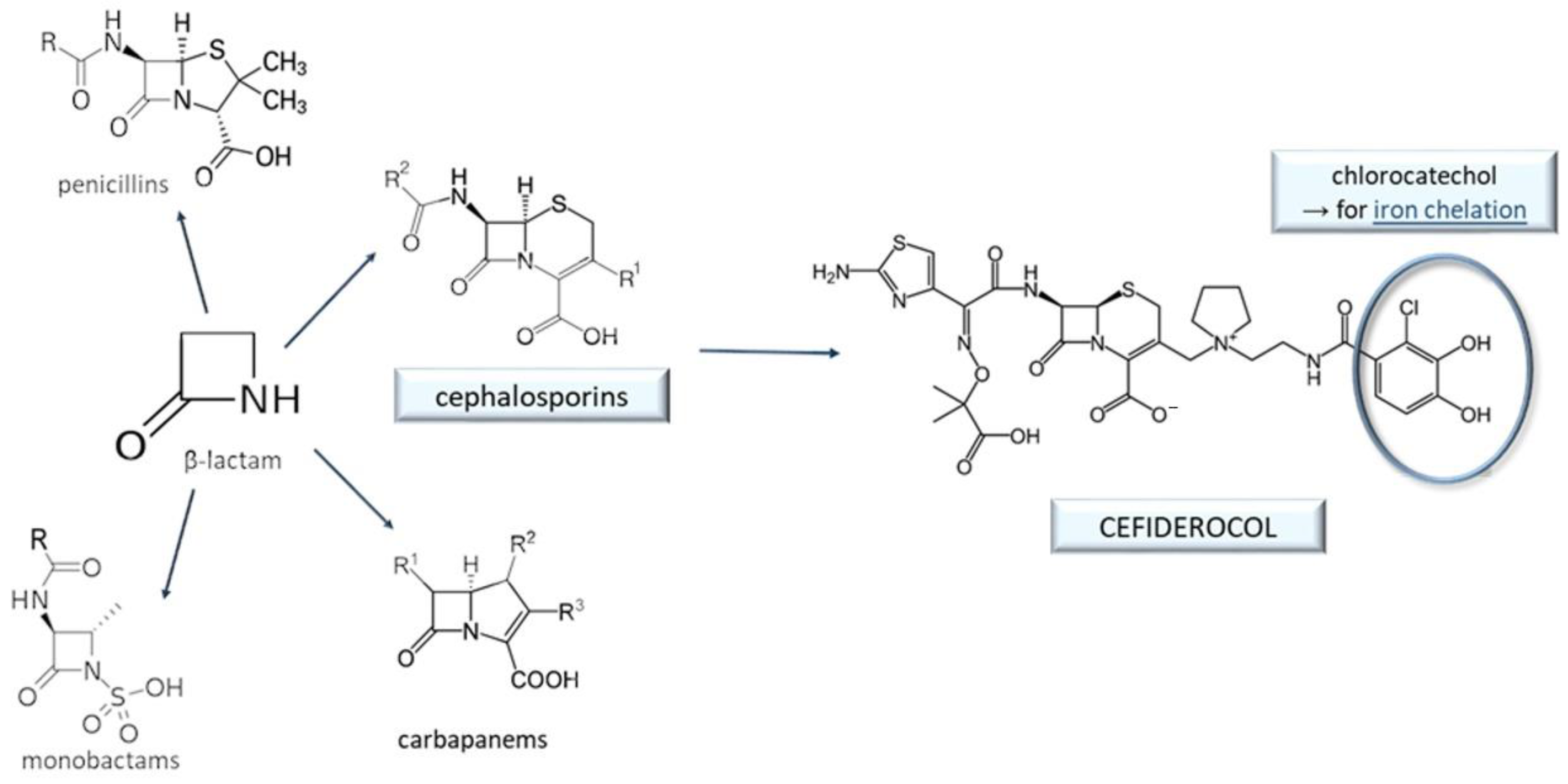
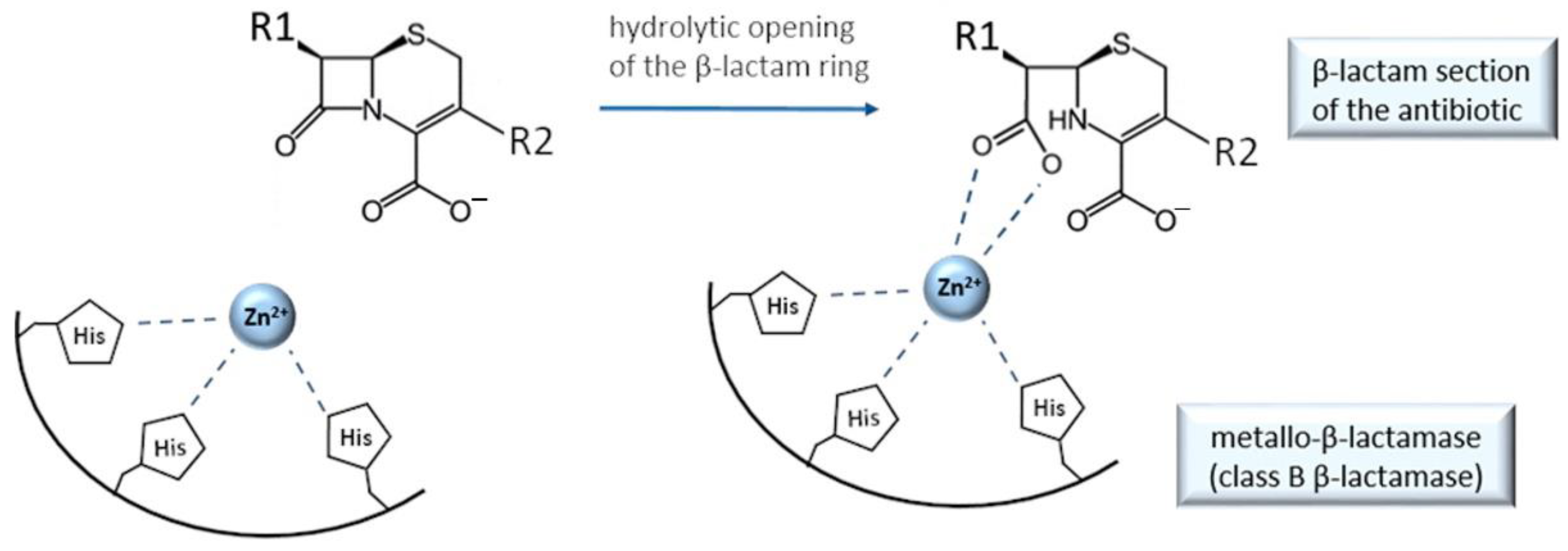
9. Cefiderocol and Other Beta-Lactam Antibacterials
10. Outlook: Chelation in Current Antibacterial Research and New Directions
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, Present and Future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Barzic, A.I.; Ioan, S. Antibacterial Drugs—From Basic Concepts to Complex Therapeutic Mechanisms of Polymer Systems. In Concepts, Compounds and the Alternatives of Antibacterials; Bobbarala, V., Ed.; IntechOpen: London, UK, 2015; ISBN 978-953-51-2232-6. [Google Scholar]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and Resistance Mechanisms of Antibiotics: A Guide for Clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300. [Google Scholar] [CrossRef] [PubMed]
- Blanusa, M.; Varnai, V.M.; Piasek, M.; Kostial, K. Chelators as Antidotes of Metal Toxicity: Therapeutic and Experimental Aspects. Curr. Med. Chem. 2005, 12, 2771–2794. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.D.; Skaar, E.P. Transition Metals and Virulence in Bacteria. Annu. Rev. Genet. 2016, 50, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges Associated with Metal Chelation Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 17, 457–468. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Stevenson, S.W.; Silove, N.; Williams, K. Chelation for Autism Spectrum Disorder (ASD). Cochrane Database Syst. Rev. 2015, 5, CD010766. [Google Scholar] [CrossRef]
- Buss, J.; Torti, F.; Torti, S. The Role of Iron Chelation in Cancer Therapy. Curr. Med. Chem. 2003, 10, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial Resistance: A Global Multifaceted Phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, F.; Wille, J.; Hamprecht, A.; Parcina, M.; Lehmann, C.; Schwarze-Zander, C.; Seifert, H.; Higgins, P.G. In Vitro Activity of Mecillinam and Nitroxoline against Neisseria Gonorrhoeae–Re-Purposing Old Antibiotics in the Multi-Drug Resistance Era. J. Med. Microbiol. 2019, 68, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Proschak, A.; Martinelli, G.; Frank, D.; Rotter, M.J.; Brunst, S.; Weizel, L.; Burgers, L.D.; Fürst, R.; Proschak, E.; Sosič, I.; et al. Nitroxoline and Its Derivatives Are Potent Inhibitors of Metallo-β-Lactamases. Eur. J. Med. Chem. 2022, 228, 113975. [Google Scholar] [CrossRef] [PubMed]
- Abouelhassan, Y.; Yang, Q.; Yousaf, H.; Nguyen, M.T.; Rolfe, M.; Schultz, G.S.; Huigens, R.W. Nitroxoline: A Broad-Spectrum Biofilm-Eradicating Agent against Pathogenic Bacteria. Int. J. Antimicrob. Agents 2017, 49, 247–251. [Google Scholar] [CrossRef]
- Hof, H.; Juretschke, C. Nitroxoline: An Option for the Treatment of Urinary Tract Infection with Multi-Resistant Uropathogenic Bacteria. Infection 2019, 47, 493–495. [Google Scholar] [CrossRef]
- Dobrindt, U.; Wami, H.T.; Schmidt-Wieland, T.; Bertsch, D.; Oberdorfer, K.; Hof, H. Compared with Cotrimoxazole Nitroxoline Seems to Be a Better Option for the Treatment and Prophylaxis of Urinary Tract Infections Caused by Multidrug-Resistant Uropathogens: An In Vitro Study. Antibiotics 2021, 10, 645. [Google Scholar] [CrossRef]
- Van de Walle, T.; Briand, M.; Mitrović, A.; Sosič, I.; Gobec, S.; Kos, J.; Persoons, L.; Daelemans, D.; De Jonghe, S.; Ubiparip, Z.; et al. Synthesis of Novel Nitroxoline Analogs with Potent Cathepsin B Exopeptidase Inhibitory Activity. Chem. Med. Chem. 2020, 15, 2477–2490. [Google Scholar] [CrossRef]
- Naber, K.G.; Niggemann, H.; Stein, G.; Stein, G. Review of the Literature and Individual Patients’ Data Meta-Analysis on Efficacy and Tolerance of Nitroxoline in the Treatment of Uncomplicated Urinary Tract Infections. BMC Infect. Dis. 2014, 14, 628. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez de Garayo, M.; Liu, W.; Rondeau, N.C.; Damoci, C.B.; Miranda, J.L. Rationally Repurposed Nitroxoline Inhibits Preclinical Models of Epstein–Barr Virus-Associated Lymphoproliferation. J. Antibiot. 