
Cefonicid Benzathine Salt: A Convenient, Lean, and High-Performance Protocol to Make an Old Cephalosporin Shine

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
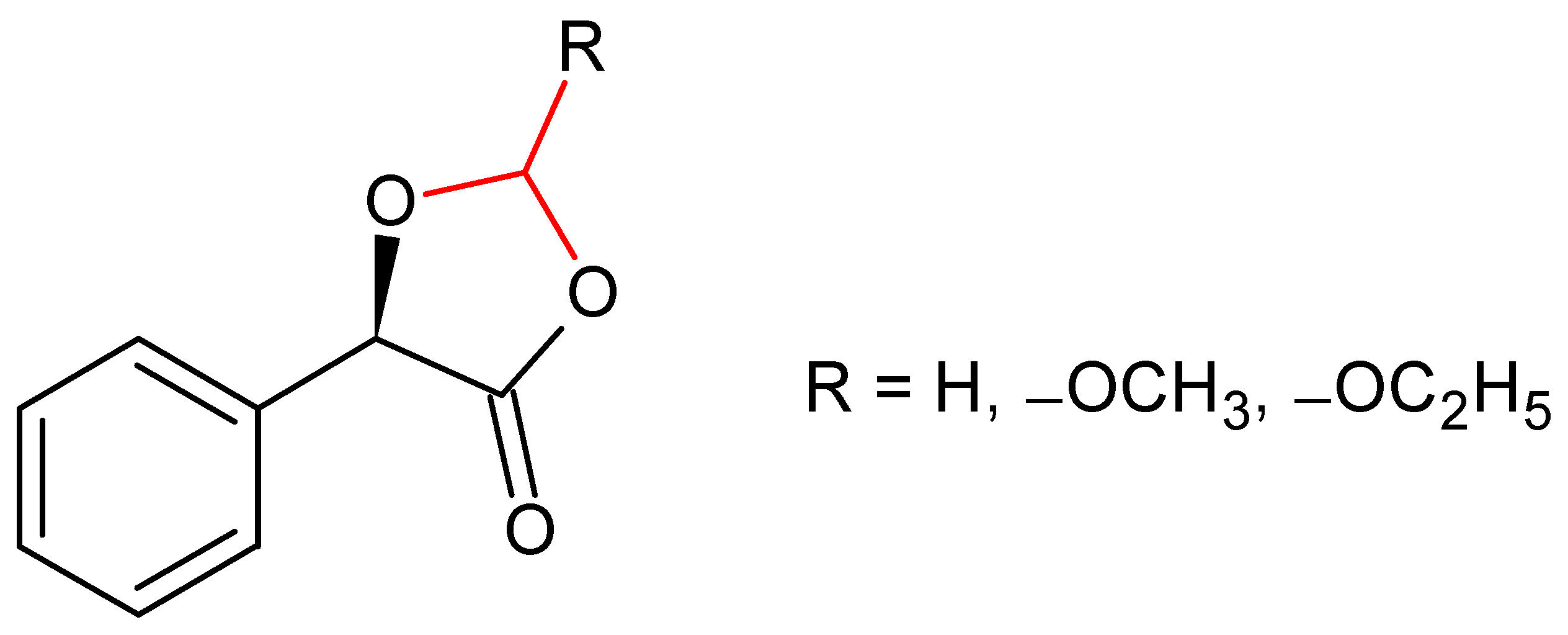
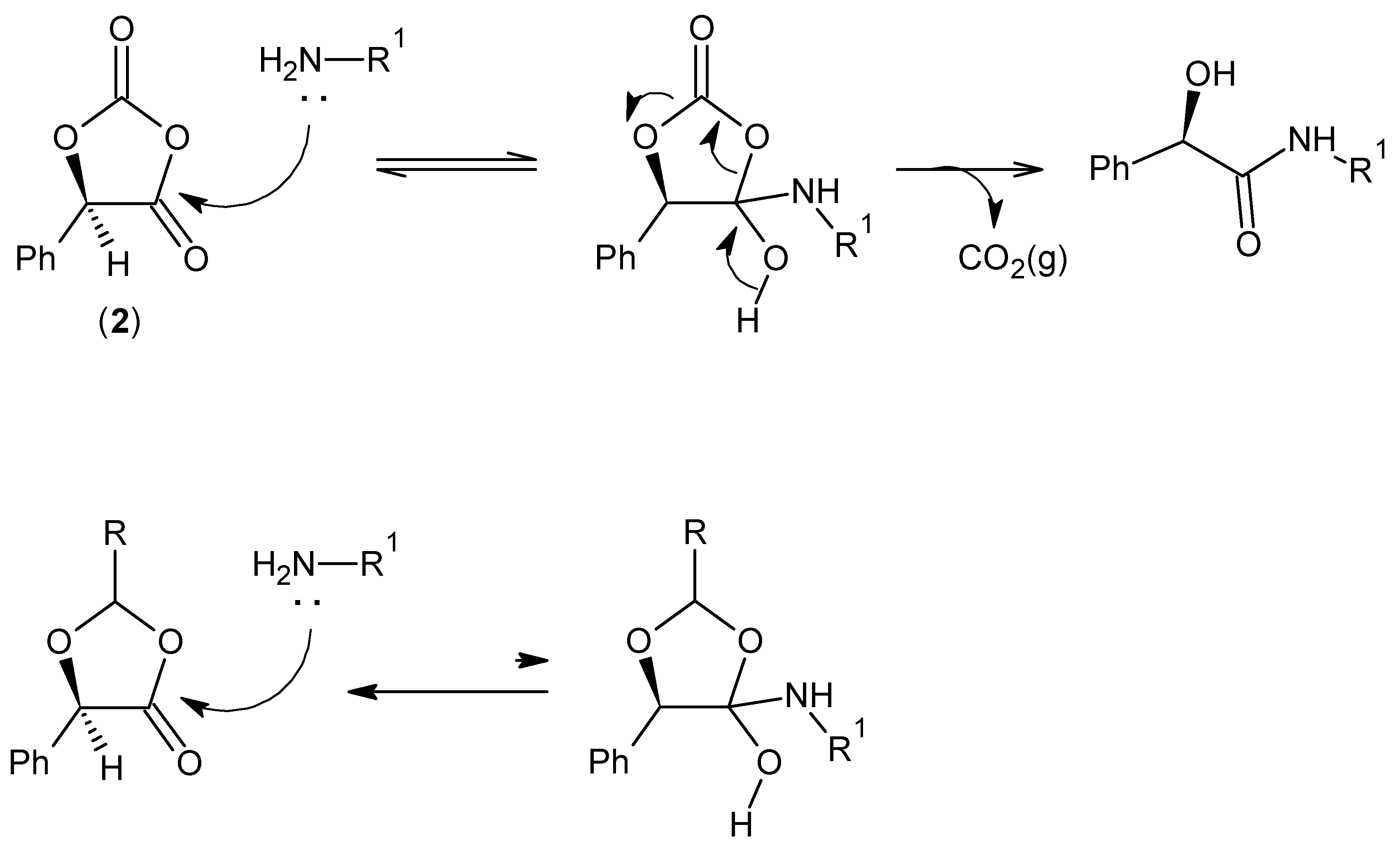
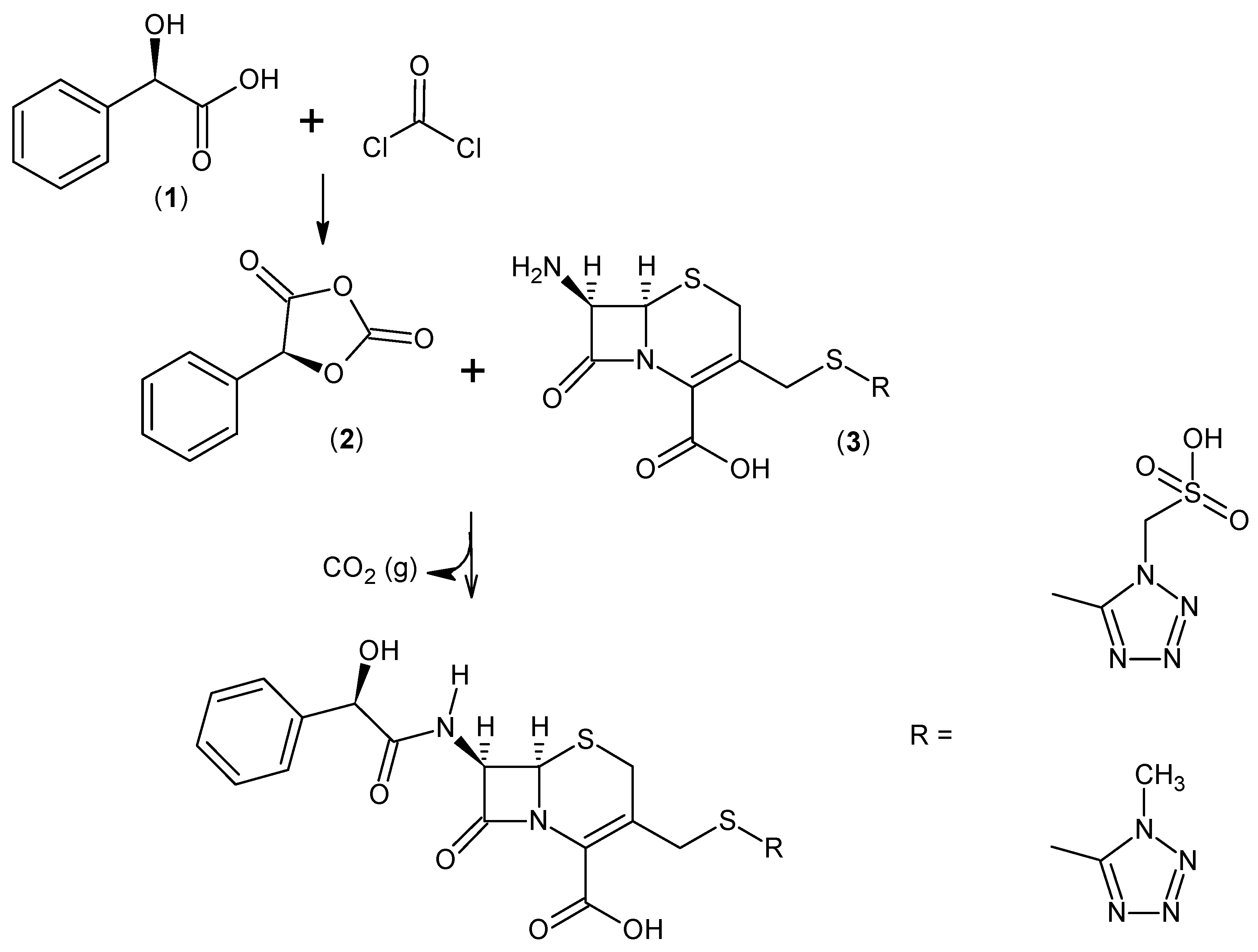
