2. Materials and Methods
2.1. General
Solvents and chemical reagents were purchased from Sigma-Aldrich (Steinheim, Germany) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA) and were used without further purification. Both
1H and
13C NMR experiments were measured on a Bruker 500 MHz instrument with chemical shifts given in parts per million, ppm (δ-values) relative to the internal tetramethylsilane standard. NMR data were processed by using the academic edition of the ACD NMR software. The NMR data were presented as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (double doublet), or bs (broad singlet); coupling constants, J, are reported in hertz (Hz). The structures of intermediates and the final MGB products were confirmed by NMR spectroscopy and mass spectrometry (
Supplementary Materials). Electrospray ionization–mass spectrometry (ESI–MS) experiments were carried out on a triple-quadrupole tandem mass spectrometer (MicromassW Quattro microTM Waters Corp., Milford, MA, USA) coupled with an electrospray ionization (ESI) interface operated in positive ionization mode. Thin-layer chromatography (TLC) experiments were performed to monitor the reactions, using silica gel plates (0.25 mm, E. Merck, 60 F254) and a UV lamp for visualization. HPLC purification was carried out for all final products, using Agilent 1260 infinity liquid chromatography (Agilent Technologies, Palo Alto, CA, USA), a Zorbax-SB-C18 column (5 μm; 25 cm × 9.4 mm i.d) with the general gradient elution described below, and a detection wavelength of 260 nm.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 80 | 20 |
| 25 | 5 | 50 | 50 |
| 26 | 5 | 0 | 100 |
| 30 | 5 | 0 | 100 |
| 31 | 5 | 80 | 20 |
| 35 | 5 | 80 | 20 |
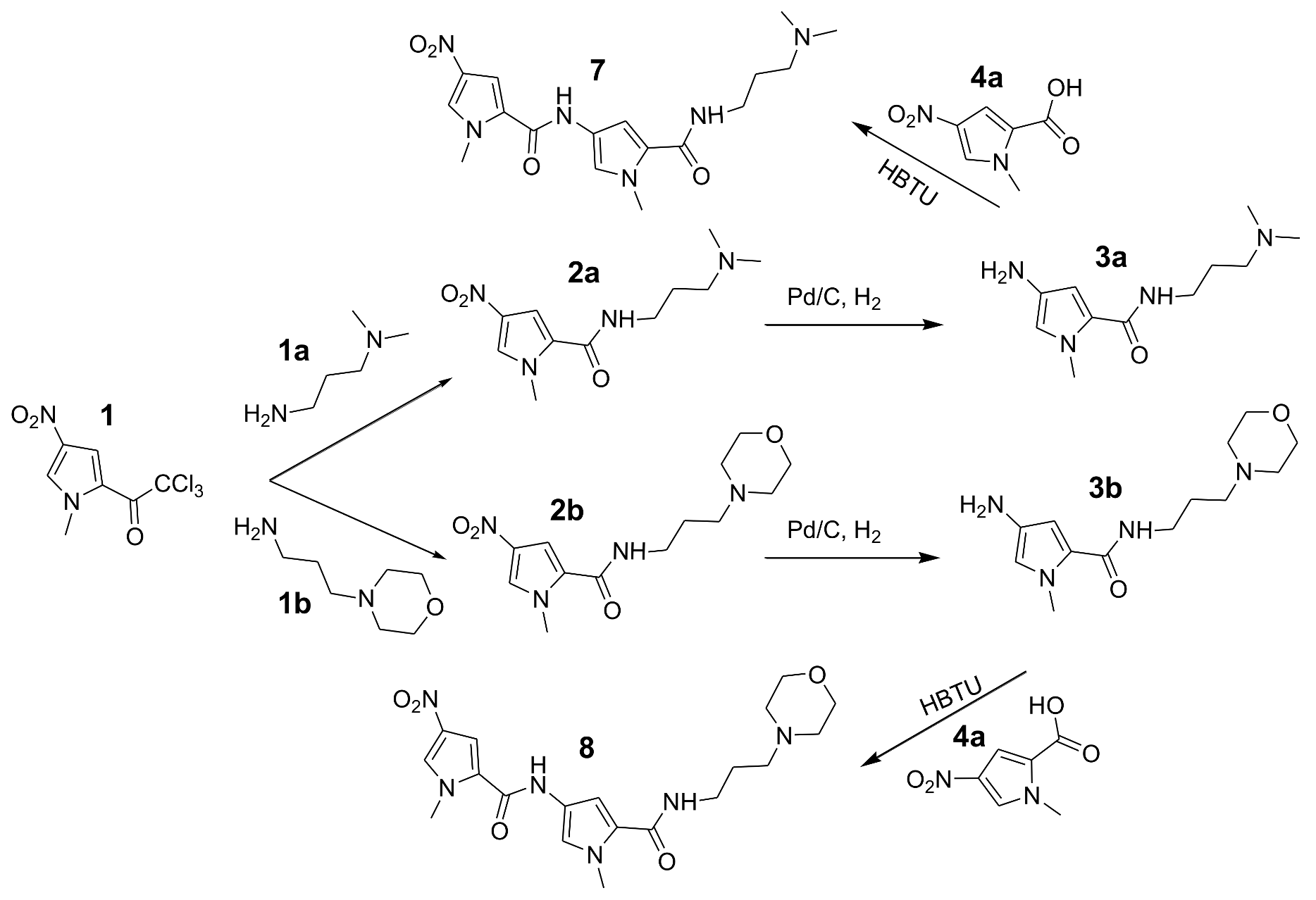
2.2. Synthesis of 1-methyl-N-(3-morpholinopropyl)-4-nitro-1H-pyrrole-2-carboxamide (2a)
A solution of 4-(3-aminopropyl) morpholine (2.93 g, 20.40 mmol) in dry DCM (10 mL) was cooled in an ice-water bath and stirred while a solution of 4-nitro-1-methyl-2-trichloroacetylpyrrole 1 (5.0 g, 0.01852 mol) in dry DCM (15 mL) was added slowly, over a period of five minutes. Then the mixture was stirred at room temperature for four hours, during which a precipitate formed. The solvent was evaporated under reduced pressure. Diethyl ether (30 mL) was added, and the product was collected by filtration, washed with ether, and dried under reduced pressure to produce the product as white solid crystals. Yield (75%); 1H NMR (CHLOROFORM-d) δ 1.77 (2H, m, CH2), 2.37 (6H, s, NCH3), 2.56 (2H, t, NCH2), 3.50 (2H, q, CONH—CH2), 4.01 (3H, s, NCH3), 6.98 (1H, d, Ar—H), 7.53 (1H, d, Ar—H), 8.67 (1H, s, CONH); 13C NMR (CHLOROFORM-d): δ 24.60, 37.88, 40.19, 45.25, 59.37, 106.51, 126.28, 126.92, 134.91, 160.26; LC–MS (ESI): m/z calcd for C11H18N4O3, 254.14, found 255.21 [M + H]+.
2.3. Synthesis of N-(3-(dimethylamino) propyl)-1-methyl-4-nitro-1H-pyrrole-2-carboxamide (2b)
A solution of 4-nitro-1-methyl-2-trichloroacetylpyrrole (1) (5.0 g, 18.52 mmol) in dry DCM (15 mL) was cooled in an ice-water bath and stirred while a solution of dimethylaminopropyl amine (2.705 g, 26.48 mmol) in dry DCM (5 mL) was added slowly, during a period of 5 min; the mixture was then stirred at room temperature overnight. The solvent was evaporated under reduced pressure, and the crude product was re-crystallized by using water:ethanol (80:20%), filtered, and washed with hexane to obtain the product in the form of white solid crystals. Yield (70%); 1H NMR (CHLOROFORM-d) δ 1.79 (2H, m, CH2), 2.53 (4H, s, NCH2), 2.57 (2H, t, NCH2), 3.50 (2H, q, CONH--CH2), 3.76 (4H, t, OCH2), 4.00 (3H, s, NCH3), 7.10 (1H, d, Ar—H), 7.54 (1H, d, Ar—H), 7.94 (1H, s, CONH); 13C NMR (CHLOROFORM-d): δ 23.55, 37.51, 39.77, 53.61, 58.39, 66.58, 106.45, 126.06, 126.49, 134.62, 160.02; LC−MS (ESI): m/z calcd for C13H20N4O4, 296.15, found 297.14 [M + H]+.
2.4. Synthesis of 1-methyl-N-[1-methyl-5-(carbonyl)-1H-pyrrol-3-yl]-4-nitro-1H-pyrrole-2-carboxamide (8)
First, 1-methyl-N-[2-(4-morpholinyl)ethyl]-4-nitro-1H-pyrrole-2-carboxamide (3.0 g, 10.13 mmol) was dissolved in THF (12 mL) and cooled to 0 °C. Then 10%-Pd/C (0.5 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was then stirred under hydrogen for 6 h. The suspension was then filtered, and the THF was removed under reduced pressure to produce an oily amine product which was then diluted with 2 mL THF and added dropwise to a solution of 1-methyl-4-nitro-1H-pyrrole-2-carboxylic acid 4a (1.2918 g, 7.545 mmol), triethylamine (5.585 mL, 45.31 mmol), and HBTU (0.331 g, 8.73 mmol) in THF (10 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, and the crude product obtained was purified on basified silica, using a 5:94:1 solution of MeOH/EA/TEA to obtain the product as white solid. Yield (68%); 1H NMR (ACETIC ACID-d4) δ 2.01 (2H, t, NCH2), 3.04 (4H, s, NCH2), 3.51 (2H, q, CONH-CH2), 3.90 (4H, s, OCH2) 3.93 (3H, s, NCH3), 4.07 (3H, s, NCH3), 6.98 (1H, d, Ar—H), 7.28 (1H, d, Ar—H), 7.34 (1H, d, Ar—H), 7.94 (1H, d, Ar—H), 9.52 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ15.16, 17.54, 25.98, 26.77, 27.14, 33.12, 45.90, 94.87, 97.26, 112.50, 114.20, 117.02, 117.18, 137.53, 149.71; LC−MS (ESI): m/z calcd for C19H26N6O5, 418.2, found 419.12 [M + H]+.