2021, 74, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Sobke, A.; Klinger, M.; Hermann, B.; Sachse, S.; Nietzsche, S.; Makarewicz, O.; Keller, P.M.; Pfister, W.; Straube, E. The Urinary Antibiotic 5-Nitro-8-Hydroxyquinoline (Nitroxoline) Reduces the Formation and Induces the Dispersal of Pseudomonas Aeruginosa Biofilms by Chelation of Iron and Zinc. Antimicrob. Agents Chemother. 2012, 56, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Mirković, B.; Renko, M.; Turk, S.; Sosič, I.; Jevnikar, Z.; Obermajer, N.; Turk, D.; Gobec, S.; Kos, J. Novel Mechanism of Cathepsin B Inhibition by Antibiotic Nitroxoline and Related Compounds. Chem. Med. Chem. 2011, 6, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Wijma, R.A.; Huttner, A.; Koch, B.C.P.; Mouton, J.W.; Muller, A.E. Review of the Pharmacokinetic Properties of Nitrofurantoin and Nitroxoline. J. Antimicrob. Chemother. 2018, 73, 2916–2926. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, C.; Prognon, P.; Bourlioux, P. Roles of Divalent Cations and pH in Mechanism of Action of Nitroxoline against Escherichia Coli Strains. Antimicrob. Agents Chemother 1995, 39, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A Privileged Structure with a Broad-Ranging Pharmacological Potential. Med. Chem. Commun. 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Nolan, E.M. Beyond Iron: Non-Classical Biological Functions of Bacterial Siderophores. Dalton Trans. 2015, 44, 6320–6339. [Google Scholar] [CrossRef] [PubMed]
- El-Lababidi, R.M.; Rizk, J.G. Cefiderocol: A Siderophore Cephalosporin. Ann. Pharmacother. 2020, 54, 1215–1231. [Google Scholar] [CrossRef]
- Yao, J.; Wang, J.; Chen, M.; Cai, Y. Cefiderocol: An Overview of Its in-vitro and in-vivo Activity and Underlying Resistant Mechanisms. Front. Med. 2021, 8, 741940. [Google Scholar] [CrossRef]
- Santos, A.L.S.; Sodre, C.L.; Valle, R.S.; Silva, B.A.; Abi-chacra, E.A.; Silva, L.V.; Souza-Goncalves, A.L.; Sangenito, L.S.; Goncalves, D.S.; Souza, L.O.P.; et al. Antimicrobial Action of Chelating Agents: Repercussions on the Microorganism Development, Virulence and Pathogenesis. Curr. Med. Chem. 2012, 19, 2715–2737. [Google Scholar] [CrossRef] [PubMed]
- Negash, K.H.; Norris, J.K.S.; Hodgkinson, J.T. Hodgkinson Siderophore–Antibiotic Conjugate Design: New Drugs for Bad Bugs? Molecules 2019, 24, 3314. [Google Scholar] [CrossRef] [PubMed]
- Coraça-Huber, D.C.; Dichtl, S.; Steixner, S.; Nogler, M.; Weiss, G. Iron Chelation Destabilizes Bacterial Biofilms and Potentiates the Antimicrobial Activity of Antibiotics against Coagulase-Negative Staphylococci. Pathog. Dis. 2018, 76, fty052. [Google Scholar] [CrossRef] [PubMed]
- Ezraty, B.; Barras, F. The ‘Liaisons Dangereuses’ between Iron and Antibiotics. FEMS Microbiol. Rev. 2016, 40, 418–435. [Google Scholar] [CrossRef]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and Natural Iron Chelators: Therapeutic Potential and Clinical Use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef]
- Kos, J.; Mitrović, A. Nitroxoline: Repurposing Its Antimicrobial to Antitumor Application. Acta. Biochim. Pol. 2019, 66, 521–531. [Google Scholar] [CrossRef]
- Fraser, R.S.S.; Creanor, J. Rapid and Selective Inhibition of RNA Synthesis in Yeast by 8-Hydroxyquinoline. Eur. J. Biochem. 1974, 46, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Fraser, R.S.; Creanor, J. The Mechanism of Inhibition of Ribonucleic Acid Synthesis by 8-Hydroxyquinoline and the Antibiotic Lomofungin. Biochem. J. 1975, 147, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Latrache, H.; Bourlioux, P.; Karroua, M.; Zahir, H.; Hakkou, A. Effects of Subinhibitory Concentrations of Nitroxoline on the Surface Properties Of Escherichia Coli. Folia. Microbiol. 2000, 45, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Joaquim, A.R.; Gionbelli, M.P.; Gosmann, G.; Fuentefria, A.M.; Lopes, M.S.; Fernandes de Andrade, S. Novel Antimicrobial 8-Hydroxyquinoline-Based Agents: Current Development, Structure–Activity Relationships, and Perspectives. J. Med. Chem. 2021, 64, 16349–16379. [Google Scholar] [CrossRef] [PubMed]
- Lambs, L.; Venturim, M.; Révérend, B.D.-L.; Kozlowski, H.; Berthon, G. Metal Ion-Tetracycline Interactions in Biological Fluids. J. Inorg. Biochem. 1988, 33, 193–209. [Google Scholar] [CrossRef]