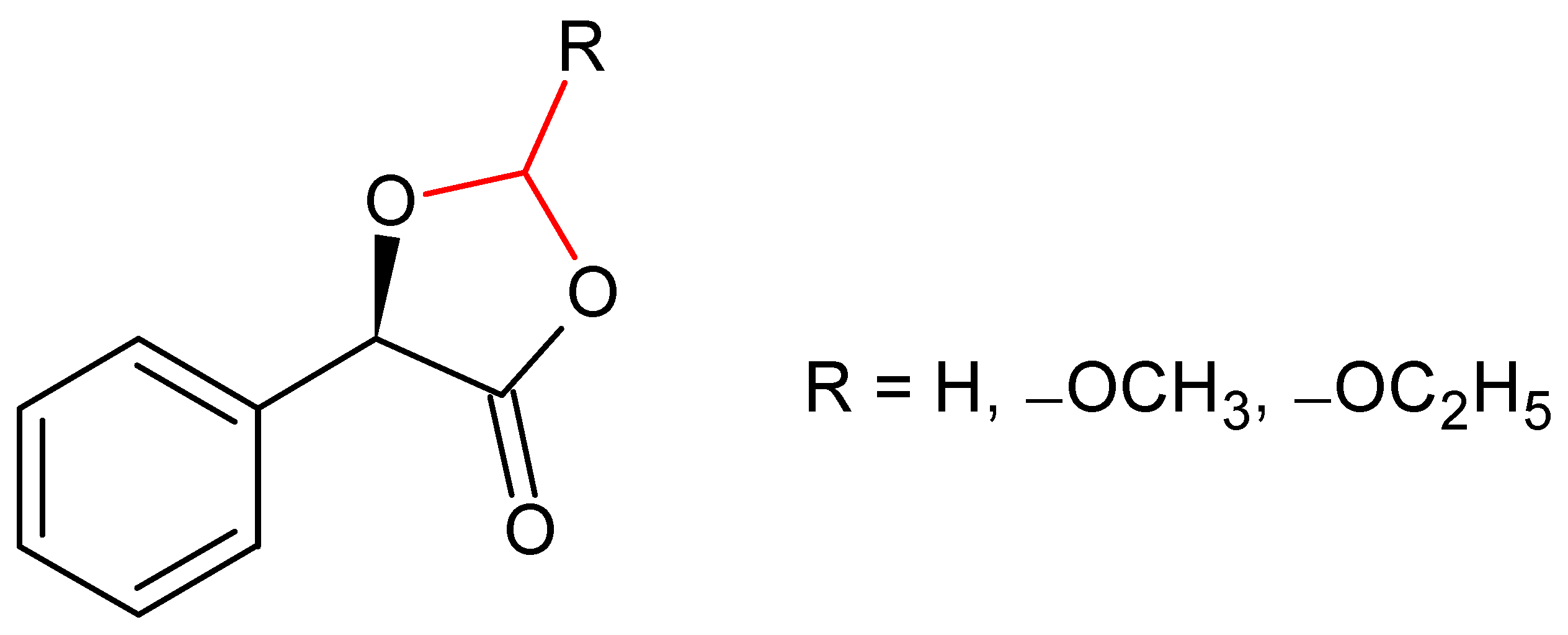
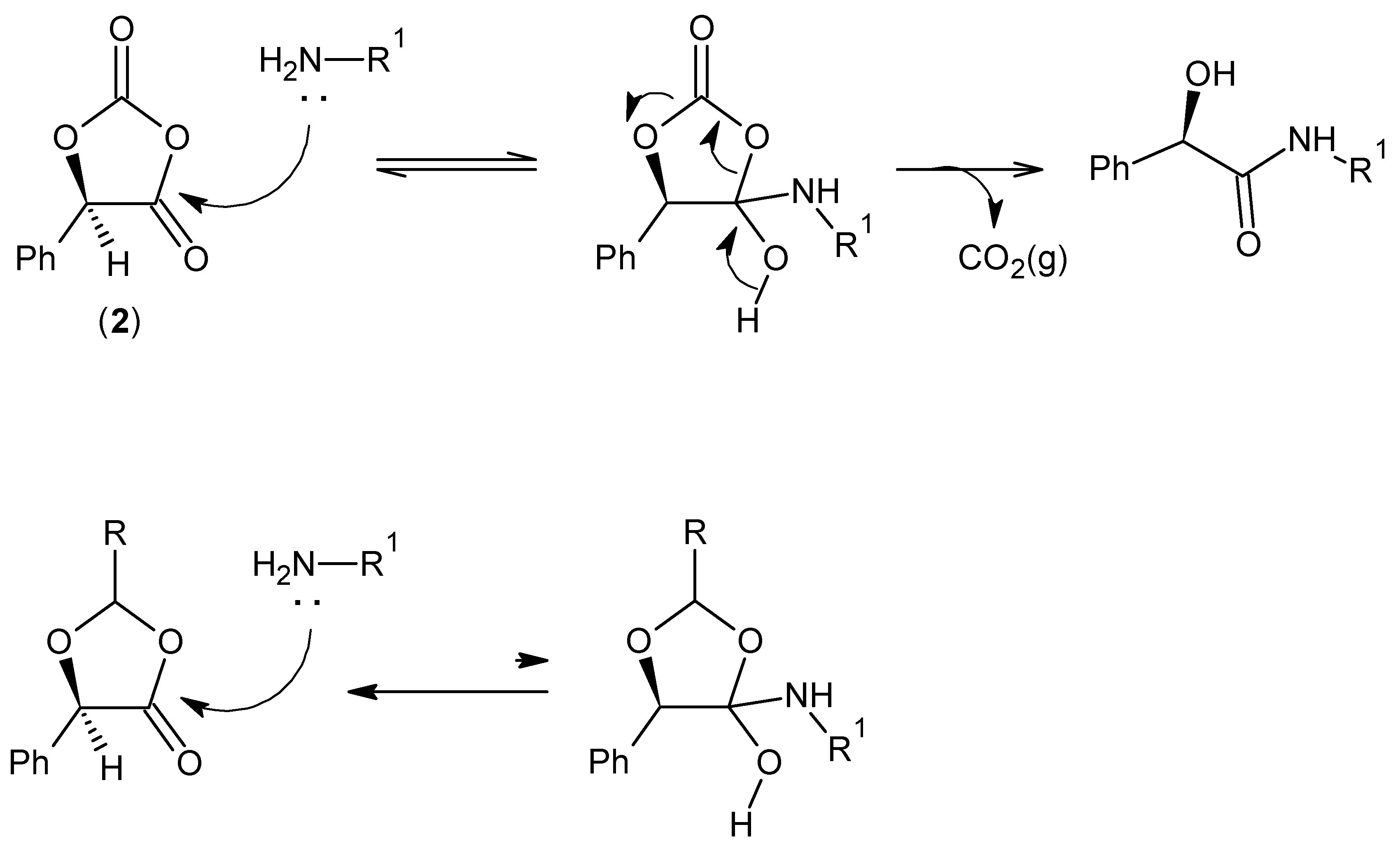
2.1. (R)-5-Phenyl-1,3-Dioxolane-2,4-Dione
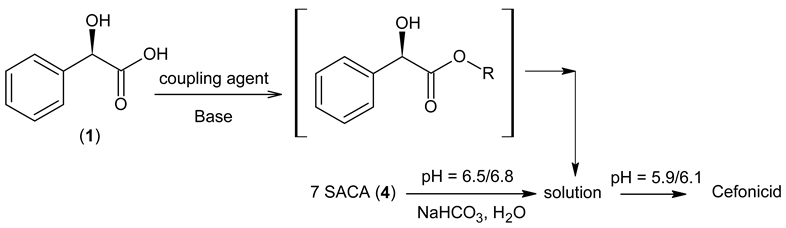
2.2. Amidation in Water and Organic Solvents
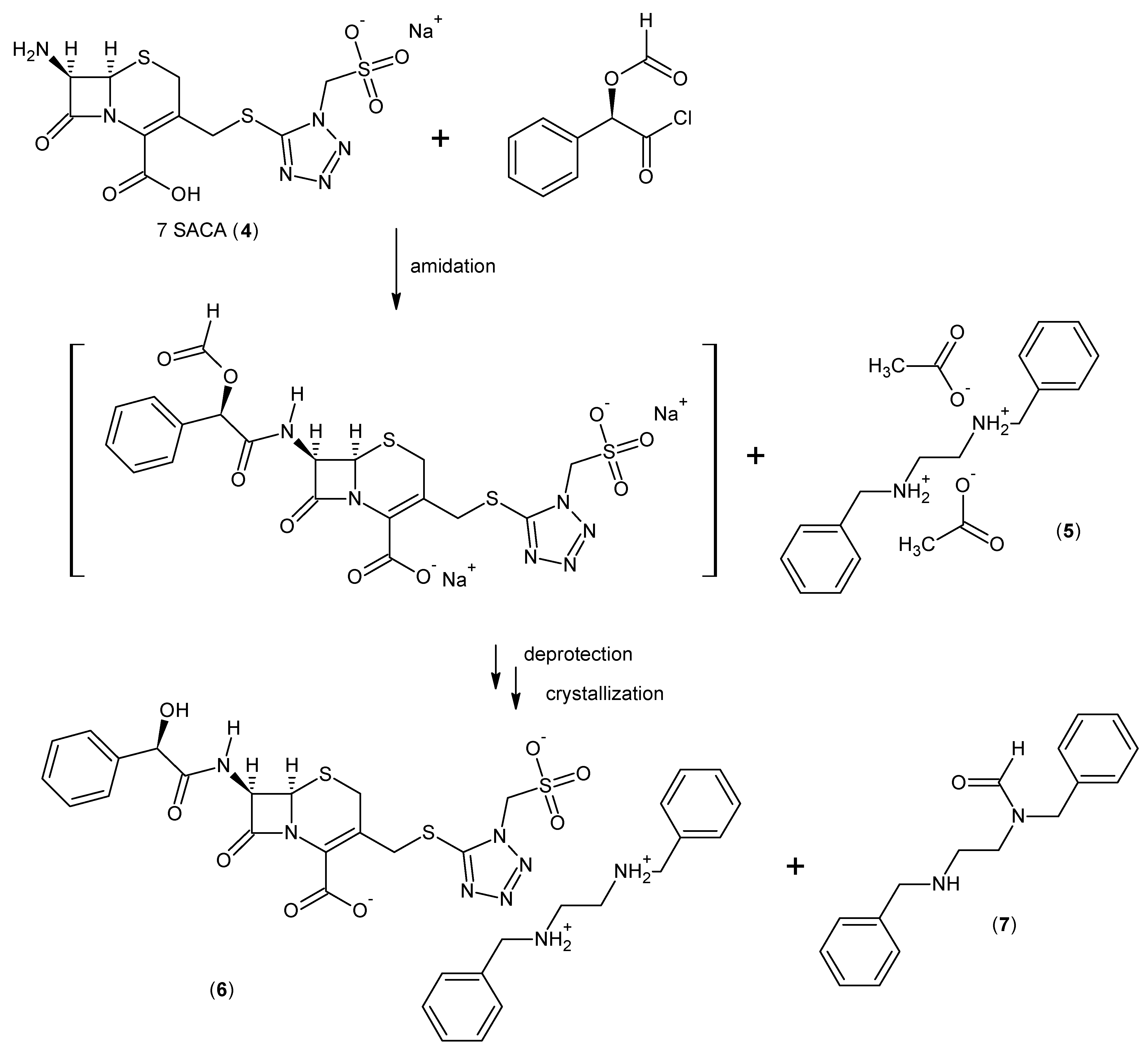
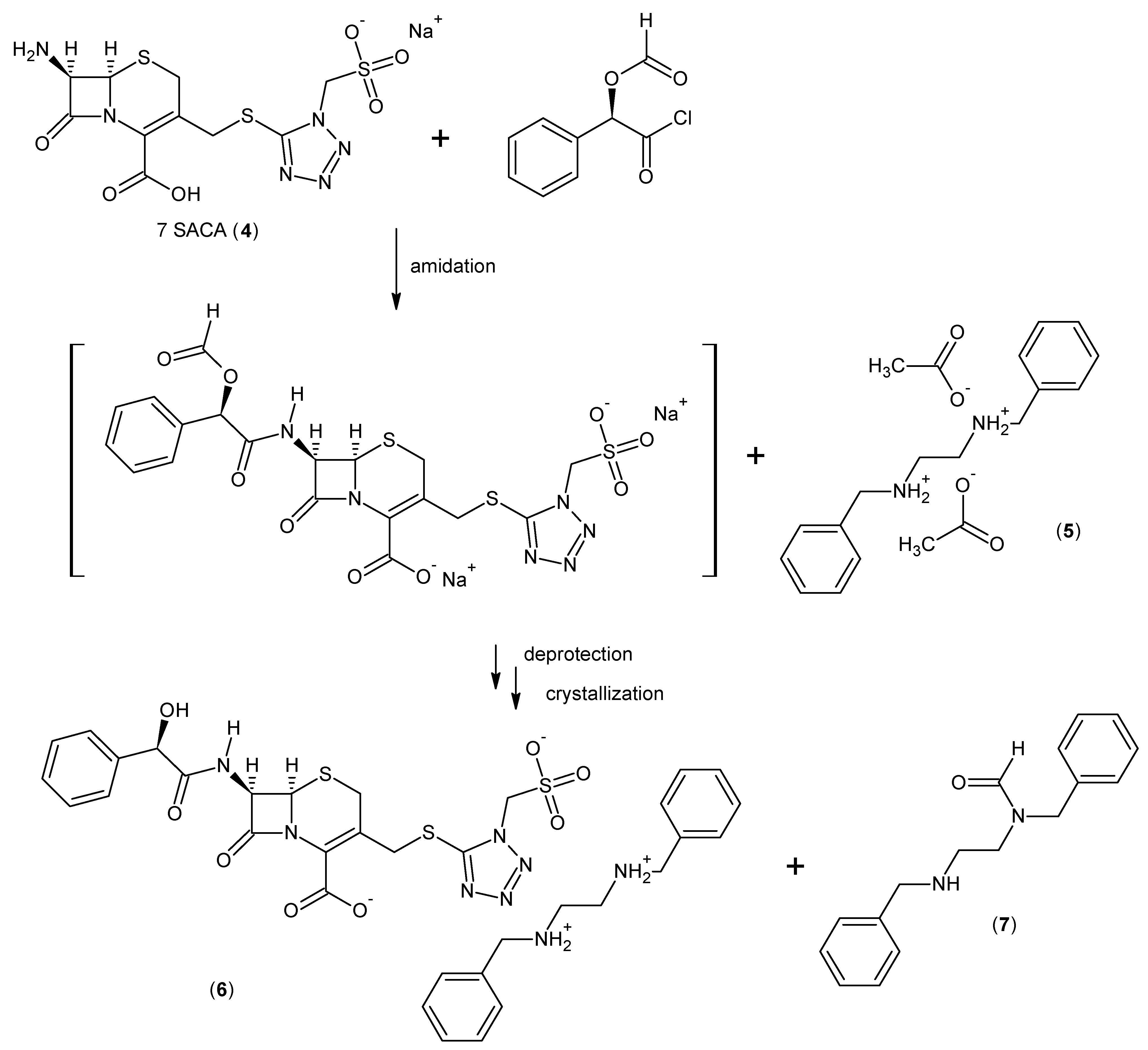
2.3. Cefonicid Benzathine Salt
