
Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the ‘Maximum Tolerable Dose’


Abstract
:1. Introduction
2. How PK/PD Is Currently Used to Optimize Dosing of Beta-Lactam Antibiotics in the Critically Ill
3. Why We Need to Rethink the Use of Prolonged Infusion of Beta-Lactam Antibiotics to Improve the Outcome of Infection
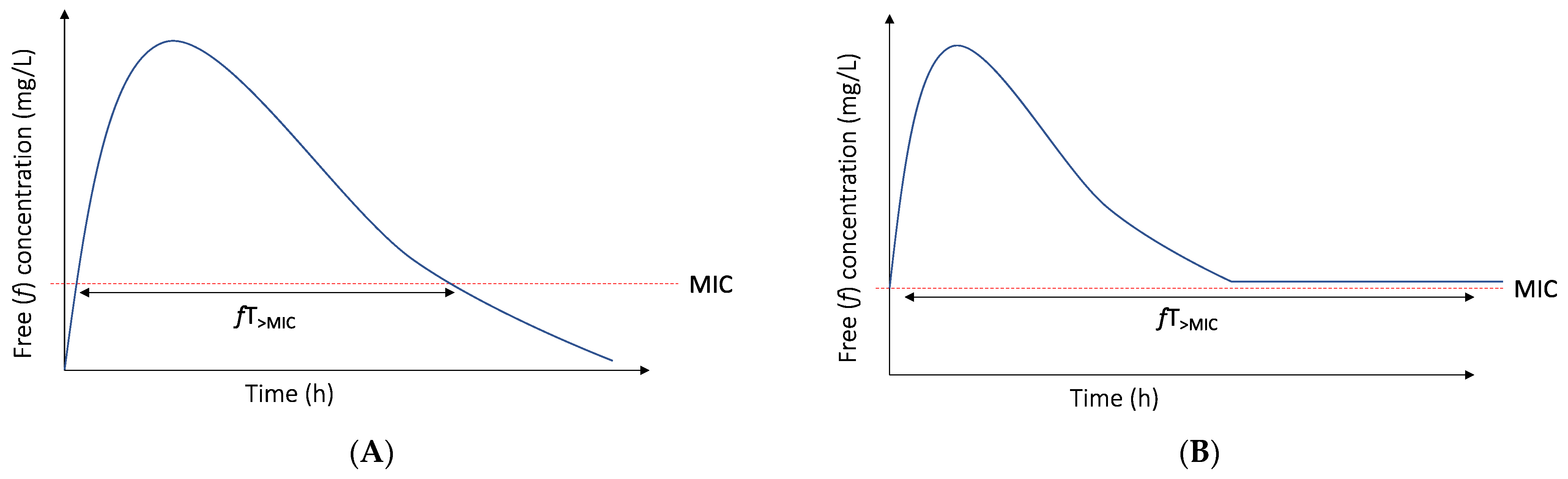
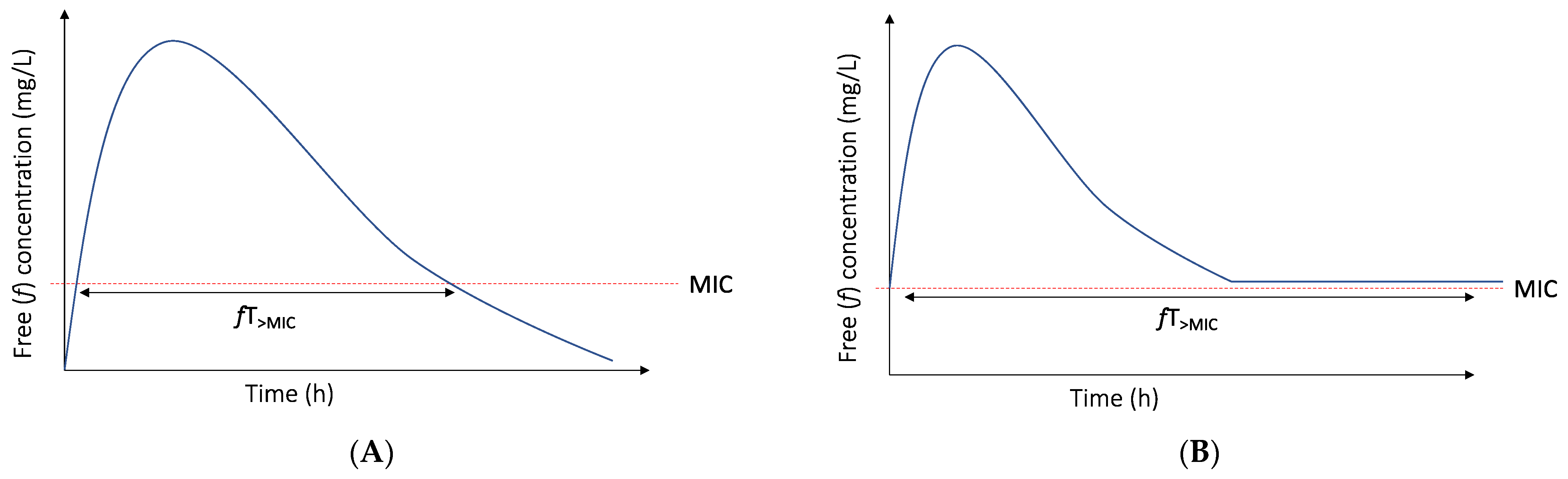
3.1. The PK/PD Index and Target of Choice for Beta-Lactam Antibiotics Are Not Static Entities
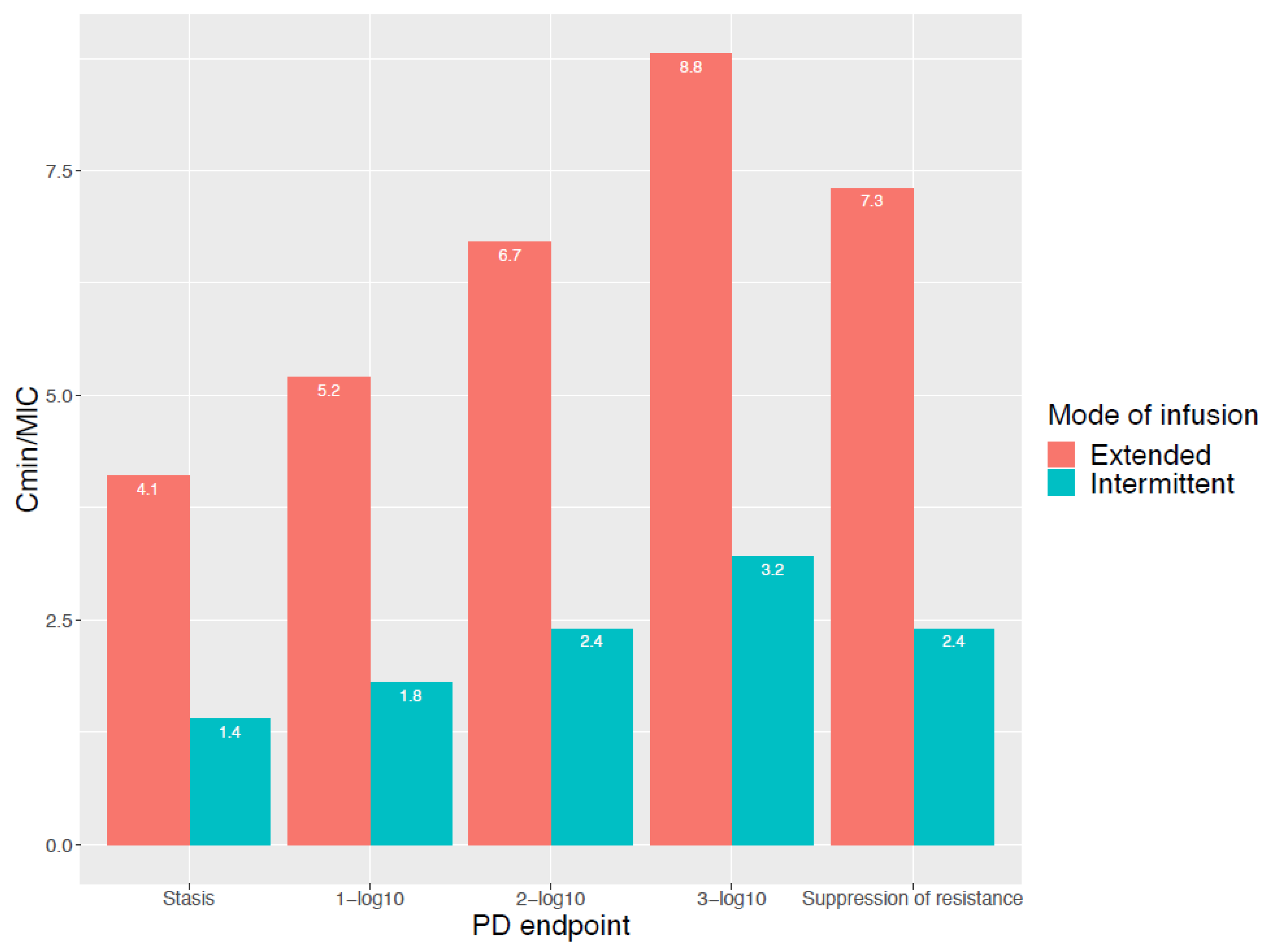
3.2. Bacterial Cell Kill Is Not the Only Goal
4. Introducing the ‘Maximum Tolerable Dose’ to Overcome the above Limitations

{kind=link}
{kind=link}
| Beta-Lactam Antibiotic | Neurotoxicity Levels Reported | References |
|---|---|---|
| Cefepime | 20 mg/dL (II, t), 21.6 mg/dL (II, t), 22 mg/dL (II, t), 36 mg/dL (II, t), 63.2 mg/dL (CI, ss) | [49,50,51,58,59] |
| Piperacillin/tazobactam | 361.4 mg/dL (II, t),157 mg/dL (CI, ss) | [47,60] |
| Meropenem | 64.2 mg/dL (II, t) | [47] |
| Flucloxacillin | 125.1 mg/dL (II, t) | [47] |