- Speer, B.S.; Shoemaker, N.B.; Salyers, A.A. Bacterial Resistance to Tetracycline: Mechanisms, Transfer, and Clinical Significance. Clin. Microbiol. Rev. 1992, 5, 387–399. [Google Scholar] [CrossRef]
- Guerra, W.; Silva-Caldeira, P.P.; Terenzi, H.; Pereira-Maia, E.C. Impact of Metal Coordination on the Antibiotic and Non-Antibiotic Activities of Tetracycline-Based Drugs. Coord. Chem. Rev. 2016, 327–328, 188–199. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Ohmori, H.; Kaneko-Ohdera, M.; Nomura, T.; Sawai, T. Delta PH-Dependent Accumulation of Tetracycline in Escherichia Coli. Antimicrob. Agents Chemother. 1991, 35, 53–56. [Google Scholar] [CrossRef]
- Saenger, W.; Hinrichs, W.; Orth, P.; Schnappinger, D.; Hillen, W. Structural basis of gene regulation by the tetracycline inducible Tet repressor–operator system. Nat. Struct. Biol. 2000, 7, 215–219. [Google Scholar] [CrossRef]
- McMurry, L.; Petrucci, R.E.; Levy, S.B. Active Efflux of Tetracycline Encoded by Four Genetically Different Tetracycline Resistance Determinants in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1980, 77, 3974–3977. [Google Scholar] [CrossRef]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Dönhöfer, A.; Wilson, D.N. Tetracycline Antibiotics and Resistance Mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Eljaaly, K.; Helal, A.; Almandeel, T.; Algarni, R.; Alshehri, S. Multivalent Cations Interactions with Fluoroquinolones or Tetracyclines: A Cross-Sectional Study. Saudi. J. Bio. Sci. 2021, 28, 6929–6932. [Google Scholar] [CrossRef] [PubMed]
- Sissi, C.; Perdonà, E.; Domenici, E.; Feriani, A.; Howells, A.J.; Maxwell, A.; Palumbo, M. Ciprofloxacin Affects Conformational Equilibria of DNA Gyrase A in the Presence of Magnesium Ions. J. Mol. Bio. 2001, 311, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Wohlkonig, A.; Chan, P.F.; Fosberry, A.P.; Homes, P.; Huang, J.; Kranz, M.; Leydon, V.R.; Miles, T.J.; Pearson, N.D.; Perera, R.L.; et al. Structural Basis of Quinolone Inhibition of Type IIA Topoisomerases and Target-Mediated Resistance. Nat. Struct. Mol. Biol. 2010, 17, 1152–1153. [Google Scholar] [CrossRef]
- Aldred, K.J.; McPherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N. Topoisomerase IV-Quinolone Interactions Are Mediated through a Water-Metal Ion Bridge: Mechanistic Basis of Quinolone Resistance. Nucleic Acids Res. 2013, 41, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Chohan, Z.H. Metal-Based Sulfonamides: Their Preparation, Characterization and in-Vitro Antibacterial, Antifungal & Cytotoxic Properties. X-ray Structure of 4-[(2-Hydroxybenzylidene) Amino] Benzenesulfonamide. J. Enzyme Inhib. Med. Chem. 2008, 23, 120–130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chohan, Z.H.; Hernandes, M.Z.; Sensato, F.R.; Moreira, D.R.M.; Alves Pereira, V.R.; de Neves, J.K.A.L.; de Oliveira, A.P.; de Oliveira, B.C.; Lima Leite, A.C. Sulfonamide–Metal Complexes Endowed with Potent Anti- Trypanosoma Cruzi Activity. J. Enzyme Inhib. Med. Chem. 2014, 29, 230–236. [Google Scholar] [CrossRef]
- Hassan, A.U.; Sumrra, S.H. Exploring the Bioactive Sites of New Sulfonamide Metal Chelates for Multi-Drug Resistance: An Experimental Versus Theoretical Design. J. Inorg. Organomet. Polym. 2022, 32, 513–535. [Google Scholar] [CrossRef]
- Yarlagadda, V.; Sarkar, P.; Samaddar, S.; Haldar, J. A Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria. Angew. Chem. Int. Ed. 2016, 55, 7836–7840. [Google Scholar] [CrossRef]
- Guan, D.; Chen, F.; Faridoon; Liu, J.; Li, J.; Lan, L.; Huang, W. Design and Synthesis of Pyrophosphate-Targeting Vancomycin Derivatives for Combating Vancomycin-Resistant Enterococci. Chem. Med. Chem. 2018, 13, 1644–1657. [Google Scholar] [CrossRef]
- Nair, U.B.; Chang, S.S.C.; Armstrong, D.W.; Rawjee, Y.Y.; Eggleston, D.S.; McArdle, J.V. Elucidation of Vancomycin’s Enantioselective Binding Site Using Its Copper Complex. Chirality 1996, 8, 590–595. [Google Scholar] [CrossRef]
- Kucharczyk, M.; Brzezowska, M.; Maciąg, A.; Lis, T.; Jeżowska-Bojczuk, M. Structural Features of the Cu2+–Vancomycin Complex. J. Inorg. Biochem. 2008, 102, 936–942. [Google Scholar] [CrossRef]