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of O-Formyl-(R)-Mandelic Acid
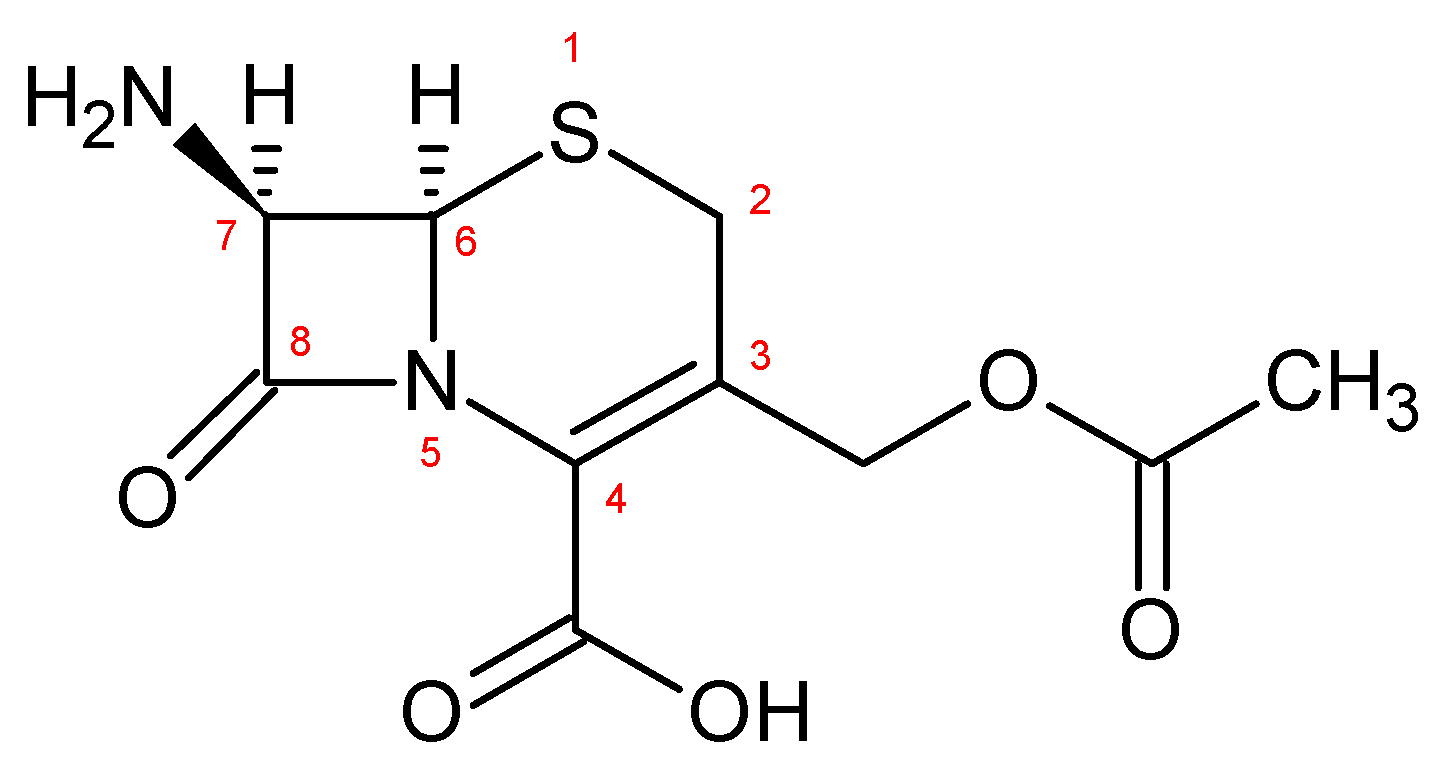
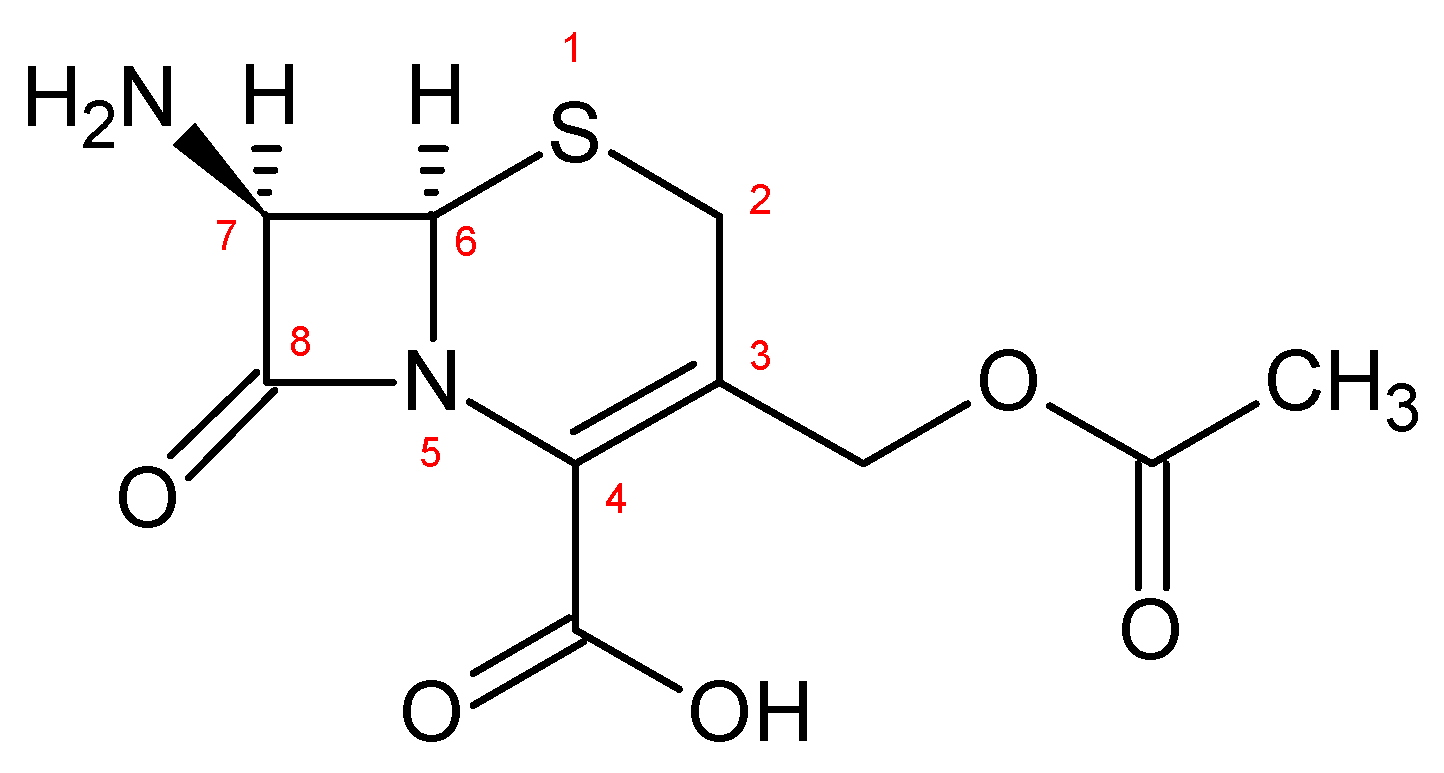
3.1.2. General Procedure for the Synthesis of 7-Amino-3-[sulphomethyl-1H-tetrazol-5-yl-thiomethyl]-3-cephem-4-carboxylate Monosodium Salt [7-SACA, 4]
3.1.3. General Procedure for the Synthesis of Cefonicid Benzathine Salt (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischman, J. Nine for Ninety. Chem. Eng. News 2013, 91, 26–27. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Manufacturing Routes to Fifth-Generation Cephalosporin Drugs. Part 1, Ceftolozane. Org. Process. Res. Dev. 2017, 21, 430–443. [Google Scholar] [CrossRef]
- Top Therapeutic Classes Global Pharmaceutical Sales 2018|Statista. Available online: https://www.statista.com/statistics/279916/top-10-therapeutic-classes-by-global-pharmaceutical-sales/ (accessed on 31 March 2022).