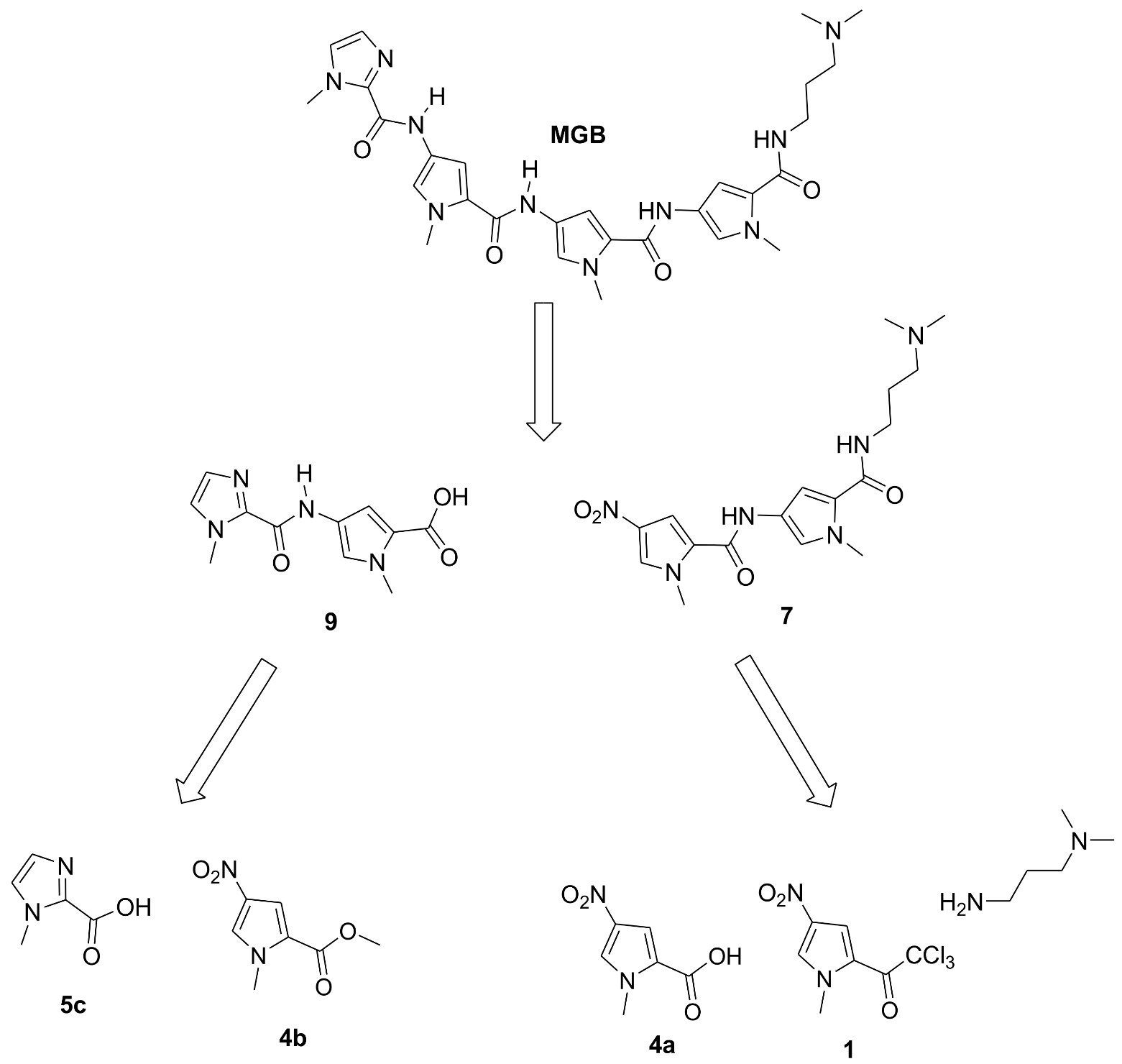
2.5. Synthesis of N-[3-(dimethylamino) propyl]-1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl]amino}-1H-pyrrole-2-carboxamide (7)
N-[3-(Dimethylamino)propyl]-1-methyl-4-nitro-1H-pyrrole-2-carboxamide (0.40 g, 1.5739 mmol) was dissolved in methanol (10 mL) and cooled to 0 °C. Then 10%-Pd/C (0.1 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was then stirred under hydrogen for 4 h The suspension was then filtered, and the solvent was removed under reduced pressure to produce an oily amine product which was diluted with 2 mL THF and added dropwise to 1-methyl-4-nitro-1H-pyrrole-2-carboxylic acid 4a (0.2406 g, 1.411 mmol), triethylamine (1.179 g, 11.65 mmol), and HBTU (0.716 g, 1.887 mmol) in THF (10 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, and sodium bicarbonate solution (10 mL) was added to the crude product, which was then left stirring for 10 min. The desired product was then precipitated as yellow solid and collected by filtration and dried under vacuum. Yield (55%); 1H NMR (CHOLOROFORM-d) δ 1.84 (2H, m, CH2), 2.44 (6H, s, NCH3), 2.65 (2H, t, NCH2), 3.48 (2H, q, CONH--CH2), 3.93 (3H, s, NCH3), 4.04 (3H, s, NCH3), 6.58 (1H, d, Ar—H), 7.25 (1H, d, Ar—H), 7.32 (1H, d, Ar—H), 7.60 (1H, d, Ar—H), 7.76 (1H, s, CONH), 8.11 (1H, s, CONH); 13C NMR (CHLOROFORM-d): δ 14.27, 22.84, 25.58, 29.51, 29.85, 32.08, 36.88, 38.10, 45.19, 103.1, 107.15, 118.89, 120.64, 124.22, 127.03, 161.75; LC−MS (ESI): m/z calcd for C17H24N6O4, 376.09, found 377.02 [M + H]+.
2.6. Synthesis of methyl 1-methyl-4-nitro-1H-pyrrole-2-carboxylate (4b)
First, 4-nitro-1-methyl-2-trichloroacetylpyrrole 1 (2.0 g, 7.4 mmol) was dissolved in methanol (14 mL). Sodium methoxide (0.48 g, 8.8 mmol) was dissolved in methanol (10 mL) and then added to 1 while stirring, and the reaction was refluxed (70 °C) for 1 h. The solvent was evaporated under reduced pressure; then cold distilled water was added, and the product was precipitated as white powder, which was then filtered, washed with a 1:1 water:methanol solution, and dried under vacuum. Yield (45%); 1H NMR (CHLOROFORM-d) δ 3.87 (3H, s, OCH3), 4.00 (3H, s, NCH3), 7.42 (1H, d, Ar—H), 7.61 (1H, d, Ar—H); 13C NMR (CHLOROFORM-d): δ 37.97, 51.86, 112.78, 122.84, 127.53, 160.62; LC−MS (ESI): m/z calcd for C7H8N2O4, 184.85, found 184.89 [M + H]+.
2.7. Synthesis of methyl 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylate (6a)
Methyl 1-methyl-4-nitro-1H-pyrrole-2-carboxylate 4b (0.40 g, 2.1733 mmol) was dissolved in THF (4 mL) and methanol (4 mL) and cooled to 0 °C. Then 10%-Pd/C (150 mg) was added portion-wise, while stirring. The flask was repeatedly evacuated and refilled with hydrogen gas, and the reaction was left stirring for 4 h. The Palladium on carbon was filtered through celite, and the solvent was then removed under reduced pressure to yield oily amine product 5a, which was then diluted with 2 mL THF and added dropwise to a solution of 1-Methyl-Imdazole-5-Carboxylic Acid 5c (0.274 g, 2.1733 mmol), triethylamine (1.305 g, 12.9 mmol), and HBTU (0.989 g, 2.608 mmol) in dry DMF (5 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, and a solution of sodium carbonate (15 mL) was added and left stirring for 10 min. The desired product was precipitated, filtered, and then washed with distilled water and dried under vacuum. Yield (60%); 1H NMR (ACETIC ACID-d4) δ 3.75 (3H, s, OCH3), 3.90 (3H, s, NCH3), 3.94 (3H, s, NCH3), 6.88 (1H, d, Ar—H), 7.45 (1H, d, Ar—H), 7.59 (1H, d, Ar—H), 7.65 (1H, d, Ar—H); 13C NMR (ACETIC ACID-d4): δ 24.43, 27.07, 41.53, 99.30, 99.36, 110.66, 111.82, 113.89, 123.00, 133.27, 148.66, 152.25; LC−MS (ESI): m/z calcd for C12H14N4O3, 262.11, found 263.11 [M + H]+.
2.8. Synthesis of methyl 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylic Acid (9)
Methyl 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylate 6a (0.280 g, 1.0697 mmol) was dissolved in THF (3 mL) and methanol (1 mL). Lithium hydroxide (0.1537 g, 6.4186 mmol) was dissolved in distilled water (2 mL) and added to 6a solution; the reaction was then left stirring for 4 h. The organic solvent was evaporated under reduced pressure, and the aqueous layer was acidified by using 1N HCl solution. The desired product was precipitated, filtered, and washed with distilled water; it was then dried under vacuum to obtain the desired product as an off-white solid. Yield (90%); 1H NMR (DMSO-d6) δ 3.82 (3H, s, NCH3), 3.98 (3H, s, NCH3), 6.98 (1H, d, Ar—H), 7.03 (1H, d, Ar—H), 7.38 (1H, d, Ar—H), 7.47 (1H, d, Ar—H), 10.46 (1H, s, COOH); 13C NMR (DMSO-d6): δ 35.10, 36.20, 99.53, 108.85, 119.81, 120.40, 121.91, 126.41, 127.02, 138.64, 156.09, 161.99; LC−MS (ESI): m/z calcd for C11H12N4O3, 248.09, found 249.04 [M + H]+.
2.9. Synthesis of methyl 1-methyl-4-(1,3-thiazole-4-amido)-1H-pyrrole-2-carboxylate (6b)
Methyl 1-methyl-4-nitro-1H-pyrrole-2-carboxylate (4b) (0.50 g, 2.7166 mmol) was dissolved in THF (4 mL) and LCMS methanol (4 mL) and cooled to 0 °C. Then 10%-Pd/C (200 mg) was added portion-wise, while stirring. The flask was repeatedly evacuated and refilled with H2 gas, and the reaction was left stirring for 4 h. The Palladium on carbon was filtered through celite, and the solvent was removed under reduced pressure to yield an oily amine product which was then diluted with 2 mL DCM and added dropwise to a solution of 4-Thizole-Carboxylic Acid 5b (0.280 g, 2.173 mmol,), triethylamine (1.042 g, 10.299 mmol), and HBTU (1.236 g, 3.26 mmol) in dry DCM (5 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, and a solution of sodium carbonate (15 mL) was added and left stirring for 10 min. The desired product precipitated, was filtered, and then was washed with distilled water and dried under vacuum to obtain the product as a white powder. Yield (65%); 1H NMR (CHLOROFORM-d) δ 3.83 (3H, s, OCH3), 3.94 (3H, s, NCH3), 6.85 (1H, d, Ar—H), 7.54 (1H, d, Ar—H), 8.24 (1H, d, Ar—H), 8.81 (1H, d, Ar—H), 9.10 (1H, s, CONH); 13C NMR (CHLOROFORM-d): δ 36.86, 51.14, 108.20, 120.03, 121.02, 121.22, 123.35, 150.80, 152.79, 157.91, 161.48; LC−MS (ESI): m/z calcd for C11H11N3O3S, 265.15, found 266.18 [M + H]+.