- Howden, B.P.; Davies, J.K.; Johnson, P.D.R.; Stinear, T.P.; Grayson, M.L. Reduced Vancomycin Susceptibility in Staphylococcus Aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clin. Microbiol. Rev. 2010, 23, 99–139. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.M.; Aghamollaei, H.; Kooshki, H.; Barjini, K.A.; Mirnejad, R.; Choopani, A. The Development of Antimicrobial Peptides as an Approach to Prevention of Antibiotic Resistance. Rev. Med. Microbiol. 2015, 26, 98–110. [Google Scholar] [CrossRef]
- Ayoub Moubareck, C. Polymyxins and Bacterial Membranes: A Review of Antibacterial Activity and Mechanisms of Resistance. Membranes 2020, 10, 181. [Google Scholar] [CrossRef]
- Velkov, T.; Roberts, K.D.; Nation, R.L.; Thompson, P.E.; Li, J. Pharmacology of Polymyxins: New Insights into an ‘Old’ Class of Antibiotics. Future Microbiol. 2013, 8, 711–724. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure—Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef]
- Schindler, M.; Osborn, M.J. Interaction of Divalent Cations and Polymyxin B with Lipopolysaccharide. Biochemistry 1979, 18, 4425–4430. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, A.; Maeda, A.; Itto, K.; Arimoto, H. Deciphering the Mode of Action of Cell Wall-Inhibiting Antibiotics Using Metabolic Labeling of Growing Peptidoglycan in Streptococcus Pyogenes. Sci. Rep. 2017, 7, 1129. [Google Scholar] [CrossRef] [PubMed]
- Economou, N.J.; Cocklin, S.; Loll, P.J. High-Resolution Crystal Structure Reveals Molecular Details of Target Recognition by Bacitracin. Proc. Natl. Acad. Sci. USA 2013, 110, 14207–14212. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-D.; Lin, Y.; Zhou, B.; Ren, X.-D.; Pang, D.-W.; Liu, Y. Characterization of the Mechanism of the Staphylococcus Aureus Cell Envelope by Bacitracin and Bacitracin-Metal Ions. J. Membr. Biol. 2008, 225, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Yoshizawa, H.; Yamawaki, K.; Yokoo, K.; Sato, J.; Hisakawa, S.; Hasegawa, Y.; Kusano, H.; Sano, M.; Sugimoto, H.; et al. Cefiderocol (S-649266), A New Siderophore Cephalosporin Exhibiting Potent Activities against Pseudomonas Aeruginosa and Other Gram-Negative Pathogens Including Multi-Drug Resistant Bacteria: Structure Activity Relationship. Eur. J. Med. Chem. 2018, 155, 847–868. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; El Tabei, L.; Büsker, S.; Krauss, C.; Fuhr, U.; Taubert, M. Clinical Pharmacokinetics and Pharmacodynamics of Cefiderocol. Clin. Pharmacokinet. 2021, 60, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.S.; Ahmad, A.R.; Sabir, M.; Asad, S.M. Preparation, Characterization and Biological Evaluation of Copper(II) and Zinc(II) Complexes with Cephalexin. J. Pharm. Pharmacol. 2010, 51, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Auda, S.H.; Mrestani, Y.; Fetouh, M.I.; Neubert, R.H.H. ChemInform Abstract: Characterization and Activity of Cephalosporin Metal Complexes. ChemInform 2008, 63, 555–561. [Google Scholar] [CrossRef]
- Principe, L.; Vecchio, G.; Sheehan, G.; Kavanagh, K.; Morroni, G.; Viaggi, V.; di Masi, A.; Giacobbe, D.R.; Luzzaro, F.; Luzzati, R.; et al. Zinc Chelators as Carbapenem Adjuvants for Metallo-β-Lactamase-Producing Bacteria: In Vitro and In Vivo Evaluation. Microb. Drug Resist. 2020, 26, 1133–1143. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kuroki, T.; Yasuzawa, H.; Higashi, T.; Jin, W.; Kawanami, A.; Yamagata, Y.; Arakawa, Y.; Goto, M.; Kurosaki, H. Probing the Role of Asp-120(81) of Metallo-β-Lactamase (IMP-1) by Site-Directed Mutagenesis, Kinetic Studies, and X-ray Crystallography. J. Biol. Chem. 2005, 280, 20824–20832. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Lei, J.; Pan, X.; Huang, X.; Zhao, Y. The Hydrolytic Water Molecule of Class A β-Lactamase Relies on the Acyl-Enzyme Intermediate ES* for Proper Coordination and Catalysis. Sci. Rep. 2020, 10, 10205. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, S.-D.; Haidar, A.K.; Bajimaya, M.; Guo, Y.; Larsen, T.O.; Gram, L. Polycyclic Tetramate Macrolactams—A Group of Natural Bioactive Metallophores. Front. Chem. 2021, 9, 772858. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Genov, M.; Pretsch, A.; Pretsch, D.; Moloney, M.G. Metal Binding and Its Amelioration in Tetramates. J. Org. Chem. 2021, 86, 12886–12907. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, J.A.V.; Oh, D.-C.; Cao, S.; Currie, C.R.; Kolter, R.; Clardy, J. Common Biosynthetic Origins for Polycyclic Tetramate Macrolactams from Phylogenetically Diverse Bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 11692–11697. [Google Scholar] [CrossRef]