- Beta-Lactam and Beta-Lactamase Inhibitors Market Size and Share-2028. Available online: https://www.alliedmarketresearch.com/beta-lactam-and-beta-lactamase-inhibitors-market (accessed on 31 March 2022).
- Von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Habich, D. Antibacterial Natural Products in Medicinal Chemistry-Exodus or Revival? Angew. Chem. Int. Ed. 2006, 45, 5072–5129. [Google Scholar] [CrossRef]
- Duplessis, C.; Crum-Cianflone, N.F. Ceftaroline: A new cephalosporin with activity against methicillin-resistant Staphylococcus aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3, 1–17. [Google Scholar] [CrossRef]
- Cluck, D.; Lewis, P.; Stayer, B.; Spivey, J.; Moorman, J. Ceftolozane-Tazobactam: A new-generation cephalosporin. Am. J. Health-Syst. Pharm. 2015, 72, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Basilea Pharmaceutica-News and Media. Available online: https://web.archive.org/web/20190331140355/http:/www.basilea.com/News-and-Media/Basilea-to-launch-Zevtera-Mabelio-ceftobiprole-medocaril-in-Europe-through-a-commercial-services-provider/7dcfb10b-c4ef-5a82-8359-f88b1f36cb96/ (accessed on 31 March 2022).
- Aoki, T.; Yoshizawa, H.; Yamawaki, K.; Yokoo, K.; Sato, J.; Hisakawa, S.; Hasegawa, Y.; Kusano, H.; Sano, M.; Sugimoto, H.; et al. Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship. Eur. J. Med. Chem. 2018, 155, 847–868. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves New Antibacterial Drug|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug (accessed on 31 March 2022).
- FDA Approves New Treatment for Complicated Urinary Tract and Complicated Intra-Abdominal Infections|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-complicated-urinary-tract-and-complicated-intra-abdominal-infections (accessed on 31 March 2022).
- Press Announcements > FDA Approves New Antibacterial Drug Avycaz. Available online: https://web.archive.org/web/20151117082448/https:/www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm435629.htm (accessed on 31 March 2022).
- Actor, P.; Uri, J.V.; Zajac, I.; Gaurini, J.R.; Phillips, L.; Pitkin, D.H.; Berges, D.A.; Dunn, G.L.; Hoover, J.R.E.; Weisbach, J.A. SK&F 75073, New parenteral broad-spectrum cephalosporin with high and prolonged serum level. Antimicrob. Agents Chemother. 1978, 13, 784–790. [Google Scholar] [CrossRef]
- Liras, P.; Martin, J.F. Gene clusters for β-lactam antibiotics and control of their expression: Why have clusters evolved, and from where did they originate? Int. Microbiol. 2006, 9, 9–19. [Google Scholar] [PubMed]
- Tartaglione, T.A.; Polk, R.E. Review of the new second-generation cephalosporins: Cefonicid, ceforanide, and cefuroxime. Drug Intell. Clin. Pharm. 1985, 19, 188–198. [Google Scholar] [CrossRef]
- Saltiel, E.; Brogden, R. Cefonicid: A review of its antibacterial activity, pharmacological properties and therapeutic use. Drugs 1986, 32, 222–259. [Google Scholar] [CrossRef]
- Gröger, H.; Pieper, M.; König, B.; Bayer, T.; Schleich, H. Industrial landmarks in the development of sustainable production processes for the β-lactam antibiotic key intermediate 7-aminocephalosporanic acid (7-ACA). Sustain. Chem. Pharm. 2017, 5, 72–79. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Hatfield, L.D. Cephalosporins. Compr. Heterocycl. Chem. II 1996, 1B, 591–622. [Google Scholar] [CrossRef]
- Berges, D.A. 7-Acyl-3-(sulfonic acid and sulfamoyl substituted tetrazolyl thiomethyl) Cepahlosporins. U.S. Patent 4,048,311, 13 September 1977. [Google Scholar]
- Berges, D.A. 7-Acyl-3-(sulfonic acid and sulfamoyl substituted tetrazolyl thiomethyl) Cephalosporins. U.S. Patent 4,093,723, 6 June 1978. [Google Scholar]
- Berges, D.A. 7-Acyl-3-(sulfonic acid and sulfamoyl substituted tetrazolyl thiomethyl) Cephalosporins. U.S. Patent 4,159,373, 26 June 1979. [Google Scholar]
- Comito, M.; Monguzzi, R.; Tagliapietra, S.; Palmisano, G.; Cravotto, G. Efficient pilot-scale synthesis of the key cefonicid intermediate at room temperature. Green Process. Synth. 2022, 11, 96–105. [Google Scholar] [CrossRef]
- Bryan, M.C.; Dunn, P.J.; Entwistle, D.; Gallou, F.; Koenig, S.G.; Hayler, J.D.; Hickey, M.R.; Hughes, S.; Kopach, M.E.; Moine, G.; et al. Key green chemistry research areas from a pharmaceutical manufacturers’ perspective revisited. Green Chem. 2018, 20, 5082–5103. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L., Jr.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key green chemistry research areas-a perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, C.; Poechlauer, P.; Broxterman, Q.B.; Yang, B.S.; Ende, D.; Baird, J.; Bertsch, C.; Hannah, R.E.; Dell’Orco, P.; Noorman, H.; et al. Key green engineering research areas for sustainable manufacturing: A perspective from pharmaceutical and fine chemicals manufacturers. Org. Process Res. Dev. 2011, 15, 900–911. [Google Scholar] [CrossRef]
- Fuller, W.D.; Verlander, M.S.; Murray, G. A procedure for the facile synthesis of amino-acid N-carboxy anhydrides. Biopolymers 1976, 15, 1869–1871. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Jonte, J.M. New polymer syntheses. 8. Synthesis and polymerization of L-lactic acid O-carobxyanhydride (5-methyl-dioxolan-2,4-dione). Polym. Bull. 1983, 9, 276–283. [Google Scholar] [CrossRef]