2.10. Synthesis of methyl 1-methyl-4-(1,3-thiazole-4-amido)-1H-pyrrole-2-carboxylic Acid (10)
Methyl 1-methyl-4-(1,3-thiazole-4-amido)-1H-pyrrole-2-carboxylate 6b (0.223 g, 9.303 mmol) was dissolved in Tetrahydrofuran (3 mL) and HPLC-grade methanol (1 mL). Lithium Hydroxide (0.223 g, 9.30 mmol) was dissolved in distilled water (2 mL) and added to 6b solution, and the reaction was left stirring for 4 h. The organic solvents were evaporated under reduced pressure, and the aqueous layer was acidified by using 1N HCl solution. The desired product was precipitated, filtered, and washed with distilled water; it was then dried under vacuum to obtain the desired product as an off-white powder. Yield (85%); 1H NMR (DMSO-d6) δ 3.83 (3H, s, NCH3), 7.02 (1H, d, Ar—H), 7.50 (1H, d, Ar—H), 8.39 (1H, d, Ar—H), 9.23 (1H, d, Ar—H), 10.49 (1H, s, COOH), 12.21 (1H, s, CONH); 13C NMR (DMSO-d6): δ 25.13, 36.23, 109.04, 120.61, 121.99, 124.58, 150.71, 154.93, 157.76, 161.97; LC−MS (ESI): m/z calcd for C10H9N3O3S, 251.04, found 251.89 [M + H]+.
2.11. Synthesis of methyl 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylic Acid (4a)
Methyl 1-methyl-4-nitro-1H-pyrrole-2-carboxylate 4b (5.0 g, 27.16 mmol) was dissolved in THF (10 mL). Sodium hydroxide (4.5 g, 112.5 mmol) was dissolved in distilled water (15 mL) and added to 4b solution, and the reaction was left stirring overnight. The organic solvent was evaporated under reduced pressure, and the aqueous layer was acidified by using 1N HCl solution. The desired product was precipitated, filtered, and washed with distilled water; it was then dried under vacuum to obtain the desired product as a white solid. Yield (81%); 1H NMR (DMSO-d6) δ 3.91 (3H, s, NCH3), 7.26 (1H, d, Ar—H), 8.23 (1H, d, Ar—H); 13C NMR (DMSO-d6): δ 37.50, 111.41, 123.81, 129.21, 134.02, 160.99; LC−MS (ESI): m/z calcd for C6H6N2O4, 170.03, found 169.01 [M − H]−.
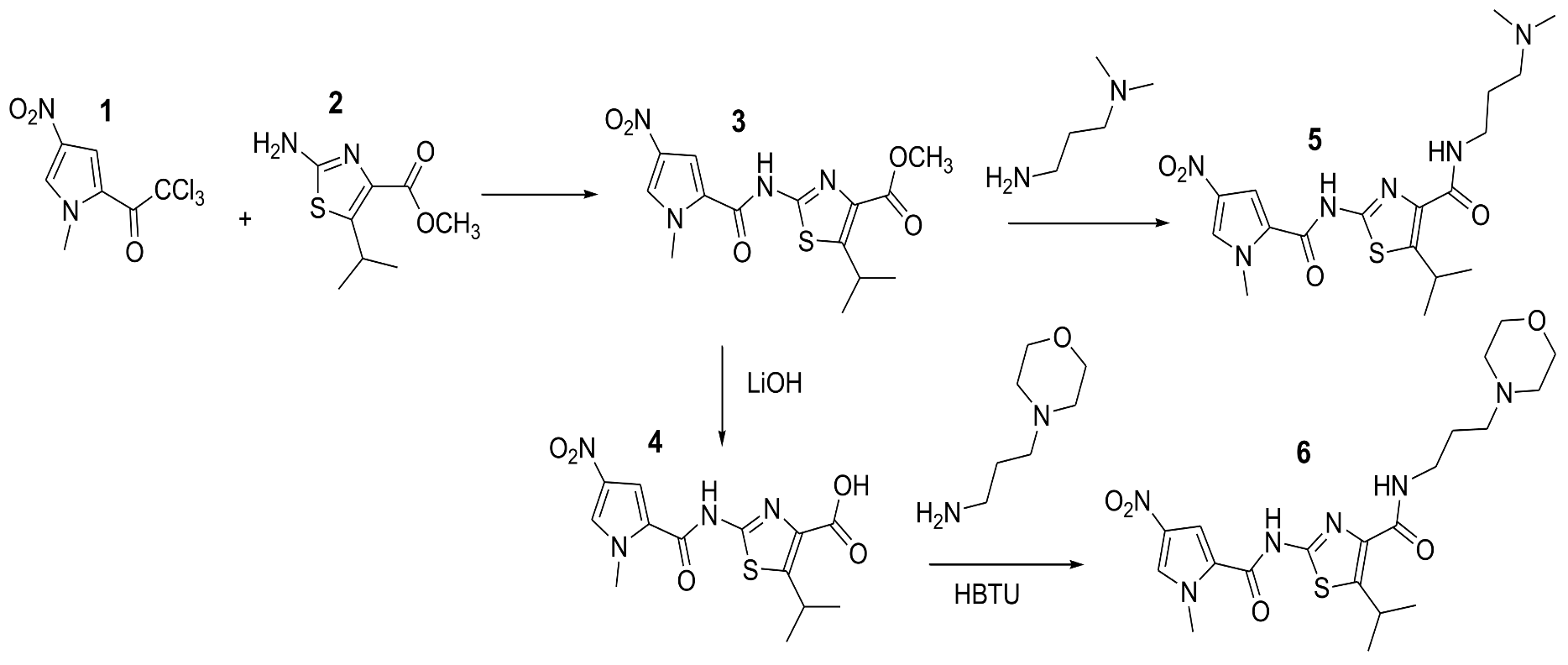
2.12. Synthesis of methyl 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-5-isopropylthiazole-4-carboxylate (3)
Methhyl-2-amino-5-isopropyl thiazole-4-carboxylate 2 (0.400 g, 1.9994 mmol) was dissolved in anhydrous DCM (3–4 mL) and TEA (0.607 g, 5.998 mmol). This solution was added dropwise to a solution of N-Methyl-4-Nitro-2-trichloroacetylpyrrole 1 (0.404 g, 1.499 mmol) dissolved in anhydrous DCM (3 mL), and the reaction was left stirring under nitrogen gas for 1 h. The solvent was removed under reduced pressure, a solution of sodium carbonate (10 mL) was added, and the product was then filtered and washed with 40:60 MeOH:water and dried under reduced pressure. Yield (70%); 1H NMR (CHOLOROFORM-d) δ 1.38 (6H, d, CH3), 3.89 (3H, s, OCH3), 4.09 (3H, d, NCH3), 4.14 (1H, m, CH), 7.30 (1H, d, AR—H), 7.68 (1H, d, Ar—H); 13C NMR (CHLOROFORM-d): δ 24.85, 27.79, 38.52, 52.41, 100.13, 109.95, 123.69, 128.58, 135.69, 153.30, 154.46, 157.85, 161.83; LC−MS (ESI): m/z calcd for C14H16N4O4S, 352.08, found 353.02 [M + H]+.
2.13. Synthesis of 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-5-isopropylthiazole-4-carboxylic Acid (4)
Methyl 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-5-isopropylthiazole-4-carboxylate 3 (3.8 g, 10.793 mmol) was dissolved in THF (10 mL). Lithium hydroxide (1.551 g, 64.758 mmol) was dissolved in distilled water (6 mL) and added to 4b solution, and the reaction was left stirring for 6 h. The organic solvent was evaporated under reduced pressure, and the aqueous layer was acidified by using 1N HCl solution. The desired product was precipitated, filtered, and washed with distilled water; it was then dried under vacuum to obtain the desired product as white solid. 1H NMR (DMSO-d6) δ 1.28 (6H, d, CH3), 3.98 (3H, s, NCH3), 4.06 (1H, m, CH), 7.98 (1H, s, AR—H), 8.28 (1H, s, Ar—H); 13C NMR (DMSO-d6): δ 24.72, 26.90, 37.99, 110.44, 123.88, 129.73, 134.18, 134.92, 149.92, 153.42, 158.34, 163.54; (LC−MS (ESI): m/z calcd for C13H14N4O5S, 338.70, found 339.23 [M + H]+.
2.14. Synthesis of 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-N(3(dimethylamino)propyl)-5-isopropylthiazole-4-carboxamide (5)
First, 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-5-isopropylthiazole-4-carboxylate 3 (0.402 g, 1.163 mmol) was dissolved in 4 mL of dimethylaminopropylamine, and the reaction was left stirring overnight at 90 °C. The solvent was removed under reduced pressure, and basic water was added; the crude product was then precipitated, collected by filtration, and purified by silica column chromatography, using 1:98:1 MeOH/EA/TEA as mobile phase. Yield (90%); 1H NMR (CHOLOROFORM-d) δ 1.33 (6H, d, CH3), 1.8 (2H, m, CH2), 2.26 (6H, s, NCH3), 2.41 (2H, t, NCH2), 3.45 (2H, q, CONH--CH2), 4.10 (3H, d, NCH3), 4.41 (1H, m, CH), 7.55 (1H, t, CONH), 7.64 (1H, d, Ar—H), 7.70 (1H, d, Ar—H); 13C NMR (CHLOROFORM-d): δ 25.12, 27.23, 27.47, 37.71, 38.59, 45.61, 57.56, 109.71, 124.19, 128.38, 135.55, 136.07, 148.99, 152.78, 157.92, 162.86; LC−MS (ESI): m/z calcd for C18H26N6O4S, 422.17, found 423.08 [M + H]+.