- Van Hau, T.; Ruankham, W.; Suwanjang, W.; Songtawee, N.; Wongchitrat, P.; Pingaew, R.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Repurposing of Nitroxoline Drug for the Prevention of Neurodegeneration. Chem. Res. Toxicol. 2019, 32, 2182–2191. [Google Scholar] [CrossRef]
- Odingo, J.O.; Early, J.V.; Smith, J.; Johnson, J.; Bailey, M.A.; Files, M.; Guzman, J.; Ollinger, J.; Korkegian, A.; Kumar, A.; et al. 8-Hydroxyquinolines Are Bactericidal against Mycobacterium Tuberculosis. Drug Dev. Res. 2019, 80, 566–572. [Google Scholar] [CrossRef]
- Sureshkumar, B.; Mary, Y.S.; Mary, Y.S.; Suma, S. Spectroscopic and DFT Investigations of 8-Hydroxy Quinoline-5-Sulfonic Acid-5-Chloro-8-Hydroxyquinoline Cocrystal. Chem. Pap. 2021, 75, 3387–3399. [Google Scholar] [CrossRef]
- El-Wakiel, N.; El-Ghamry, H. Nitroxoline Azo Dye Complexes as Effective Heterogeneous Catalysts for Color Removal and Degradation of Some Organic Textile Dyes: Nitroxoline azo dye complexes as effective heterogeneous catalysts. Int. J. Chem. Kinet. 2017, 49, 464–476. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A Review of Their Metal Chelating Properties and Medicinal Applications. Drug Des. Devel. Ther. 2013, 7, 1157–1178. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Taggart, J.E.; Zhang, X.; Benbrook, D.M.; Lind, S.E.; Ding, W.-Q. Nitroxoline (8-Hydroxy-5-Nitroquinoline) Is More a Potent Anti-Cancer Agent than Clioquinol (5-Chloro-7-Iodo-8-Quinoline). Cancer Lett. 2011, 312, 11–17. [Google Scholar] [CrossRef]
- Kresken, M.; Körber-Irrgang, B. In Vitro Activity of Nitroxoline against Escherichia Coli Urine Isolates from Outpatient Departments in Germany. Antimicrob. Agents Chemother 2014, 58, 7019–7020. [Google Scholar] [CrossRef] [PubMed]
- Albert, A.; Magrath, D. The Choice of a Chelating Agent for Inactivating Trace Metals: II. Derivatives of Oxine (8-Hydroxyquinoline). Biochem. J. 1947, 41, 534–545. [Google Scholar] [CrossRef] [PubMed]
- McElroy, J. The Treatment of Pulmonary Tuberculosis by Intravenous Injections of Chinosol with Formaldehyde. Lancet 1910, 176, 1408–1409. [Google Scholar] [CrossRef]
- Bourlioux, P.; Karam, D.; Amgar, A.; Perdiz, M. Relation of the chelating property of nitroxoline, the surface hydrophobicity and the inhibition of bacterial adherence. Pathol. Biol. 1989, 37, 600–604. [Google Scholar]
- Kim, J.; Kim, H.; Park, S.B. Privileged Structures: Efficient Chemical “Navigators” toward Unexplored Biologically Relevant Chemical Spaces. J. Am. Chem. Soc. 2014, 136, 14629–14638. [Google Scholar] [CrossRef] [PubMed]
- Veschi, S.; Carradori, S.; De Lellis, L.; Florio, R.; Brocco, D.; Secci, D.; Guglielmi, P.; Spano, M.; Sobolev, A.P.; Cama, A. Synthesis and Evaluation of a Large Library of Nitroxoline Derivatives as Pancreatic Cancer Antiproliferative Agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 1331–1344. [Google Scholar] [CrossRef]
- Sosič, I.; Mirković, B.; Arenz, K.; Štefane, B.; Kos, J.; Gobec, S. Development of New Cathepsin B Inhibitors: Combining Bioisosteric Replacements and Structure-Based Design To Explore the Structure–Activity Relationships of Nitroxoline Derivatives. J. Med. Chem. 2013, 56, 521–533. [Google Scholar] [CrossRef]
- Begić, G.; Petković Didović, M.; Lučić Blagojević, S.; Jelovica Badovinac, I.; Žigon, J.; Perčić, M.; Cvijanović Peloza, O.; Gobin, I. Adhesion of Oral Bacteria to Commercial D-PTFE Membranes: Polymer Microstructure Makes a Difference. Int. J. Mol. Sci. 2022, 23, 2983. [Google Scholar] [CrossRef]