- Komori, T.; Sakane, K.; Setoi, H.; Kawai, Y.; Teraji, T.; Kohsaka, M.; Imanaka, H. Structure activity relationships of synthetic antibiotic analogs of chryscandin. J. Antibiot. 1985, 38, 1182–1203. [Google Scholar] [CrossRef]
- Toyooka, K.; Takeuchi, Y.; Ohto, H.; Shibuya, M.; Kubota, S. Synthesis and absolute configuration of optically active 2,3-disubstituted 2,3-dihydro-1,3,4-thia-diazoles. Chem. Pharm. Bulletin. 1991, 39, 777–779. [Google Scholar] [CrossRef]
- Naito, T.; Okumura, J.; Kamachi, H. 7-(D-α-hydroxy-2-arylacetamido)-3-(2-carboxyalkyl-2,3-dihydro-s-triazolo-[4,3-b]pyridazine-3-on-6-ylthiomethyl)-3-cephem-4-carboxylic Acids and Derivates. U.S. Patent 4,112,228, 5 September 1978. [Google Scholar]
- Yin, Q.; Yin, L.; Wang, H.; Cheng, J. Synthesis and biomedical applications of functional poly(α-hydroxy acids) via ring-opening polymerization of O-carboxyanhydrides. Acc. Chem. Res. 2015, 48, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.M. Process for Preparing 7-(α-Hydroxy-α-phenyl)acetamido-3-(1-methyl-1H-tetrazol-5-xylthiomethyl)-3-cephem-4-carboxylic Acid and Derivates. CA 994,327, 3 August 1976. [Google Scholar]
- Prager, B.C. Cephalosporins. EP 0,156,771, 2 October 1985. [Google Scholar]
- Hoover, J.R.E. Derivates of 7-Aminocephalospoanic Acid. U.S. Patent 3,167,549, 26 January 1965. [Google Scholar]
- Greene, J.M. Process for Preparing 7-(α-Hydroxy-α-phenyl)acetamido-3-(1-methyl-1H-tetrazol-5-ylthiomethyl)-3-cephem-4-carboxylic Acid and Derivates Thereof. U.S. Patent 3,840,531, 8 October 1974. [Google Scholar]
- Masayuki, N.; Tadashi, Y.; Hiroshi, O.; Mitsuaki, O.; Tetsuo, O.; Teruji, T.; Ikuo, K.; Nobuhiro, H.; Hisashi, S. Synthetic studies on β-lactam antibiotics. Part 10. Synthesis of 7β-[2-carboxy-2-(4-hydroxyphenyl)acetamido]-7α-methoxy-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-1-oxa-1-dethia-3-cephem-4-carboxylic acid disodium salt (6059-S) and its related 1-oxacephems. J. Med. Chem. 1979, 22, 757–759. [Google Scholar] [CrossRef]
- Masayuki, N.; Tadashi, Y.; Hiroshi, O.; Mitsuaki, O.; Tetsuo, O.; Wataru, N. Synthesis and antibacterial activity of 1-oxacephem derivates. J. Antibiot. 1982, 35, 463–482. [Google Scholar] [CrossRef]
- Davies, W.H. Anhydrocarboxy-derivates of hydroxy and mercapto acids. J. Chem. Soc. Resumed 1951, 1357–1359. [Google Scholar] [CrossRef]
- Toyooka, K.; Takeuchi, Y.; Kubota, S. A novel and facile synthesis of 5-substituted 1,3-dioxolane-2,4-diones using trichloromethyl chloroformate. Heterocycles 1989, 29, 975–978. [Google Scholar] [CrossRef]
- Oya, M.; Ryoichi, K.; Katakai, R.; Nakai, H. Novel synthesis of N-carboxy-α-amino acid anhydride. Chem. Lett. 1973, 11, 1143–1144. [Google Scholar] [CrossRef]
- Katakai, R.; Iizuka, Y. An improved rapid method for the synthesis of N-carboxy α-amino acid anhydride using trichloromethyl chloroformate. J. Org. Chem. 1985, 50, 715–716. [Google Scholar] [CrossRef]
- Tang, L.; Deng, L. Dynamic kinetic resolution via dual-function catalysis of modified cinchona alkaloids: Asymmetric synthesis of α-hydroxy carboxylic acids. J. Am. Chem. Soc. 2002, 124, 2870–2871. [Google Scholar] [CrossRef]
- Li, M.; Tao, Y.; Tang, J.; Wang, Y.; Zhang, X.; Tao, Y.; Wang, X. Synergetic organocatalysis for eliminating epimerization in ring-opening polymerizations enables synthesis of stereoregular isotactic polyester. J. Am. Chem. Soc. 2019, 141, 281–289. [Google Scholar] [CrossRef]
- Jiang, J.; Cui, Y.; Lu, Y.; Zhang, B.; Pan, X.; Wu, J. Weak lewis pairs as catalysts for highly isoselective ring-opening polymerization of epimerically labile rac-O-carboxyanhydride of mandelic acid. Macromolecules 2020, 53, 946–955. [Google Scholar] [CrossRef]
- Cairns, S.A.; Schultheiss, A.; Shaver, M.P. A broad scope of aliphatic polyesters prepared by elimination of small molecules from sustainable 1,3-dioxolan-4-ones. Polym. Chem. 2017, 8, 19. [Google Scholar] [CrossRef]
- Xu, Y.; Perry, M.R.; Cairns, S.A.; Shaver, M.P. Understanding the ring-opening polymerization of dioxolanones. Polym. Chem. 2019, 10, 23. [Google Scholar] [CrossRef]
- Bylikin, S.Y.; Shipov, A.G.; Kramarova, E.P.; Negrebetsky, V.V.; Korlyukov, A.A.; Baukov, Y.; Hursthouse, M.B.; Male, L.; Bassindale, A.R.; Taylor, P.G. O,O-Monochelate complexes of silicon and germanium halides: The derivates of L-mandelic N,N-dimethylamide. J. Organomet. Chem. 2009, 694, 244–248. [Google Scholar] [CrossRef]
- Shipov, A.G.; Gruener, S.V.; Korylyukov, A.A.; Kramarova, E.P.; Murasheva, T.P.; Bylikin, S.Y.; Negrebetskii, V.V.; Ivashchenko, F.A.; Airapetyan, D.V.; Zueva, G.Y.; et al. Tribromogermyl monochelates–derivates of N,N-disubstituted 2-hydroxycarboxylic amides. Russ. Chem. Bull. 2010, 59, 761–770. [Google Scholar] [CrossRef]
- Misaki, T.; Ureshino, S.; Nagase, R.; Oguni, Y.; Tanabe, Y. Improved practical asymmetric synthesis of α-alkylmandelic acids utilizing highly diastereoselective alkylation of 5-aryl-2-(1-naphthyl)-1,3-dioxolan-4-ones. Org. Process. Res. Dev. 2006, 10, 500–504. [Google Scholar] [CrossRef]
- Kunz, W. 1,3-Dioxolan-5-one Derivates. U.S. Patent 4,447,625, 8 May 1984. [Google Scholar]