5. What Other Options Might We Have to Assess Beta-Lactam Antibiotic Toxicity?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kollef, M.H.; Bassetti, M.; Francois, B.; Burnham, J.; Dimopoulos, G.; Garnacho-Montero, J.; Lipman, J.; Luyt, C.-E.; Nicolau, D.P.; Postma, M.J.; et al. The Intensive Care Medicine Research Agenda on Multidrug-Resistant Bacteria, Antibiotics, and Stewardship. Intensive Care Med. 2017, 43, 1187–1197. [Google Scholar] [CrossRef]
- Dyar, O.J.; Huttner, B.; Schouten, J.; Pulcini, C. ESGAP (ESCMID Study Group for Antimicrobial stewardshiP) What Is Antimicrobial Stewardship? Clin. Microbiol. Infect. 2017, 23, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Society for Healthcare Epidemiology of America; Infectious Diseases Society of America; Pediatric Infectious Diseases Society. Policy Statement on Antimicrobial Stewardship by the Society for Healthcare Epidemiology of America (SHEA), the Infectious Diseases Society of America (IDSA), and the Pediatric Infectious Diseases Society (PIDS). Infect. Control Hosp. Epidemiol. 2012, 33, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Roger, C.; De Waele, J.J. Personalized Antibiotic Dosing for the Critically Ill. Intensive Care Med. 2019, 45, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.I.; Friberg, L.E. Pharmacokinetic-Pharmacodynamic Modeling of Antibacterial Drugs. Pharmacol. Rev. 2013, 65, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Lipman, J.; Blot, S.; Rello, J. Better Outcomes through Continuous Infusion of Time-Dependent Antibiotics to Critically Ill Patients? Curr. Opin. Crit. Care 2008, 14, 390–396. [Google Scholar] [CrossRef]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised Antibiotic Dosing for Patients Who Are Critically Ill: Challenges and Potential Solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Veiga, R.P.; Paiva, J.-A. Pharmacokinetics–Pharmacodynamics Issues Relevant for the Clinical Use of Beta-Lactam Antibiotics in Critically Ill Patients. Crit. Care 2018, 22, 233. [Google Scholar] [CrossRef] [Green Version]
- DALI: Defining Antibiotic Levels in Intensive Care Unit Patients: Are Current β-Lactam Antibiotic Doses Sufficient for Critically Ill Patients?|Clinical Infectious Diseases|Oxford Academic. Available online: https://academic.oup.com/cid/article/58/8/1072/356400?login=true (accessed on 29 March 2022).