2.15. Synthesis of N-(3-(dimethylamino)propyl)-5-isopropyl-2-(1-methyl-4-(1-methyl-4-(1-methyl-1H-imidazole-2-carboxamido)-1H-pyrrole-2-carboxamido)-1H-pyrrole-2-carboxamido)thiazole-4-carboxamide (MGB3)
First, 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-N-(3-(dimethylamino)propyl)-5-isopropylthiazole-4-carboxamide (0.1593 g, 0.3773 mmol) was dissolved in methanol (2 mL) and THF (4 mL), and the solution then cooled to 0 °C. Then 10%-Pd/C (0.08 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was stirred under hydrogen for 4 h. The suspension was then filtered, and the solvent was removed under reduced pressure to produce an oily amine product which was then diluted with 2 mL DMF and added dropwise to a solution of 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylic acid (0.078 mg, 0.31440 mmol), triethylamine (0.191 g, 1.8864 mmol), and HBTU (0.1431 g, 0.3773 mmol) in DMF (5 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, sodium bicarbonate solution (10 mL) was added, and the crude product was extracted twice (2 × 15 mL) with ethylacetate. The organic solvent was evaporated under reduced pressure, and the crude product was purified by reverse-phase HPLC, using a gradient elution method as described below. The product peaks were collected and freeze-dried to obtain the desired product in the form of a white solid. Yield (35%); 1H NMR (ACETIC ACID-d4) δ 1.32 (6H, d, CH3), 2.12 (2H, m, CH2), 3.02 (6H, s, NCH3), 3.31 (2H, t, NCH2), 3.49 (2H, q, CONH--CH2), 3.97 (3H, s, NCH3), 4.00 (3H, s, NCH3), 4.11 (3H, s, NCH3), 4.34 (1H, m, Ar—CH), 7.07 (1H, d, Ar—H), 7.09 (1H, d, Ar—H), 7.33 (1H, d, Ar—H), 7.35 (1H, d, Ar—H), 7.39 (1H, d, Ar—H), 7.49 (1H, d, Ar—H); 13C NMR (ACETIC ACID-d4): δ 15.30, 17.71, 26.28, 26.98, 27.34, 33.33, 46.07, 95.08, 97.47, 97.53, 109.76, 112.41, 114.43, 117.22, 126.70, 129.49, 137.79, 146.05, 149.83; LC−MS (ESI): m/z calcd for C29H38N10O4S, 622.28, found 623.27 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 3 | 75 | 25 |
| 25 | 3 | 0 | 100 |
2.16. Synthesis of N-(3-(dimethylamino)propyl)-5-isopropyl-2-(1-methyl-4-(1-methyl-4-(thiazole-4-carboxamido)-1H-pyrrole-2-carboxamido)-1H-pyrrole-2-carboxamido)thiazole-4-carboxamide (MGB4)
First, 2-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-N-(3-(dimethylamino)propyl)-5-isopropylthiazole-4-carboxamide (0.1593 g, 0.37728 mmol) was dissolved in methanol (2 mL) and THF (4 mL), and the solution then cooled to 0 °C. Then 10%-Pd/C (0.08 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was then stirred under hydrogen for 4 h. The suspension was filtered, and the solvent was removed under reduced pressure to produce an oily amine product which was then diluted with 2 mL DMF and added dropwise to a solution of 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylic acid (0.078 g, 0.31440 mmol), triethylamine (0.191 g, 1.8864 mmol), and HBTU (0.1431 mg, 0.37728 mmol) in DMF (5 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, sodium bicarbonate solution (10 mL) was added, and the crude product was extracted twice (2 × 15 mL) with ethyl acetate. The organic solvent was evaporated under reduced pressure, and the crude product was purified by reverse-phase HPLC, using a gradient elution method as described below. The product peaks were collected and freeze-dried to obtain the desired product in the form of a white solid. Yield (40%); 1H NMR (ACETIC ACID-d4) δ 1.32 (6H, d, CH3), 2.12 (2H, m, CH2), 3.03 (6H, s, NCH3), 3.32 (2H, t, NCH2), 3.49 (2H, q, CONH—CH2), 3.97 (3H, s, NCH3), 3.99 (3H, s, NCH3), 4.34 (1H, m, Ar—CH), 7.15 (1H, d, Ar—H), 7.41 (1H, d, Ar—H), 7.43 (1H, d, Ar—H), 7.48 (1H, d, Ar—H), 8.32 (1H, d, Ar—H), 9.10 (1H, d, Ar—H), 9.52 (1H, s, CONH), 9.68 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ 15.14, 17.56, 26.04, 26.79, 27.15, 36.89, 45.93, 95.13, 97.42, 106.04, 108.35, 111.35, 112.65, 114.33, 126.47, 137.70, 142.24, 144.95, 148.55, 149.70, 149.78, 150.32, 154.38; LC−MS (ESI): m/z calcd for C28H35N9O4S2, 625.23, found 626.23 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 3 | 75 | 25 |
| 25 | 3 | 5 | 95 |
2.17. Synthesis of N-(5-((5-((5-((3-(dimethylamino)propyl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)carbamoyl)-1-methyl-1H-pyrrol-3-yl)-1-methyl-1H-imidazole-2-carboxamide (MGB5)
N-[3-(dimethylamino)propyl]-1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl] amino}-1H-pyrrole-2-carboxamide (0.1819 g, 0.4836 mmol) was dissolved in methanol (2 mL) and THF (4 mL), and the solution then cooled to 0 °C. Then 10%-Pd/C (0.095 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was stirred under hydrogen for 4 h. The suspension was then filtered, and the solvent was removed under reduced pressure to produce an oily amine product, which was diluted with 2 mL DMF and added dropwise to a solution of methyl 1-methyl-4-(1-methyl-1H-imidazole-5-amido)-1H-pyrrole-2-carboxylic acid 9 (0.100 g, 0.4031 mmol), triethylamine (0.2447 g, 2.4184 mmol), and HBTU (0.1834 g, 0.4836 mmol) in DMF (2 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, sodium bicarbonate solution (10 mL) was added, and the crude product was extracted twice (2 × 15 mL) with ethyl acetate. The organic solvent was evaporated under reduced pressure, and the crude product was purified by reverse-phase HPLC, using a gradient elution method, as described below. The product peaks were collected and freeze-dried to obtain the desired product as a yellow solid. Yield (41%); 1H NMR (ACETIC ACID-d4) δ 2.11 (2H, m, CH2), 3.04 (6H, s, NCH3), 3.33 (2H, t, NCH2), 3.36 (6H, s, NCH3), 3.49 (2H, q, CONH—CH2), 3.90 (3H, s, NCH3), 3.93 (3H, s, NCH3), 3.96 (3H, s, NCH3), 4.13 (1H, s, NH) 6.93 (1H, d, Ar—H), 6.97 (1H, d, Ar—H), 7.04 (1H, d, Ar—H), 7.23 (1H, d, Ar—H), 7.26 (1H, d, Ar—H), 7.34 (1H, d, Ar—H), 7.83 (1H, s, CONH), 9.26 (1H, s, CONH), 9.34 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ 16.21, 20.81, 20.90, 26.30, 26.54, 26.92, 27.01, 27.10, 33.40, 46.02, 95.02, 95.38, 109.79, 112.63, 113.78, 113.95, 114.32, 114.77, 116.53, 117.42, 145.87, 149.78, 149.92, 154.01; LC−MS (ESI): m/z calcd for C27H38N10O4, 576.29, found 577.33 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 3 | 90 | 10 |
| 25 | 3 | 5 | 95 |
2.18. Synthesis of N-(5-((5-((5-((3-(dimethylamino)propyl]carbamoyl)-1-methyl-1H-pyrrol-3-yl)carbamoyl)-1-methyl-1H-pyrrol-3-yl}carbamoyl)-1-methyl-1H-pyrrol-3-yl)-1,3-thiazole-4-carboxamide (MGB6)
N-[3-(dimethylamino)propyl]-1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl] amino}-1H-pyrrole-2-carboxamide 7 (0.2823 g, 0.750 mmol) was dissolved in methanol (2 mL) and THF (4 mL), and the solution was then cooled to 0 °C. Then 10%-Pd/C (0.141 g) was added in small portions, and the flask was repeatedly evacuated and refilled with hydrogen gas; the suspension was stirred under hydrogen for 4 h. The suspension was then filtered, and the solvent was removed under reduced pressure to produce an oily amine product, which was diluted with 2 mL DMF and added dropwise to a solution of methyl 1-methyl-4-(1,3-thiazole-4-amido)-1H-pyrrole-2-carboxylic acid 10 (0.157 g, 0.6254 mmol), triethylamine (0.3830 g, 3.785 mmol), and HBTU (0.2846 g, 0.750 mmol) in DMF (2 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, sodium bicarbonate solution (10 mL) was added, and the crude product was extracted twice (2 × 15 mL) with ethyl acetate. The organic solvent was evaporated under reduced pressure, and the crude product was purified by reverse-phase HPLC, using a gradient elution method, as described below. The product peaks were collected and freeze-dried to obtain the desired product as a white solid. Yield (31%); 1H NMR (ACETIC ACID-d4) δ 2.08 (2H, m, CH2), 2.99 (6H, s, NCH3), 3.30 (2H, t, NCH2), 3.45 (2H, m, CONH--CH2), 3.89 (3H, s, NCH3), 3.94 (3H, s, NCH3), 3.97 (3H, s, NCH3), 6.89 (1H, s, Ar—H), 6.97 (1H, s, Ar—H), 7.09 (1H, d, Ar—H), 7.15 (1H, d, Ar—H), 7.23 (1H, s, Ar—H), 7.27 (1H, d, Ar—H), 7.39 (1H, s, Ar—H), 7.41 (1H, s, Ar—H), 8.32 (1H, s, CONH), 8.34 (1H, s, CONH), 9.10 (1H, s, CONH), 9.11 (1H, s, CONH); (CHLOROFORM-d): δ 25.19, 36.62, 36.78, 36.97, 34.22, 104.10, 104.37, 104.50, 111.11, 118.46, 119.69, 121.14, 122.06, 123.43, 123.52, 124.54, 125.10, 127.76, 132.23, 143.48, 150.88, 153.11, 158.33, 159.17, 162.72; LC−MS (ESI): m/z calcd for C27H33N9O4S, 579.24, found 580.32 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 3 | 90 | 10 |
| 25 | 3 | 5 | 95 |
2.19. General Procedure for the Synthesis of MGB16, MGB20, MGB22, MGB24, MGB26, MGB28, MGB30, and MGB32
A variety of carboxylic acid compounds (stated below) were coupled with N-[3-(dimethylamino)propyl]-1-methyl-4-{[(1-methyl-4-nitro-1H-pyrrol-2-yl)carbonyl] amino}-1H-pyrrole-2-carboxamide 7 or 1-methyl-4-(1-methyl-4-nitro-1H-pyrrole-2-carboxamido)-N-(3-morpholinopropyl)-1H-pyrrole-2 carboxamide 8 to generate the final product. Equimolar amounts of 7 or 8 were dissolved in methanol (2 mL) and THF (4 mL), and the solution then cooled to 0 °C. Then 10%-Pd/C (0.1 g) was added in small portions, the flask was repeatedly evacuated and refilled with hydrogen gas, and the suspension was stirred under hydrogen for 4 h. The suspension was then filtered, and the solvent was removed under reduced pressure to produce an oily amine product which was then diluted with 1 mL DMF and added dropwise to a solution containing equimolar amount the acid, triethylamine (6 equivalent), and HBTU (1.1 equivalent) in DMF (1 mL) and left stirring overnight, at room temperature, under nitrogen gas. The solvent was removed under reduced pressure, sodium bicarbonate solution (10 mL) was added, and the crude product was extracted twice (2 × 15 mL) with ethyl acetate. The organic solvent was evaporated under reduced pressure, and the crude product was purified by reverse-phase HPLC, using a gradient elution method, as described below. The product peaks were collected and freeze-dried to obtain the desired product as solid TFA salts.