- Thillaye du Boullay, O.; Marchal, E.; Martin-Vaca, B.; Cossio, F.P.; Bourissou, D. An activated equivalent of lactide toward organocatalytic ring-opening polymerization. J. Am. Chem. Soc. 2006, 128, 16442–16443. [Google Scholar] [CrossRef]
- Pessa, A.; Lerpini, A. Process for the Preparation of Cephalosporins. U.S. Patent 5,625,058, 29 April 1997. [Google Scholar]
- Brenner, M.; Renganathan, V. Enzymatic Process for the Production of Cefonicid. WO 92/17600, 15 October 1992. [Google Scholar]
- Polansky, T.J. Cristalline Benzathine Salt of Cefonicid and Its Preparation. U.S. Patent 5,705,496, 6 January 1998. [Google Scholar]
- Terreni, M.; Ubiali, D.; Bavaro, T.; Pregnolato, M.; Fernandez-Lafuente, R.; Guisan, J.M. Enzymatic synthesis of cephalosporins. The immobilized acylase from Arthrobacter viscosus: A new useful biocatalyst. Appl. Microbiol. Biotechnol. 2007, 77, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhuang, Z.; Liu, Q.; Zhang, Z.; Zhan, F.; Zheng, G. Concise synthesis of key intermediate of mirabegron via a mixed anhydride method. Org. Process Res. Dev. 2016, 20, 1993–1996. [Google Scholar] [CrossRef]
- Deshmukh, D.G.; Bangal, M.N.; Patekar, M.R.; Medhane, V.J.; Mathad, V.T. Investigation of mechanistic pathway for trimethyl borate mediated amidation of (R)-mandelic acid for the synthesis of mirabegron, an antimuscarinic agent. Acta Chim. Slov. 2018, 65, 239–245. [Google Scholar] [CrossRef]
- Magano, J. Large-scale amidations in process chemistry: Practical considerations for reagents selection and reaction execution. Org. Process. Res. Dev. 2022, 26, 1562–1689. [Google Scholar] [CrossRef]
- Fattahi, N.; Ayubi, M.; Ramazani, A. Amidation and esterification of carboxylic acids with amines and phenols by N,N′-diisopropylcarbodiimide: A new approach for amide and ester bond formation in water. Tetrahedron 2018, 74, 4351–4356. [Google Scholar] [CrossRef]
- Sharma, S.; Buchbinder, N.W.; Braje, W.M.; Handa, S. Fast amide couplings in water: Extraction, column chromatography, and crystallization not required. Org. Lett. 2020, 22, 5737–5740. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process. Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Monguzzi, R.; Menapace, S.; Piergiorgio, A. Improved Process for the Preparation of Ceftriaxone. EP 0,399,094, 28 November 1990. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Coupling Agent | Base | Solvent | Activation Temperature (°C) | Amidation Temperature (°C) | Cefonicid Yield in Solution (%) |
|---|---|---|---|---|---|---|
| 1 | TsCl | TEA | CH3CN | reflux | 0–5 r.t. | 0.5 1.3 |
| 2 | MsCl | TEA | CH3CN | r.t. | 0–5 r.t. | 2.0 3.7 |
| 3 | PivCl | TEA | CH3CN | 0–5 °C | 0–5 r.t. | - - |
| 4 | B(OMe)3 | NaHCO3 a | CH3CN | 60 °C | 0–5 r.t. | 0.1 0.1 |
| 5 | B(OMe)3 | K2CO3 a | CH3CN | 60 °C | 0–5 r.t. | 0.1 0.1 |
| 6 | EDC b | - | water | r.t. | r.t. | 1.2 |
| 7 | DIC b | - | water | r.t. | r.t. | 0.3 |
| 8 | TsCl | TEA | CH2Cl2 c EtOAc c | reflux | r.t. r.t. | - - |
| 9 | MsCl | TEA | CH2Cl2 c EtOAc c | r.t. | r.t. r.t. | 0.1 1.0 |
| Entry | OH-Protected Moiety | Coupling Agent | Solvent | OH-Protected Cefonicid Conversion in Solution (%) a |
|---|---|---|---|---|
| 1 | 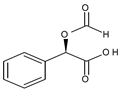 | TsCl | CH3CN b | 1.4 |
| 2 | | MsCl | CH3CN b | 4.0 |
| 3 | | PivCl | CH3CN b | 0.5 |
| 4 | | B(OMe)3 | CH3CN b | 0.4 |
| 5 | | EDC | Water c | 27.0 |
| 6 | | DIC | Water c | 32.0 |
| 7 | | SOCl2 | CH3CN d | 99.7 |
| 8 |  | SOCl2 | CH3CN d | 99.4 |
| 9 |  | SOCl2 | CH3CN d | 99.3 |
| Entry | Equivalents a | Conversion in Deformylation (%) b | Crystallization c | Yields (%) | Cefonicid Assay (%) | Benzathine Assay (%) |
|---|---|---|---|---|---|---|
| 1 | 1.3 | 52 | A | - | - | - |
| 2 | 1.7 | 84 | A | - | - | - |
| 3 | 2.0 | 94 | B | 51 | 72.8 | 25.2 |
| 4 | 2.4 | 99.3 | B | 62 | 71.5 | 27.0 |
| 5 | 2.8 | 99.1 | B | 61 | 72.2 | 26.3 |
| Time (Months) | Cefonicid Assay (%) | Benzathine Assay (%) | Total Impurities |
|---|---|---|---|
| 0 | 71.5 | 27.0 | 1.9 |
| 6 | 71.6 | 26.8 | 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comito, M.; Monguzzi, R.; Tagliapietra, S.; Palmisano, G.; Cravotto, G. Cefonicid Benzathine Salt: A Convenient, Lean, and High-Performance Protocol to Make an Old Cephalosporin Shine. Antibiotics 2022, 11, 1095. https://doi.org/10.3390/antibiotics11081095
Comito M, Monguzzi R, Tagliapietra S, Palmisano G, Cravotto G. Cefonicid Benzathine Salt: A Convenient, Lean, and High-Performance Protocol to Make an Old Cephalosporin Shine. Antibiotics. 2022; 11(8):1095. https://doi.org/10.3390/antibiotics11081095
Chicago/Turabian StyleComito, Marziale, Riccardo Monguzzi, Silvia Tagliapietra, Giovanni Palmisano, and Giancarlo Cravotto. 2022. "Cefonicid Benzathine Salt: A Convenient, Lean, and High-Performance Protocol to Make an Old Cephalosporin Shine" Antibiotics 11, no. 8: 1095. https://doi.org/10.3390/antibiotics11081095
APA StyleComito, M., Monguzzi, R., Tagliapietra, S., Palmisano, G., & Cravotto, G. (2022). Cefonicid Benzathine Salt: A Convenient, Lean, and High-Performance Protocol to Make an Old Cephalosporin Shine. Antibiotics, 11(8), 1095. https://doi.org/10.3390/antibiotics11081095