- Taccone, F.S.; Laterre, P.-F.; Dugernier, T.; Spapen, H.; Delattre, I.; Wittebole, X.; De Backer, D.; Layeux, B.; Wallemacq, P.; Vincent, J.-L.; et al. Insufficient β-Lactam Concentrations in the Early Phase of Severe Sepsis and Septic Shock. Crit. Care 2010, 14, R126. [Google Scholar] [CrossRef] [Green Version]
- Craig, W.A. Interrelationship between Pharmacokinetics and Pharmacodynamics in Determining Dosage Regimens for Broad-Spectrum Cephalosporins. Diagn. Microbiol. Infect. Dis. 1995, 22, 89–96. [Google Scholar] [CrossRef]
- Craig, W.A. Pharmacokinetic/Pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clin. Infect. Dis. 1998, 26, 1–12. [Google Scholar] [CrossRef]
- Mouton, J.W.; Muller, A.E.; Canton, R.; Giske, C.G.; Kahlmeter, G.; Turnidge, J. MIC-Based Dose Adjustment: Facts and Fables. J. Antimicrob. Chemother. 2018, 73, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Aziz, M.H.; Sulaiman, H.; Mat-Nor, M.-B.; Rai, V.; Wong, K.K.; Hasan, M.S.; Abd Rahman, A.N.; Jamal, J.A.; Wallis, S.C.; Lipman, J.; et al. Beta-Lactam Infusion in Severe Sepsis (BLISS): A Prospective, Two-Centre, Open-Labelled Randomised Controlled Trial of Continuous versus Intermittent Beta-Lactam Infusion in Critically Ill Patients with Severe Sepsis. Intensive Care Med. 2016, 42, 1535–1545. [Google Scholar] [CrossRef]
- Roberts, J.A.; Kirkpatrick, C.M.J.; Roberts, M.S.; Dalley, A.J.; Lipman, J. First-Dose and Steady-State Population Pharmacokinetics and Pharmacodynamics of Piperacillin by Continuous or Intermittent Dosing in Critically Ill Patients with Sepsis. Int. J. Antimicrob. Agents 2010, 35, 156–163. [Google Scholar] [CrossRef]
- Lyu, Y.; Yang, Y.; Li, X.; Peng, M.; He, X.; Zhang, P.; Dong, S.; Wang, W.; Wang, D. Selection of Piperacillin/Tazobactam Infusion Mode Guided by SOFA Score in Cancer Patients with Hospital-Acquired Pneumonia: A Randomized Controlled Study. Ther. Clin. Risk Manag. 2017, 14, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Shan, T.; Liu, Y.; Ding, S.; Li, C.; Zhai, Q.; Chen, X.; Du, B.; Li, Y.; Zhang, J.; et al. Comparison of 3-hour and 30-minute infusion regimens for meropenem in patients with hospital acquired pneumonia in intensive care unit: A randomized controlled clinical trial. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2014, 26, 644–649. [Google Scholar] [CrossRef]