- Shim, J.S.; Matsui, Y.; Bhat, S.; Nacev, B.A.; Xu, J.; Bhang, H.C.; Dhara, S.; Han, K.C.; Chong, C.R.; Pomper, M.G.; et al. Effect of Nitroxoline on Angiogenesis and Growth of Human Bladder Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1855–1873. [Google Scholar] [CrossRef]
- Nelson, M.L.; Levy, S.B. The History of the Tetracyclines: The History of the Tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef]
- Albert, A.; Rees, C.W. Avidity of the Tetracyclines for the Cations of Metals. Nature 1956, 177, 433–434. [Google Scholar] [CrossRef]
- Griffin, M.O.; Fricovsky, E.; Ceballos, G.; Villarreal, F. Tetracyclines: A Pleitropic Family of Compounds with Promising Therapeutic Properties. Review of the Literature. Am. J. Physiol. Cell Physiol. 2010, 299, C539–C548. [Google Scholar] [CrossRef] [PubMed]
- Jenner, L.; Starosta, A.L.; Terry, D.S.; Mikolajka, A.; Filonava, L.; Yusupov, M.; Blanchard, S.C.; Wilson, D.N.; Yusupova, G. Structural Basis for Potent Inhibitory Activity of the Antibiotic Tigecycline during Protein Synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3812–3816. [Google Scholar] [CrossRef] [PubMed]
- Pioletti, M. Crystal Structures of Complexes of the Small Ribosomal Subunit with Tetracycline, Edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.H. Tetracycline Antibiotics and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025387. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Altschmied, L.; Hillen, W. Kinetic and Equilibrium Characterization of the Tet Repressor-Tetracycline Complex by Fluorescence Measurements. J. Mol. Bio. 1986, 187, 341–348. [Google Scholar] [CrossRef]
- Ryan, M.; Usman, A.; Ramamurthy, N.; Golub, L.; Greenwald, R. Excessive Matrix Metalloproteinase Activity in Diabetes: Inhibition by Tetracycline Analogues with Zinc Reactivity. Curr. Med. Chem. 2001, 8, 305–316. [Google Scholar] [CrossRef]
- Dalhoff, A. Selective Toxicity of Antibacterial Agents—Still a Valid Concept or Do We Miss Chances and Ignore Risks? Infection 2021, 49, 29–56. [Google Scholar] [CrossRef]
- Chukwudi, C.U. RRNA Binding Sites and the Molecular Mechanism of Action of the Tetracyclines. Antimicrob. Agents Chemother 2016, 60, 4433–4441. [Google Scholar] [CrossRef]
- Neuvonen, P.J. Interactions with the Absorption of Tetracyclines. Drugs 1976, 11, 45–54. [Google Scholar] [CrossRef]
- Faure, M.; Cilibrizzi, A.; Abbate, V.; Bruce, K.; Hider, R. Effect of Iron Chelation on Anti-Pseudomonal Activity of Doxycycline. Int. J. Antimicrob. Agents 2021, 58, 106438. [Google Scholar] [CrossRef]
- Fiori, A.; Van Dijck, P. Potent Synergistic Effect of Doxycycline with Fluconazole against Candida Albicans Is Mediated by Interference with Iron Homeostasis. Antimicrob. Agents Chemother. 2012, 56, 3785–3796. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.G.J.; Flach, C.-F. Antibiotic Resistance in the Environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lan, H.; Liu, H.; Qu, J. Removal of Tetracycline Antibiotics from Aqueous Solution by Amino-Fe (III) Functionalized SBA15. Colloids Surf. A Physicochem. Eng. Asp. 2015, 471, 133–138. [Google Scholar] [CrossRef]
- Uivarosi, V. Metal Complexes of Quinolone Antibiotics and Their Applications: An Update. Molecules 2013, 18, 11153–11197. [Google Scholar] [CrossRef]
- Tarushi, A.; Lafazanis, K.; Kljun, J.; Turel, I.; Pantazaki, A.A.; Psomas, G.; Kessissoglou, D.P. First- and Second-Generation Quinolone Antibacterial Drugs Interacting with Zinc(II): Structure and Biological Perspectives. J. Inorg. Biochem. 2013, 121, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Greenaway, F.T.; Riviere, E.; Girerd, J.J.; Labouze, X.; Morgant, G.; Viossat, B.; Daran, J.C.; Roch Arveiller, M.; Dung, N.-H. Copper(II) Complexes of a Nonsteroidal Anti-Inflammatory Drug Niflumic Acid. Synthesis, Crystal Structure of Tetrakis-μ-(2-[3-(Trifluoromethyl)Phenyl]Aminonicotinato)Bis(Dimethylsulfoxide)Dicopper(II) Complex at 190 K. Anti-Inflammatory Properties. J. Inorg. Biochem. 1999, 76, 19–27. [Google Scholar] [CrossRef]
- Turel, I.; Golobič, A.; Klavžar, A.; Pihlar, B.; Buglyó, P.; Tolis, E.; Rehder, D.; Sepčić, K. Interactions of Oxovanadium(IV) and the Quinolone Family Member—Ciprofloxacin. J. Inorg. Biochem. 2003, 95, 199–207. [Google Scholar] [CrossRef]