2.20. Synthesis of 1-methyl-N-(1-methyl-5-{[3-(morpholin-4-yl)propyl]carbamoyl}-1H-pyrrol-3-yl)-4-(2-oxo-2H-chromene-3-amido)-1H-pyrrole-2-carboxamide (MGB16)
Reactants: coumarin-3-carboxylic acid and 8. Purified by silica column chromatography. Yield (55%); 1H NMR (ACETIC ACID-d4) δ 2.18 (2H, m, CH2), 3.32 (4H, t, NCH2), 3.43 (2H, m, NCH2), 3.59 (2H, q, CONH--CH2), 3.75 (4H, d, OCH2), 3.93 (3H, s, NCH3), 3.98 (3H, s, NCH3), 6.95 (1H, d, Ar—H), 7.04 (1H, d, Ar—H), 7.30 (1H, d, Ar—H), 7.44 (1H, d, Ar—H), 7.49 (1H, s, Ar—H), 7.50 (1H, d, Ar—H), 7.81 (1H, t, Ar—H), 8.00 (1H, d, Ar—H), 8.08 (1H, t, Ar—H), 8.98 (1H, s, CONH), 9.26 (1H, s, CONH), 10.57 (1H, s, CONH); 13C NMR (DMSO-d6): δ 8.48, 30.54, 35.89, 36.11, 45.61, 51.05, 54.05, 63.37, 104.27, 116.08, 118.41, 119.24, 122.44, 123.08, 125.15, 130.10, 134.02, 146.86, 153.66, 175.95, 158.04, 160.58, 161.49; LC−MS (ESI): m/z calcd for C29H32N6O6, 560.24, found 561.27 [M + H]+.
2.21. Synthesis of 1-methyl-4-(1-methyl-4-(3-(naphthalen-2-ylthio)propanamido)-1H-pyrrole-2-carboxamido)-N-(3-morpholinopropyl)-1H-pyrrole-2-carboxamide (MGB20)
Reactants: coumarin-3-carboxylic acid and 8. Yield (31%); 1H NMR (ACETIC ACID-d4) δ, 2.14 (2H, m, CH2), 2.76 (2H, t, SCH2), 3.22 (4H, t, NCH2), 3.35 (2H, t, NCH2), 3.39 (2H, t, NHCO--CH2), 3.49 (2H, q, CONH--CH2), 3.66 (4H, t, OCH2), 3.90 (3H, s, NCH3), 3.90 (3H, s, NCH3), 6.80 (1H, d, Ar—H), 6.91 (1H, s, Ar—H), 7.13 (1H, d, Ar—H), 7.26 (1H, s, Ar—H), 7.46 (1H, m, Ar—H), 7.48 (1H, d, Ar—H), 7.51 (1H, t, Ar—H), 7.86 (4H, q, Ar—H), 9.11 (1H, s, CONH), 9.20 (1H, s, CONH); 13C NMR (DMSO-d6): δ 8.48, 30.54, 35.89, 36.11, 45.61, 51.05, 54.05, 63.37, 104.27, 116.08, 118.41, 119.24, 120.62, 123.08, 125.15, 130.10, 134.02, 146.86, 153.66, 175.95, 158.04, 160.58, 161.49; LC−MS (ESI): m/z calcd for C32H38N6O4S, 602.27, found 603.29 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 3 | 75 | 25 |
| 25 | 3 | 25 | 75 |
2.22. Synthesis of N-(3-(dimethylamino)propyl)-1-methyl-4-(1-methyl-4-(3-((4-methylbenzyl)thio)propanamido)-1H-pyrrole-2-carboxamido)-1H-pyrrole-2-carboxamide (MGB22)
Reactants: 4-Methylbenzyl-3-thio-propionic acid and 7. Yield (55%); 1H NMR (ACETIC ACID-d4) δ 2.11 (2H, m, CH2), 2.28 (3H, s, CH3), 2.54 (2H, t, SCH2), 2.71 (2H, t, NHCO—CH2), 3.33 (2H, s, NCH2), 3.49 (2H, q, CONH--CH2), 3.72 (2H, s, SCH2—Ar), 3.89 (3H, s, NCH3), 3.90 (3H, s, NCH3), 6.81 (1H, s, Ar—H), 6.91 (1H, s, Ar—H), 7.10 (1H, s, Ar—H), 7.12 (1H, s, Ar—H), 7.21 (2H, d, Ar—H), 7.24 (1H, s, Ar--H), 7.84 (1H, s, CONH), 9.04 (1H, d, CONH), 9.19 (1H, d, CONH); 13C NMR (ACETIC ACID-d4): δ 11.22, 16.12, 17.83, 26.04, 26.32, 27.17, 33.29, 45.84, 94.44, 95.30, 113.32, 113.43, 114.14, 119.87, 119.99, 126.77, 127.27, 149.68, 154.01, 158.79; LC−MS (ESI): m/z calcd for C28H38N6O3S, 538.27, found 539.42 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 70 | 30 |
| 25 | 5 | 45 | 55 |
| 25.2 | 5 | 0 | 100 |
| 28 | 5 | 0 | 100 |
| 28.2 | 5 | 70 | 30 |
| 32 | 5 | 70 | 30 |
2.23. Synthesis of 1-methyl-4-(1-methyl-4-(3-(p-tolylthio)propanamido)-1H-pyrrole-2-carboxamido)-N-(3-morpholinopropyl)-1H-pyrrole-2-carboxamide (MGB24)