- Dulhunty, J.M.; Roberts, J.A.; Davis, J.S.; Webb, S.A.R.; Bellomo, R.; Gomersall, C.; Shirwadkar, C.; Eastwood, G.M.; Myburgh, J.; Paterson, D.L.; et al. Continuous Infusion of Beta-Lactam Antibiotics in Severe Sepsis: A Multicenter Double-Blind, Randomized Controlled Trial. Clin. Infect. Dis. 2013, 56, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Lv, Y.; Wang, D.; Xue, J.; Yan, Z. Clinical Outcomes of Extended versus Intermittent Administration of Piperacillin/Tazobactam for the Treatment of Hospital-Acquired Pneumonia: A Randomized Controlled Trial. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Wang, D. Experience with Extended-Infusion Meropenem in the Management of Ventilator-Associated Pneumonia Due to Multidrug-Resistant Acinetobacter Baumannii. Int. J. Antimicrob. Agents 2009, 33, 290–291. [Google Scholar] [CrossRef]
- Rafati, M.R.; Rouini, M.R.; Mojtahedzadeh, M.; Najafi, A.; Tavakoli, H.; Gholami, K.; Fazeli, M.R. Clinical Efficacy of Continuous Infusion of Piperacillin Compared with Intermittent Dosing in Septic Critically Ill Patients. Int. J. Antimicrob. Agents 2006, 28, 122–127. [Google Scholar] [CrossRef]
- Chytra, I.; Stepan, M.; Benes, J.; Pelnar, P.; Zidkova, A.; Bergerova, T.; Pradl, R.; Kasal, E. Clinical and Microbiological Efficacy of Continuous versus Intermittent Application of Meropenem in Critically Ill Patients: A Randomized Open-Label Controlled Trial. Crit. Care 2012, 16, R113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georges, B.; Conil, J.M.; Cougot, P.; Decun, J.F.; Archambaud, M.; Seguin, T.; Chabanon, G.; Virenque, C.; Houin, G.; Saivin, S. Cefepime in Critically Ill Patients: Continuous Infusion vs. an Intermittent Dosing Regimen. Int. J. Clin. Pharmacol. Ther. 2005, 43, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Vardakas, K.Z.; Voulgaris, G.L.; Maliaros, A.; Samonis, G.; Falagas, M.E. Prolonged versus Short-Term Intravenous Infusion of Antipseudomonal β-Lactams for Patients with Sepsis: A Systematic Review and Meta-Analysis of Randomised Trials. Lancet Infect. Dis. 2018, 18, 108–120. [Google Scholar] [CrossRef]
- Roberts, J.A.; Abdul-Aziz, M.-H.; Davis, J.S.; Dulhunty, J.M.; Cotta, M.O.; Myburgh, J.; Bellomo, R.; Lipman, J. Continuous versus Intermittent β-Lactam Infusion in Severe Sepsis. A Meta-Analysis of Individual Patient Data from Randomized Trials. Am. J. Respir. Crit. Care Med. 2016, 194, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, N.J.; Liu, J.; O’Donnell, J.N.; Dulhunty, J.M.; Abdul-Aziz, M.H.; Berko, P.Y.; Nadler, B.; Lipman, J.; Roberts, J.A. Prolonged Infusion Piperacillin-Tazobactam Decreases Mortality and Improves Outcomes in Severely Ill Patients: Results of a Systematic Review and Meta-Analysis. Crit. Care Med. 2018, 46, 236–243. [Google Scholar] [CrossRef]
- The George Institute. A Phase III Randomised Controlled Trial of Continuous Beta-Lactam Infusion Compared with Intermittent Beta-Lactam Dosing in Critically Ill Patients; clinicaltrials.gov, 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03213990 (accessed on 14 April 2022).