- López-Gresa, M.P.; Ortiz, R.; Perelló, L.; Latorre, J.; Liu-González, M.; García-Granda, S.; Pérez-Priede, M.; Cantón, E. Interactions of Metal Ions with Two Quinolone Antimicrobial Agents (Cinoxacin and Ciprofloxacin). J. Inorg. Biochem. 2002, 92, 65–74. [Google Scholar] [CrossRef]
- Tacic, A.; Nikolic, V.; Nikolic, L.; Savic, I. Antimicrobial Sulfonamide Drugs. Adv. Technol. 2017, 6, 58–71. [Google Scholar] [CrossRef]
- Masters, P.A.; O’Bryan, T.A.; Zurlo, J.; Miller, D.Q.; Joshi, N. Trimethoprim-Sulfamethoxazole Revisited. Arch. Intern. Med. 2003, 163, 402. [Google Scholar] [CrossRef]
- Takagi, M.; Omori, T.; Matsuo, S.; Matsuno, S.; Ueno, K.; Ide, S. Sulfonamides. A New Class of Chelating Agents of Potential Utility in Analytical and Separation Chemistry. Chem. Lett. 1980, 9, 387–390. [Google Scholar] [CrossRef]
- Alaghaz, A.-N.M.A.; Bayoumi, H.A.; Ammar, Y.A.; Aldhlmani, S.A. Synthesis, Characterization, and Antipathogenic Studies of Some Transition Metal Complexes with N,O-Chelating Schiff’s Base Ligand Incorporating Azo and Sulfonamide Moieties. J. Mol. Struct. 2013, 1035, 383–399. [Google Scholar] [CrossRef]
- Güler, S.; Soğukömeroğullari, H.G.; Özdemïr, S.; Yalçin, M.S.; Sönmez, M. New Carboxamid Ligand and Its Metal Complexes Containing Sulfonamide Group: Synthesis, Characterization, DNA Cleavage and Antimicrobial Activity. Erzincan Üniversitesi. Fen Bilimleri. Enstitüsü. Dergisi. 2021, 14, 724–736. [Google Scholar] [CrossRef]
- Sumrra, S.H.; Hassan, A.U.; Zafar, M.N.; Shafqat, S.S.; Mustafa, G.; Zafar, M.N.; Zubair, M.; Imran, M. Metal Incorporated Sulfonamides as Promising Multidrug Targets: Combined Enzyme Inhibitory, Antimicrobial, Antioxidant and Theoretical Exploration. J. Mol. Struct. 2022, 1250, 131710. [Google Scholar] [CrossRef]
- Kremer, E.; Facchin, G.; Estévez, E.; Alborés, P.; Baran, E.J.; Ellena, J.; Torre, M.H. Copper Complexes with Heterocyclic Sulfonamides: Synthesis, Spectroscopic Characterization, Microbiological and SOD-like Activities: Crystal Structure of [Cu(Sulfisoxazole)2(H2O)4]·2H2O. J. Inorg. Biochem. 2006, 100, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, H.; Chia, B.C.S.; Davies, N.L.; Zerella, R.; Williams, D.H. Cooperative Binding Interactions of Glycopeptide Antibiotics. J. Am. Chem. Soc. 2002, 124, 3914–3919. [Google Scholar] [CrossRef]
- Zarkan, A.; Macklyne, H.-R.; Truman, A.W.; Hesketh, A.R.; Hong, H.-J. The Frontline Antibiotic Vancomycin Induces a Zinc Starvation Response in Bacteria by Binding to Zn(II). Sci. Rep. 2016, 6, 19602. [Google Scholar] [CrossRef]
- Luo, G.; Spellberg, B.; Gebremariam, T.; Lee, H.; Xiong, Y.Q.; French, S.W.; Bayer, A.; Ibrahim, A.S. Combination Therapy with Iron Chelation and Vancomycin in Treating Murine Staphylococcemia. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 845–851. [Google Scholar] [CrossRef]
- Vaara, M. Novel Derivatives of Polymyxins. J. Antimicrob. Chemother. 2013, 68, 1213–1219. [Google Scholar] [CrossRef]
- Hamel, M.; Rolain, J.-M.; Baron, S.A. The History of Colistin Resistance Mechanisms in Bacteria: Progress and Challenges. Microorganisms 2021, 9, 442. [Google Scholar] [CrossRef]
- Clifton, L.A.; Skoda, M.W.A.; Le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of Divalent Cation Removal on the Structure of Gram-Negative Bacterial Outer Membrane Models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Topisirović, L.J.; Jovčić, B. Antibiotici: Molekularni Mehanizmi Delovanja i Rezistencije; Univerzitet u Beogradu Biološki Fakultet: Beograd, Serbia, 2013. [Google Scholar]
- Morar, M.; Bhullar, K.; Hughes, D.W.; Junop, M.; Wright, G.D. Structure and Mechanism of the Lincosamide Antibiotic Adenylyltransferase LinB. Structure 2009, 17, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, A.L. Antibiotic Inhibitors of the Peptidyl Transferase Center. 1. Clindamycin as a Composite Analogue of the Transfer RNA Fragments L-Pro-Met and the D-Ribosyl Ring of Adenosine. Bioorg. Med. Chem. Lett. 1998, 8, 87–92. [Google Scholar] [CrossRef]