Reactants: 4-Methylbenzyl-3-thio-propionic acid and 8. Yield (21%); 1H NMR (ACETIC ACID-d4) δ, 2.14 (2H, m, CH2), 2.28 (3H, s, CH3), 2.54 (2H, t, SCH2), 2.71 (2H, t, NHCO--CH2), 3.22 (2H, t, NCH2), 3.36 (2H, t, NCH2), 3.49 (2H, q, CONH--CH2), 3.67 (2H, d, NCH2), 3.72 (2H, s, SCH2—Ar), 3.89 (3H, s, NCH3), 3.90 (3H, s, NCH3), 3.94 (2H, d, OCH2), 4.08 (2H, d, OCH2), 6.80 (1H, s, Ar—H), 6.91 (1H, s, Ar—H), 7.10 (1H, s, Ar—H), 7.12 (1H, s, Ar—H), 7.21 (1H, s, Ar—H), 7.23 (1H, s, Ar—H), 7.25 (1H, s, Ar—H), 7.86 (1H, s, CONH), 9.04 (1H, s, CONH), 9.18 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ 11.07, 14.85, 17.67, 26.17, 26.60, 27.01, 27.06, 42.48, 45.05, 54.72, 94.30, 95.22, 108.98, 113.12, 113.26, 113.98, 119.72, 119.84, 119.98, 126.61, 127.12, 149.54, 153.87, 158.65; LC−MS (ESI): m/z calcd for C30H40N6O4S, 580.28, found 581.45 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 80 | 20 |
| 25 | 5 | 50 | 70 |
| 25.2 | 5 | 0 | 90 |
| 28 | 5 | 0 | 90 |
| 28.2 | 5 | 80 | 20 |
| 32 | 5 | 80 | 20 |
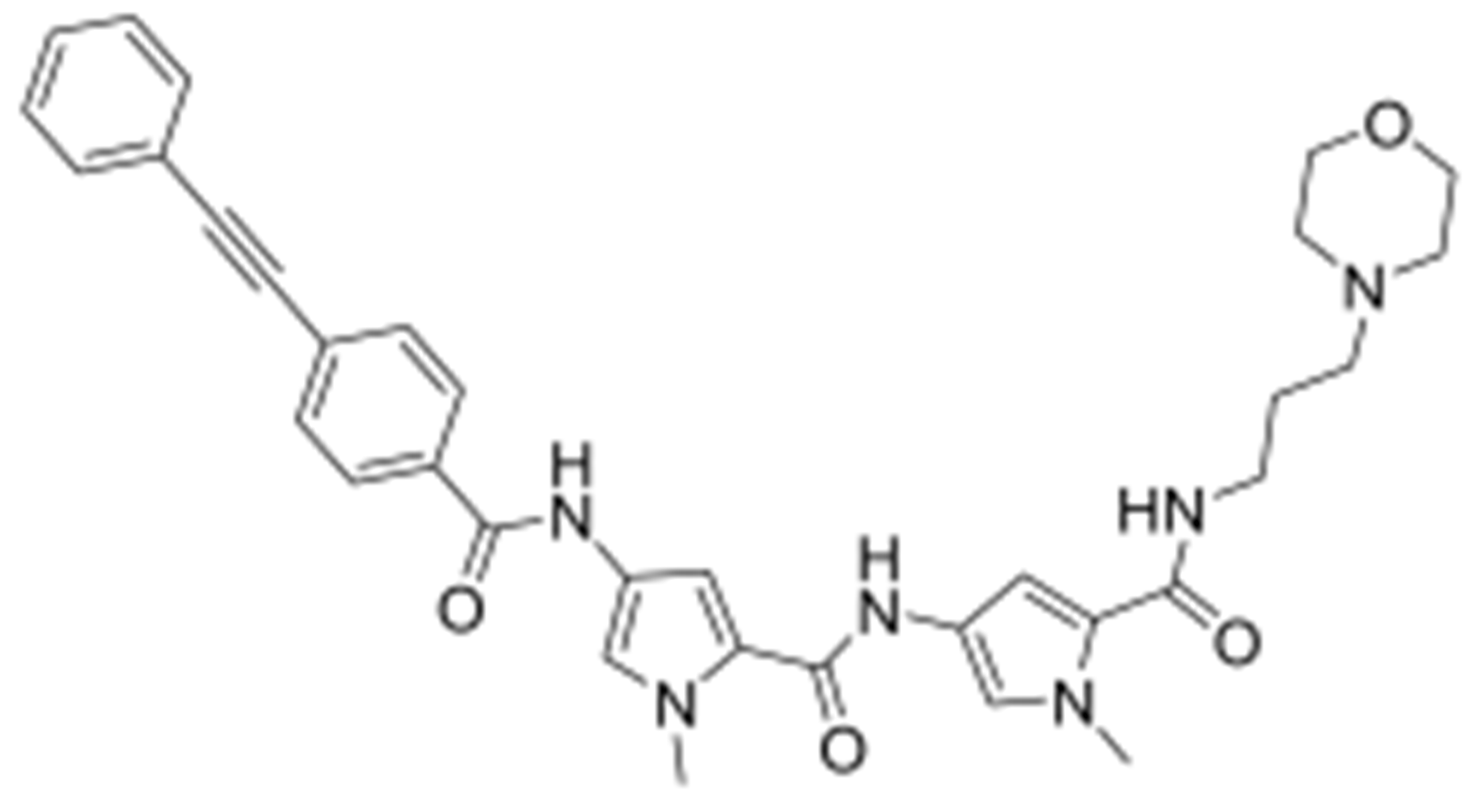
2.24. Synthesis of N-(3-(dimethylamino)propyl)-1-methyl-4-(1-methyl-4-(4(phenylethynyl)benzamido)-1H-pyrrole-2-carboxamido)-1H-pyrrole-2-carboxamide (MGB26)
Reactants: 4-(phenylethynyl)benzoic acid and 7. Yield (35%); 1H NMR (ACETIC ACID-d4) δ 2.94 (6H, s, NCH3), 3.27 (2H, t, NCH2), 3.45 (2H, q, CONH--CH2), 3.89 (3H, s, NCH3), 3.95 (3H, s, NCH3), 6.87 (1H, s, Ar—H), 7.02 (1H, s, Ar—H), 7.27 (1H, d, Ar—H), 7.34 (1H, d, Ar—H), 7.43 (2H, d, Ar—H), 7.44 (1H, d, Ar—H), 7.57 (2H, m, Ar--H), 7.64 (2H, d, Ar—H), 7.98 (2H, d, Ar—H), 9.25 (1H, s, CONH), 9.71 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ 15.94, 26.92, 27.03, 33.18, 46.04, 79.70, 82.37, 95.06, 95.18, 109.53, 109.94, 113.64, 113.79, 113.95, 117.05, 118.70, 119.86, 122.59, 122.75, 125.87, 149.84, 153.38, 154.10; LC−MS (ESI): m/z calcd for C32H34N6O3, 550.27, found 551.25 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 80 | 20 |
| 25 | 5 | 55 | 45 |
| 25.2 | 5 | 0 | 100 |
| 28 | 5 | 0 | 100 |
| 28.2 | 5 | 80 | 20 |
| 32 | 5 | 80 | 20 |
2.25. Synthesis of 1-methyl-4-(1-methyl-4-(4-(phenylethynyl)benzamido)-1H-pyrrole-2-carboxamido)-N-(3-morpholinopropyl)-1H-pyrrole-2-carboxamide (MGB28)
Reactants: 4-(phenylethynyl)benzoic acid and 8. Yield (46%); 1H NMR (ACETIC ACID-d4) δ 2.10 (2H, m, CH2), 3.16 (4H, s, NCH2), 3.29 (2H, t, NCH2), 3.46 (2H, q, CONH—CH2), 3.58 (2H, s, OCH2), 3.89 (3H, s, NCH3), 3.95 (3H, s, NCH3), 4.00 (2H, s, OCH2), 6.88 (1H, s, Ar—H), 7.01 (1H, s, Ar—H), 7.27 (1H, d, Ar—H), 7.33 (1H, s, Ar—H), 7.43 (2H, d, Ar—H), 7.44 (1H, d, Ar—H), 7.57 (2H, m, Ar--H), 7.64 (2H, d, Ar—H), 7.98 (2H, d, Ar—H), 9.25 (1H, s, CONH), 9.70 (1H, d, CONH); 13C NMR (ACETIC ACID-d4): δ10.99, 14.69, 26.49, 26.65, 26.69, 27.01, 42.41, 45.38, 54.55, 79.38, 82.07, 94.80, 94.85, 103.79, 109.29, 109.61, 113.32, 113.43, 113.63, 116.75, 118.38, 119.56, 122.29, 122.44, 125.55, 128.94, 149.53, 153.14, 153.79; LC−MS (ESI): m/z calcd for C34H36N6O4, 592.28, found 593.49 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 80 | 20 |
| 25 | 5 | 50 | 50 |
| 25.2 | 5 | 0 | 100 |
| 28 | 5 | 0 | 100 |
| 28.2 | 5 | 80 | 20 |
| 32 | 5 | 80 | 20 |
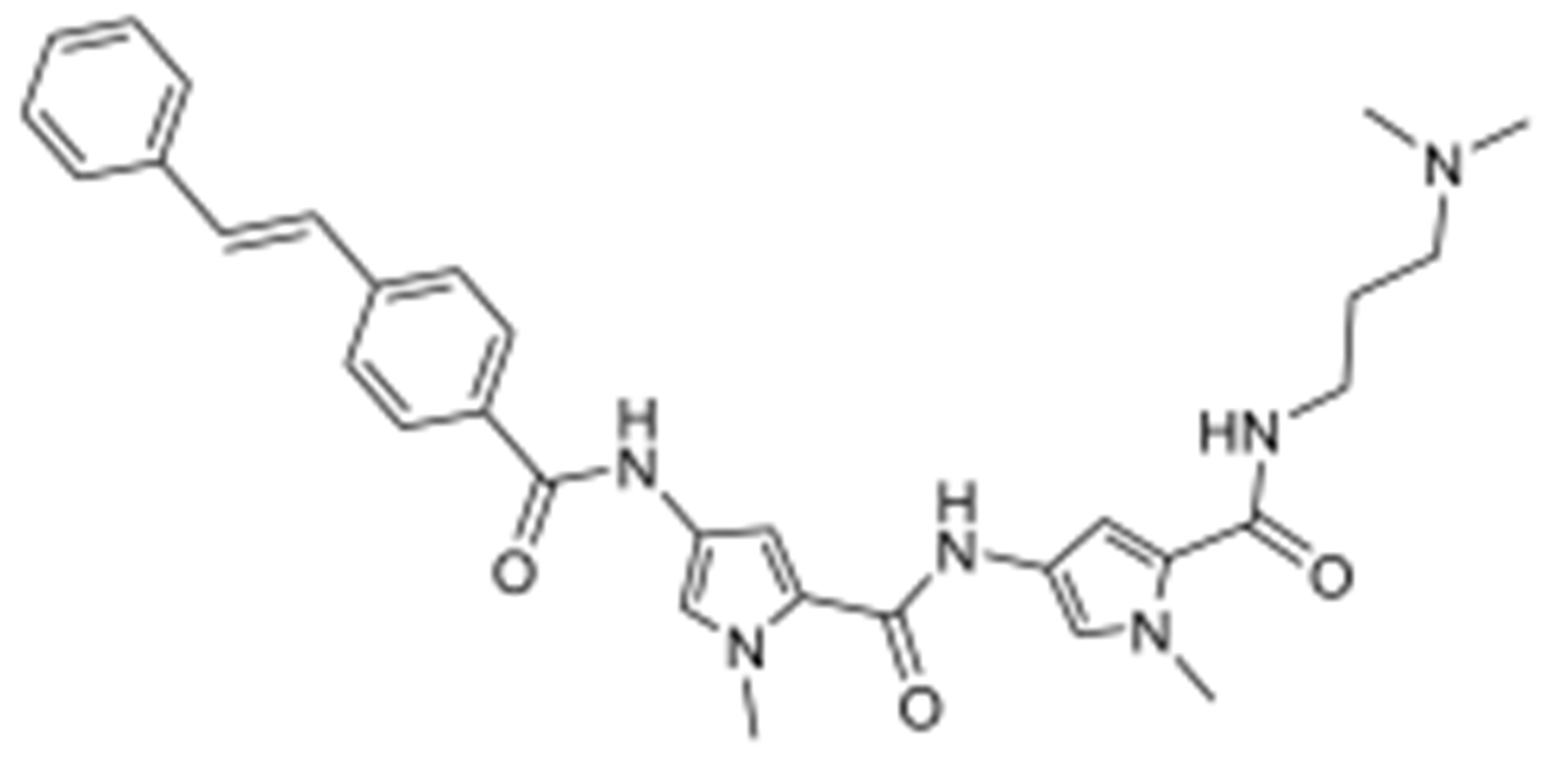
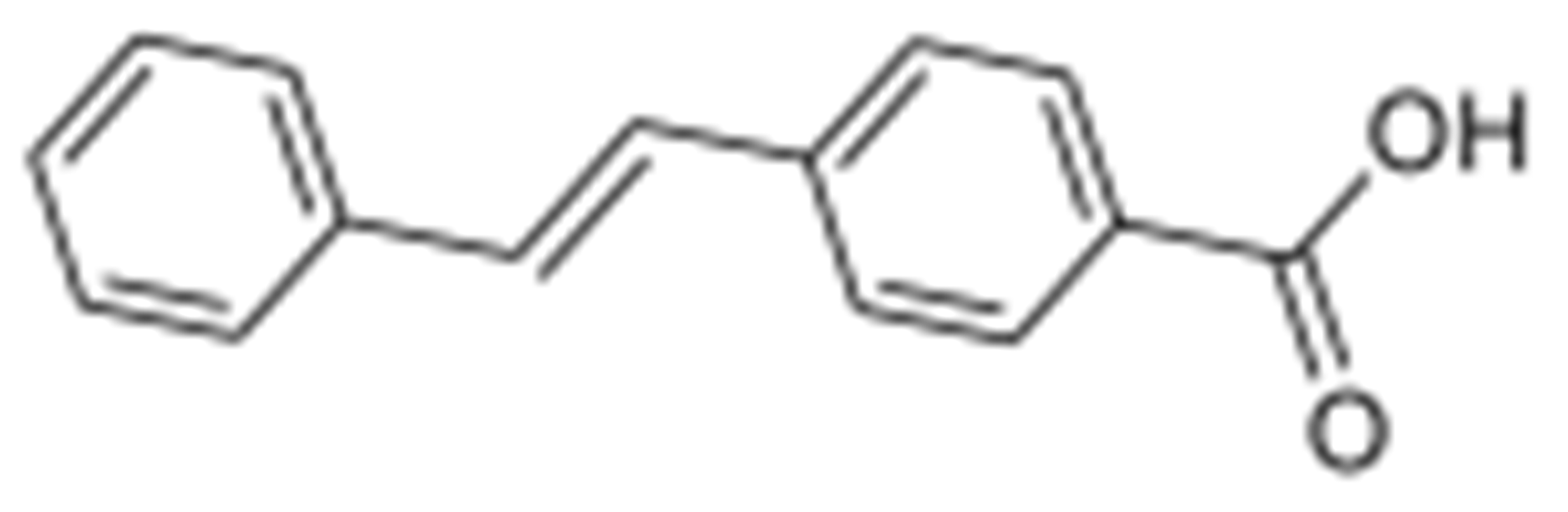
2.26. Synthesis of (E)-N-(3-(dimethylamino)propyl)-1-methyl-4-(1-methyl-4-(4-styrylbenzamido)-1H-pyrrole-2-carboxamido)-1H-pyrrole-2-carboxamide (MGB30)
Reactants: 4-stilbenecarboxylic acid and 7. Yield (48%); 1H NMR (ACETIC ACID-d4) δ 2.08 (2H, m, CH2), 2.95 (6H, s, NCH3), 3.26 (2H, t, NCH2), 3.44 (2H, q, CONH--CH2), 3.89 (3H, s, NCH3), 3.95 (3H, s, NCH3), 6.88 (1H, s, Ar—H), 7.02 (1H, s, Ar—H), 7.28 (2H, d, Ar—H), 7.30 (1H, d, Ar—H), 7.35 (2H, m, Ar—H), 7.37 (1H, s, HC = CH), 7.38 (1H, d, HC = CH), 7.63 (2H, d, Ar—H), 7.71 (2H, d, Ar—H), 7.97 (2H, d, Ar—H), 9.27 (1H, d, CONH), 9.67 (1H, d, CONH); 13C NMR (ACETIC ACID-d4): δ 15.65, 26.28, 26.62, 26.67, 32.89, 45.74, 94.79, 94.89, 113.46, 113.48, 114.20, 117.26, 117.61, 119.62, 121.29, 124.67, 128.06, 131.21, 149.57, 153.13, 154.22; LC−MS (ESI): m/z calcd for C32H36N6O3, 552.28, found 553.29 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 70 | 30 |