- Dhaese, S.; Heffernan, A.; Liu, D.; Abdul-Aziz, M.H.; Stove, V.; Tam, V.H.; Lipman, J.; Roberts, J.A.; De Waele, J.J. Prolonged Versus Intermittent Infusion of β-Lactam Antibiotics: A Systematic Review and Meta-Regression of Bacterial Killing in Preclinical Infection Models. Clin. Pharmacokinet. 2020, 59, 1237–1250. [Google Scholar] [CrossRef]
- Felton, T.W.; Goodwin, J.; O’Connor, L.; Sharp, A.; Gregson, L.; Livermore, J.; Howard, S.J.; Neely, M.N.; Hope, W.W. Impact of Bolus Dosing versus Continuous Infusion of Piperacillin and Tazobactam on the Development of Antimicrobial Resistance in Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 5811–5819. [Google Scholar] [CrossRef] [Green Version]
- Sumi, C.D.; Heffernan, A.J.; Naicker, S.; Islam, K.; Cottrell, K.; Wallis, S.C.; Lipman, J.; Harris, P.N.A.; Sime, F.B.; Roberts, J.A. Pharmacodynamic Evaluation of Intermittent versus Extended and Continuous Infusions of Piperacillin/Tazobactam in a Hollow-Fibre Infection Model against Klebsiella Pneumoniae. J. Antimicrob. Chemother. 2020, 75, 2633–2640. [Google Scholar] [CrossRef]
- Sinnollareddy, M.G.; Roberts, M.S.; Lipman, J.; Roberts, J.A. β-Lactam Pharmacokinetics and Pharmacodynamics in Critically Ill Patients and Strategies for Dose Optimization: A Structured Review. Clin. Exp. Pharmacol. Physiol. 2012, 39, 489–496. [Google Scholar] [CrossRef]
- Bergen, P.J.; Bulitta, J.B.; Kirkpatrick, C.M.J.; Rogers, K.E.; McGregor, M.J.; Wallis, S.C.; Paterson, D.L.; Nation, R.L.; Lipman, J.; Roberts, J.A.; et al. Substantial Impact of Altered Pharmacokinetics in Critically Ill Patients on the Antibacterial Effects of Meropenem Evaluated via the Dynamic Hollow-Fiber Infection Model. Antimicrob. Agents Chemother. 2017, 61, e02642-16. [Google Scholar] [CrossRef] [Green Version]
- Kristoffersson, A.N.; David-Pierson, P.; Parrott, N.J.; Kuhlmann, O.; Lave, T.; Friberg, L.E.; Nielsen, E.I. Simulation-Based Evaluation of PK/PD Indices for Meropenem Across Patient Groups and Experimental Designs. Pharm. Res. 2016, 33, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Bulitta, J.B.; Kirkpatrick, C.M.J.; Rogers, K.E.; McGregor, M.J.; Wallis, S.C.; Paterson, D.L.; Lipman, J.; Roberts, J.A.; Landersdorfer, C.B. Effect of Different Renal Function on Antibacterial Effects of Piperacillin against Pseudomonas Aeruginosa Evaluated via the Hollow-Fibre Infection Model and Mechanism-Based Modelling. J. Antimicrob. Chemother. 2016, 71, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Sjövall, F.; Alobaid, A.S.; Wallis, S.C.; Perner, A.; Lipman, J.; Roberts, J.A. Maximally Effective Dosing Regimens of Meropenem in Patients with Septic Shock. J. Antimicrob. Chemother. 2018, 73, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Van Herendael, B.; Jeurissen, A.; Tulkens, P.M.; Vlieghe, E.; Verbrugghe, W.; Jorens, P.G.; Ieven, M. Continuous Infusion of Antibiotics in the Critically Ill: The New Holy Grail for Beta-Lactams and Vancomycin? Ann. Intensive Care 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taccone, F.S.; Laupland, K.B.; Montravers, P. Continuous Infusion of β-Lactam Antibiotics for All Critically Ill Patients? Intensive Care Med. 2016, 42, 1604–1606. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.I.; Cars, O.; Friberg, L.E. Pharmacokinetic/Pharmacodynamic (PK/PD) Indices of Antibiotics Predicted by a Semimechanistic PKPD Model: A Step toward Model-Based Dose Optimization. Antimicrob. Agents Chemother. 2011, 55, 4619–4630. [Google Scholar] [CrossRef] [Green Version]