- Schlünzen, F.; Zarivach, R.; Harms, J.; Bashan, A.; Tocilj, A.; Albrecht, R.; Yonath, A.; Franceschi, F. Structural Basis for the Interaction of Antibiotics with the Peptidyl Transferase Centre in Eubacteria. Nature 2001, 413, 814–821. [Google Scholar] [CrossRef]
- Gaggelli, E.; Gaggelli, N.; Valensin, D.; Valensin, G.; Jeżowska-Bojczuk, M.; Kozłowski, H. Structure and Dynamics of the Lincomycin−Copper(II) Complex in Water Solution by 1H and 13C NMR Studies. Inorg. Chem. 2002, 41, 1518–1522. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves New Antibacterial Drug to Treat Complicated Urinary Tract Infections as Part of Ongoing Efforts to Address Antimicrobial Resistance; F.N. Release: Silver Spring, MD, USA, 2019. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-complicated-urinary-tract-and-complicated-intra-abdominal-infections (accessed on 19 July 2022).
- Goldberg, J.A.; Kumar, V.; Spencer, E.J.; Hoyer, D.; Marshall, S.H.; Hujer, A.M.; Hujer, K.M.; Bethel, C.R.; Papp-Wallace, K.M.; Perez, F.; et al. A γ-Lactam Siderophore Antibiotic Effective against Multidrug-Resistant Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter spp. Eur. J. Med. Chem. 2021, 220, 113436. [Google Scholar] [CrossRef]
- Tillotson, G.S. Trojan Horse Antibiotics–A Novel Way to Circumvent Gram-Negative Bacterial Resistance? Infect. Dis. 2016, 9, IDRT.S31567. [Google Scholar] [CrossRef]
- Zervosen, A.; Sauvage, E.; Frère, J.-M.; Charlier, P.; Luxen, A. Development of New Drugs for an Old Target—The Penicillin Binding Proteins. Molecules 2012, 17, 12478–12505. [Google Scholar] [CrossRef]
- Wang, Z.; Fast, W.; Valentine, A.M.; Benkovic, S.J. Metallo-β-Lactamase: Structure and Mechanism. Curr. Opin. Chem. Biol. 1999, 3, 614–622. [Google Scholar] [CrossRef]
- Ghuysen, J.-M. Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 1991, 45, 37–67. [Google Scholar] [CrossRef]
- Butler, M.S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex, J.H.; Fernandes, P.; Arias, C.A.; Paul, M.; Thwaites, G.E.; et al. Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed. Antimicrob. Agents Chemother. 2022, 66, e01991-21. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/show/NCT05475444 (accessed on 10 August 2022).
- Hakobyan, S.; Rzhepishevska, O.; Björn, E.; Boily, J.F.; Ramstedt, M. Influence of chelation strength and bacterial uptake of gallium salicylidene acylhydrazide on biofilm formation and virulence of Pseudomonas aeruginosa. J. Inorg. Biochem. 2016, 160, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Corbin, B.D.; Seeley, E.H.; Raab, A.; Feldmann, J.; Miller, M.R.; Torres, V.J.; Anderson, K.L.; Dattilo, B.M.; Dunman, P.M.; Gerads, R.; et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 2008, 15, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, V.; McConnell, M.J. Recent Advances in Iron Chelation and Gallium-Based Therapies for Antibiotic Resistant Bacterial Infections. Int. J. Mol. Sci. 2021, 22, 2876. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Sousa, C.A.; Simões, M. Harnessing microbial iron chelators to develop innovative therapeutic agents. J. Adv. Res. 2022, 39, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zaghouani, M.; Nay, B. 3-Acylated Tetramic and Tetronic Acids as Natural Metal Binders: Myth or Reality? Nat. Prod. Rep. 2016, 33, 540–548. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repac Antić, D.; Parčina, M.; Gobin, I.; Petković Didović, M. Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond. Antibiotics 2022, 11, 1105. https://doi.org/10.3390/antibiotics11081105
Repac Antić D, Parčina M, Gobin I, Petković Didović M. Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond. Antibiotics. 2022; 11(8):1105. https://doi.org/10.3390/antibiotics11081105
Chicago/Turabian StyleRepac Antić, Davorka, Marijo Parčina, Ivana Gobin, and Mirna Petković Didović. 2022. "Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond" Antibiotics 11, no. 8: 1105. https://doi.org/10.3390/antibiotics11081105
APA StyleRepac Antić, D., Parčina, M., Gobin, I., & Petković Didović, M. (2022). Chelation in Antibacterial Drugs: From Nitroxoline to Cefiderocol and Beyond. Antibiotics, 11(8), 1105. https://doi.org/10.3390/antibiotics11081105