| 25 | 5 | 45 | 55 |
| 25.2 | 5 | 0 | 100 |
| 28 | 5 | 0 | 100 |
| 28.2 | 5 | 70 | 30 |
| 32 | 5 | 70 | 30 |
2.27. Synthesis of (E)-1-methyl-4-(1-methyl-4-(4-styrylbenzamido)-1H-pyrrole-2-carboxamido)-N-(3-morpholinopropyl)-1H-pyrrole-2-carboxamide (MGB32)
Reactants: 4-stilbenecarboxylic acid and 8. Yield (36%); 1H NMR (ACETIC ACID-d4) δ 2.11 (2H, m, CH2), 3.10 (4H, s, NCH2), 3.30 (2H, t, NCH2), 3.46 (2H, q, CONH—CH2), 3.59 (2H, s, OCH2), 3.89 (3H, s, NCH3), 3.95 (3H, s, NCH3), 4.01 (2H, s, OCH2), 6.88 (1H, s, Ar—H), 7.01 (1H, s, Ar—H), 7.27 (1H, s, Ar—H), 7.31 (1H, d, Ar—H), 7.33 (2H, s, Ar—H), 7.36 (1H, d, Ar—H), 7.39 (1H, s, HC = CH), 7.40 (1H, d, HC = CH), 7.62 (2H, d, Ar--H), 7.71 (2H, d, Ar—H), 7.97 (2H, d, Ar—H), 9.25 (1H, s, CONH), 9.60 (1H, s, CONH); 13C NMR (ACETIC ACID-d4): δ 14.84, 26.64, 26.81, 26.83, 42.57, 45.54, 54.70, 94.96, 95.00, 113.62, 113.63, 114.36, 117.42, 117.77, 119.78, 121.46, 124.83, 128.21, 131.38, 149.72, 153.31, 154.36; LC−MS (ESI): m/z calcd for C34H38N6O4, 594.30, found 595.44 [M + H]+.
| Time (min) | Flow Rate (mL/min) | % Water (0.1% TFA) | % Acetonitrile (0.1% TFA) |
| 0 | 5 | 70 | 30 |
| 25 | 5 | 45 | 55 |
| 25.2 | 5 | 0 | 100 |
| 28 | 5 | 0 | 100 |
| 28.2 | 5 | 70 | 30 |
| 32 | 5 | 70 | 30 |
2.28. Acanthamoeba Culturing
Acanthamoeba castellanii genotype T4 (ATCC 50492) was cultivated in 10 mL growth medium of peptone yeast glucose (PYG) in tissue flasks (Baig, et al., 2021; Anwar, et al., 2019). The growth medium, PYG, consists of 0.75% proteose peptone, 0.75% yeast extract, and 1.5% glucose. Next, the amoebae were kept in tissue flasks and incubated at 30 °C. After 48 h, the flasks were found to reach approximately 90% confluency, and the amoebae were utilized in experiments [
18].
2.29. Henrietta Lacks (HeLa) Cervical Cancer Cells
HeLa cells were acquired from American Type Culture Collection (ATCC) of identification ATCC CCL-2, Singapore. The cells are utilized to conduct cytotoxicity and cytopathogenicity assays [
19]. Through their cultivation in complete media, the cells were grown, and then they were placed in a 95% humidifying incubator at 37 °C and 5% CO
2. Complete medium comprises Roswell Park Memorial Media (RPMI), 1% penicillin–streptomycin, 1% minimum essential medium amino acids, 1% L-glutamine, and 10% fetal bovine serum (FBS) [
19].
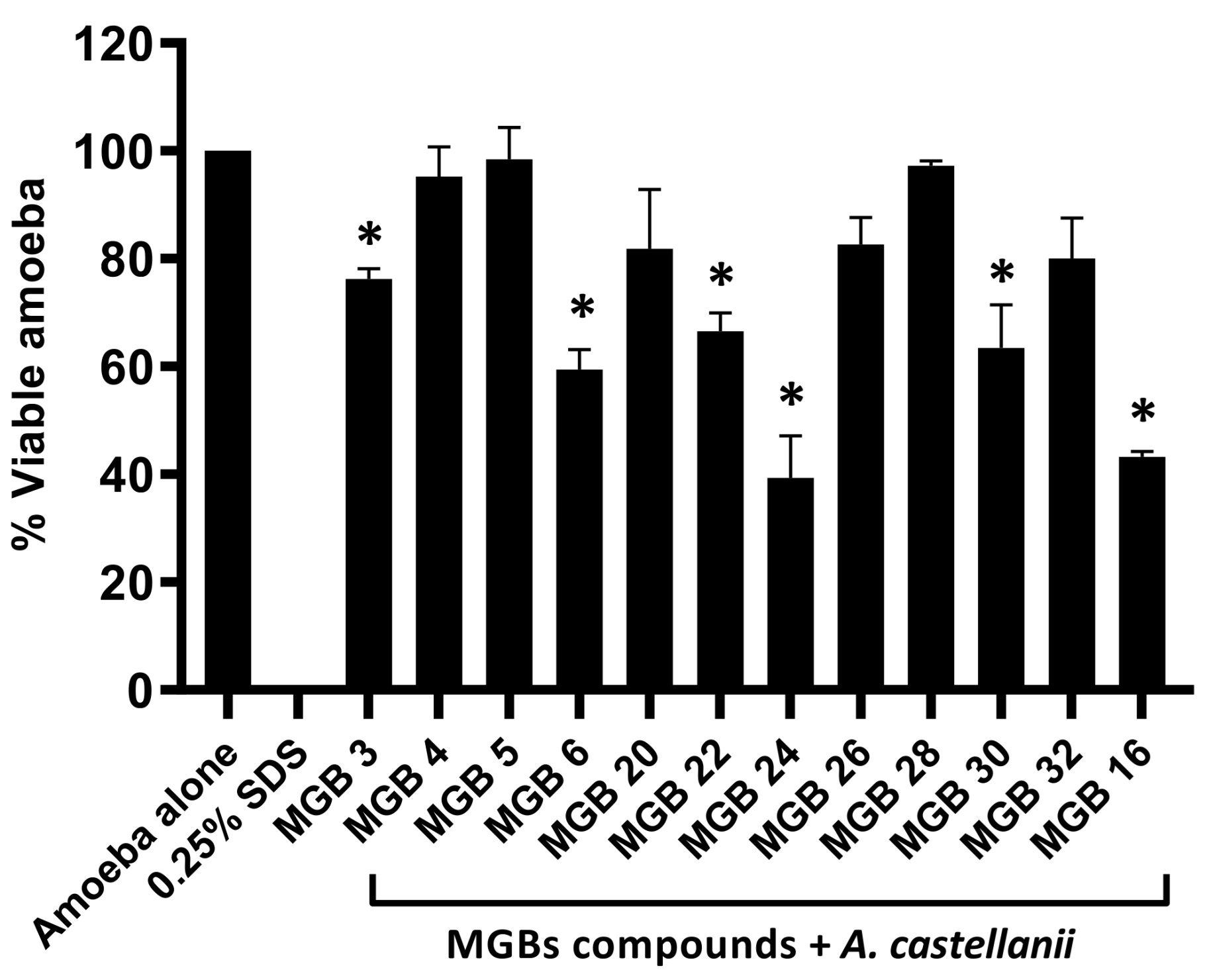
2.30. Amoebicidal Assay
To determine the compounds’ antiamoebic effects, amoebicidal assays were conducted. The 5 × 10
5 amoebae were placed in a 24-well plate and brought to a final volume of 500 µL. The amoebae were then treated with the compounds at 50 µM. The 24-well plate was then placed in a 30 °C incubator for 24 h. Positive and negative controls were added to the assay in order to ensure reliable results. Serving as the negative control was the amoebae alone, with RPMI [
4]. Meanwhile, amoebae, along with 0.25% sodium dodecyl sulphate (SDS), were used as the positive control. Through the addition of 0.1% methylene blue, live and dead amoebae were distinguished. Additionally, significant compounds were identified by conducting a Student’s
t-test with a two-tailed distribution [
19]. Furthermore, the MIC
50 values of three compounds were determined by using concentrations of 50, 75, and 100 µM.