- Craig, W.A.; Redington, J.; Ebert, S.C. Pharmacodynamics of Amikacin in Vitro and in Mouse Thigh and Lung Infections. J. Antimicrob. Chemother. 1991, 27, 29–40. [Google Scholar] [CrossRef]
- Tam, V.H.; Schilling, A.N.; Poole, K.; Nikolaou, M. Mathematical Modelling Response of Pseudomonas Aeruginosa to Meropenem. J. Antimicrob. Chemother. 2007, 60, 1302–1309. [Google Scholar] [CrossRef]
- Sumi, C.D.; Heffernan, A.J.; Lipman, J.; Roberts, J.A.; Sime, F.B. What Antibiotic Exposures Are Required to Suppress the Emergence of Resistance for Gram-Negative Bacteria? A Systematic Review. Clin. Pharmacokinet. 2019, 58, 1407–1443. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Zhang, X.-J.; Yang, Y.; Gong, W.-T.; Xu, B.; Zhu, Y.-Q.; Liu, W. Evaluation of Meropenem Regimens Suppressing Emergence of Resistance in Acinetobacter Baumannii with Human Simulated Exposure in an in Vitro Intravenous-Infusion Hollow-Fiber Infection Model. Antimicrob. Agents Chemother. 2014, 58, 6773–6781. [Google Scholar] [CrossRef] [Green Version]
- Drusano, G.L.; Hope, W.; MacGowan, A.; Louie, A. Suppression of Emergence of Resistance in Pathogenic Bacteria: Keeping Our Powder Dry, Part 2. Antimicrob. Agents Chemother. 2015, 60, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Firsov, A.A.; Vostrov, S.N.; Lubenko, I.Y.; Drlica, K.; Portnoy, Y.A.; Zinner, S.H. In Vitro Pharmacodynamic Evaluation of the Mutant Selection Window Hypothesis Using Four Fluoroquinolones against Staphylococcus Aureus. Antimicrob. Agents Chemother. 2003, 47, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugel, J.; Dos Santos Pereira, A.; Pignatari, A.C.C.; Gales, A.C. Beta-Lactam MICs Correlate Poorly with Mutant Prevention Concentrations for Clinical Isolates of Acinetobacter Spp. and Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2276–2277. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.; Marriott, D.; Schultz, H.B.; Gould, M.; Andresen, D.; Wicha, S.G.; Alffenaar, J.-W.; Penm, J.; Reuter, S.E. Assessment of Cefepime Toxicodynamics: Comprehensive Examination of Pharmacokinetic/Pharmacodynamic Targets for Cefepime-Induced Neurotoxicity and Evaluation of Current Dosing Guidelines. Int. J. Antimicrob. Agents 2021, 58, 106443. [Google Scholar] [CrossRef] [PubMed]
- Imani, S.; Buscher, H.; Marriott, D.; Gentili, S.; Sandaradura, I. Too Much of a Good Thing: A Retrospective Study of β-Lactam Concentration-Toxicity Relationships. J. Antimicrob. Chemother. 2017, 72, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Beumier, M.; Casu, G.S.; Hites, M.; Wolff, F.; Cotton, F.; Vincent, J.L.; Jacobs, F.; Taccone, F.S. Elevated β-Lactam Concentrations Associated with Neurological Deterioration in ICU Septic Patients. Minerva Anestesiol 2015, 81, 497–506. [Google Scholar] [PubMed]
- Huwyler, T.; Lenggenhager, L.; Abbas, M.; Ing Lorenzini, K.; Hughes, S.; Huttner, B.; Karmime, A.; Uçkay, I.; von Dach, E.; Lescuyer, P.; et al. Cefepime Plasma Concentrations and Clinical Toxicity: A Retrospective Cohort Study. Clin. Microbiol. Infect. 2017, 23, 454–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boschung-Pasquier, L.; Atkinson, A.; Kastner, L.K.; Banholzer, S.; Haschke, M.; Buetti, N.; Furrer, D.I.; Hauser, C.; Jent, P.; Que, Y.A.; et al. Cefepime Neurotoxicity: Thresholds and Risk Factors. A Retrospective Cohort Study. Clin. Microbiol. Infect. 2020, 26, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.; Marriott, D.; Gould, M.; Andresen, D.; Reuter, S.E.; Penm, J. A Retrospective Study to Determine the Cefepime-Induced Neurotoxicity Threshold in Hospitalized Patients. J. Antimicrob. Chemother. 2020, 75, 718–725. [Google Scholar] [CrossRef]