2.31. Encystation Assay
The effects of the compounds on amoeba encystation were determined through the conduction of encystation assays. Briefly, 1 × 10
6 trophozoites were incubated with the compounds in a final concentration of 16% filter-sterilized glucose [
4]. Following a 48 h incubation period, 0.1% SDS was added per well for 20 min. The cysts were then counted by using a haemocytometer. To ensure accuracy,
Acanthamoeba castellanii was cultured in 16% glucose, serving as the negative control.
2.32. Excystation Assay
Amoeba culture was suspended in 3 mL phosphate-buffered saline (PBS) and placed on non-nutrient bacteriological agar plates to allow for the growing of
Acanthamoeba castellanii cysts [
4]. After two weeks, PBS was applied to plates containing amoeba cysts, which were then scraped off. The amoebae were centrifuged at 3000×
g for 10 min, and the resulting pellet was resuspended in RPMI. The 5 × 10
5 cysts were then incubated in PYG, along with 50 µM of the compounds. To ensure accuracy, amoeba cysts in PYG alone were considered as the negative control, whereas amoeba cysts in RPMI alone were taken as the positive control, as the cysts would not be able to revert back to trophozoites. The amoeba trophozoites were then numbered by using a haemocytometer.
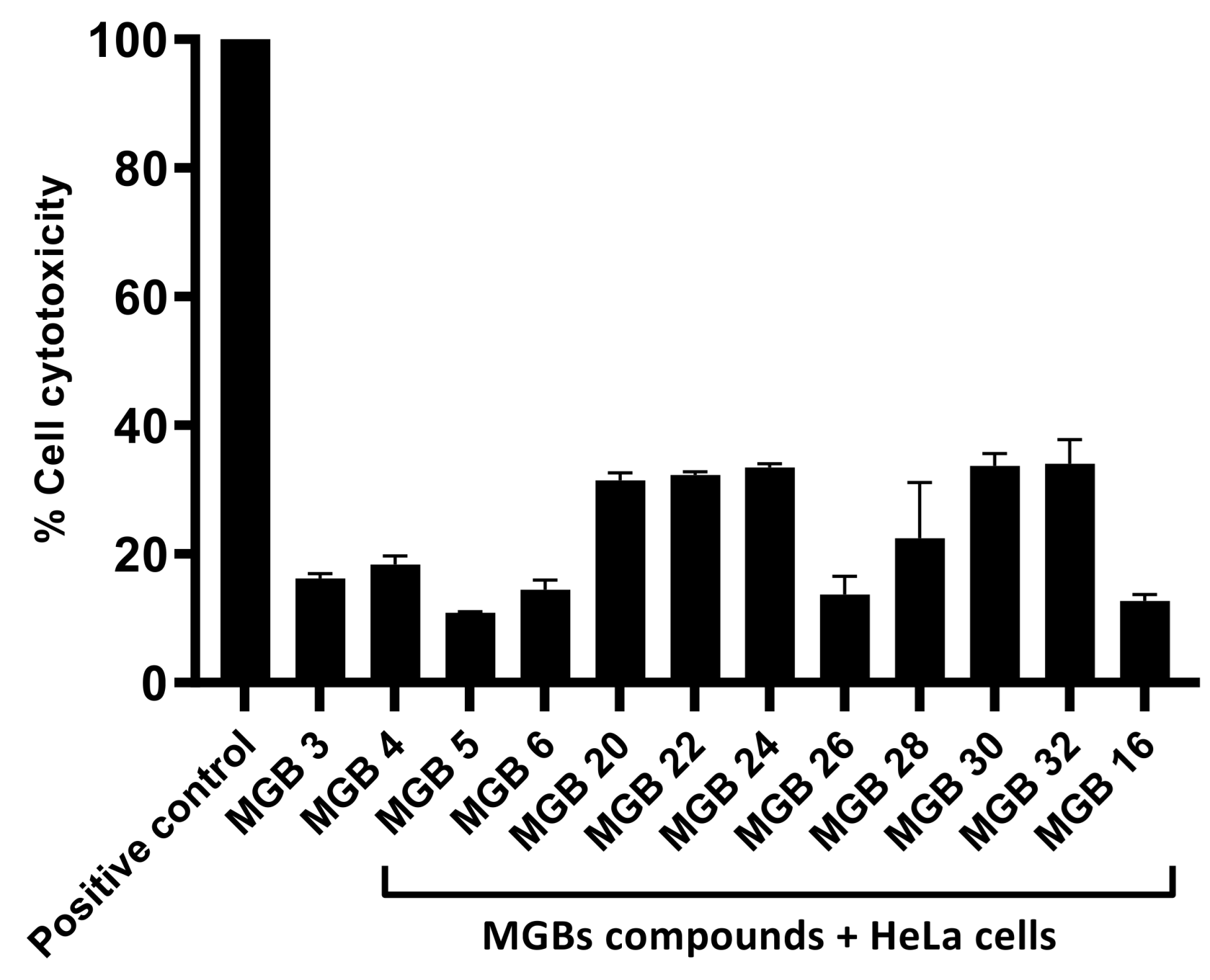
2.33. Cytotoxicity Assay
To determine the toxicity of the compounds against human HeLa cells, cytotoxicity assays were conducted. In short, HeLa cells were cultured in 96-well plates and treated with the compounds. The plate was then placed in a humidifying incubator with 95% humidity and 5% CO
2, at 37 °C, for 24 h [
19]. After 24 h, the supernatant was collected, and the cytotoxicity of the compounds was determined through a cytotoxicity detection kit that measured the quantity of lactate dehydrogenase (LDH) release [
20]. Accurate results were ensured through the addition of positive and negative controls. Untreated HeLa cells served as the negative control, whereas HeLa cells treated with 1% Triton X-100 served as the positive control. Finally, the cytotoxic properties were numerically determined by following the necessary calculations: (absorbance of media from cells treated with the drugs—absorbance of media from cells of negative control)/(absorbance of media from cells of positive control—absorbance of media from cells of negative control) × 100 [
19].
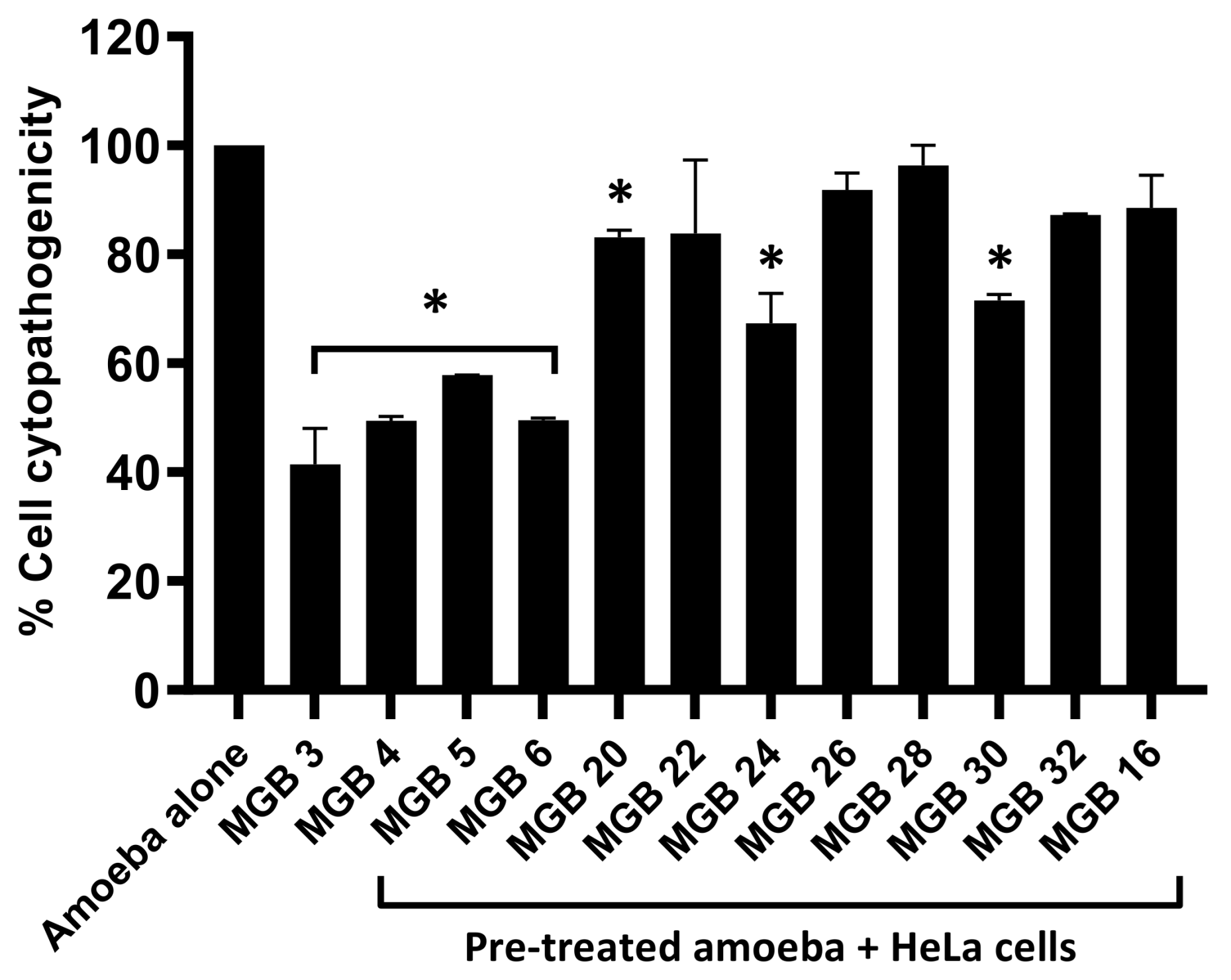
2.34. Cytopathogenicity Assay
Amoeba-mediated host-cell death was determined by treating 5 × 10
5 amoebae with 50 µM of compounds. The amoeba-containing plate was then placed in the incubator for 2 h at 30 °C. Following the 2 h incubation period, the treated amoebae were put on confluent HeLa cell monolayers [
19]. The plate containing the treated amoebae and HeLa cells was incubated for 24 h [
19]. Progressing the incubation period, the supernatant was collected, and the cytotoxicity was determined [
19]. Accurate results were ensured through the addition of negative and positive controls. The untreated amoebae, alone, on the cells served as the negative control, while the cells treated with Triton X-100 served as the positive control [
19].
2.35. Statistical Analysis
The data presented are illustrative of the mean ± standard error of at least three different independent experiments. A two-tailed distribution
t-test was conducted to determine the statistical significance of the results. Additionally,
p-values were determined to further examine and elaborate the results [
19].


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}