- Lodise, T.P.; Sorgel, F.; Melnick, D.; Mason, B.; Kinzig, M.; Drusano, G.L. Penetration of Meropenem into Epithelial Lining Fluid of Patients with Ventilator-Associated Pneumonia. Antimicrob. Agents Chemother. 2011, 55, 1606–1610. [Google Scholar] [CrossRef] [Green Version]
- Vardakas, K.Z.; Kalimeris, G.D.; Triarides, N.A.; Falagas, M.E. An Update on Adverse Drug Reactions Related to β-Lactam Antibiotics. Expert Opin. Drug Saf. 2018, 17, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Roger, C.; Louart, B. Beta-Lactams Toxicity in the Intensive Care Unit: An Underestimated Collateral Damage? Microorganisms 2021, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Mousseaux, C.; Rafat, C.; Letavernier, E.; Frochot, V.; Kerroumi, Y.; Zeller, V.; Luque, Y. Acute Kidney Injury After High Doses of Amoxicillin. Kidney Int. Rep. 2021, 6, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Demotier, S.; Limelette, A.; Charmillon, A.; Baux, E.; Parent, X.; Mestrallet, S.; Pavel, S.; Servettaz, A.; Dramé, M.; Muggeo, A.; et al. Incidence, Associated Factors, and Effect on Renal Function of Amoxicillin Crystalluria in Patients Receiving High Doses of Intravenous Amoxicillin (The CRISTAMOX Study): A Cohort Study. eClinicalMedicine 2022, 45, 101340. [Google Scholar] [CrossRef] [PubMed]
- Barreto, E.F.; Webb, A.J.; Pais, G.M.; Rule, A.D.; Jannetto, P.J.; Scheetz, M.H. Setting the Beta-Lactam Therapeutic Range for Critically Ill Patients: Is There a Floor or Even a Ceiling? Crit. Care Explor. 2021, 3, e0446. [Google Scholar] [CrossRef]
- Lamoth, F.; Buclin, T.; Pascual, A.; Vora, S.; Bolay, S.; Decosterd, L.A.; Calandra, T.; Marchetti, O. High Cefepime Plasma Concentrations and Neurological Toxicity in Febrile Neutropenic Patients with Mild Impairment of Renal Function. Antimicrob. Agents Chemother. 2010, 54, 4360–4367. [Google Scholar] [CrossRef] [Green Version]
- Vercheval, C.; Sadzot, B.; Maes, N.; Denooz, R.; Damas, P.; Frippiat, F. Continuous Infusion of Cefepime and Neurotoxicity: A Retrospective Cohort Study. Clin. Microbiol. Infect. 2021, 27, 731–735. [Google Scholar] [CrossRef]
- Quinton, M.-C.; Bodeau, S.; Kontar, L.; Zerbib, Y.; Maizel, J.; Slama, M.; Masmoudi, K.; Lemaire-Hurtel, A.-S.; Bennis, Y. Neurotoxic Concentration of Piperacillin during Continuous Infusion in Critically Ill Patients. Antimicrob. Agents Chemother. 2017, 61, e00654-17. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef]
- Rosner, M.H.; Reis, T.; Husain-Syed, F.; Vanholder, R.; Hutchison, C.; Stenvinkel, P.; Blankestijn, P.J.; Cozzolino, M.; Juillard, L.; Kashani, K.; et al. Classification of Uremic Toxins and Their Role in Kidney Failure. CJASN 2021, 16, 1918–1928. [Google Scholar] [CrossRef]
- Masereeuw, R.; Mutsaers, H.A.M.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The Kidney and Uremic Toxin Removal: Glomerulus or Tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of Uremic Toxin Transport by Organic Anion Transporters in the Kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in Vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Miners, J.O.; Yang, X.; Knights, K.M.; Zhang, L. The Role of the Kidney in Drug Elimination: Transport, Metabolism, and the Impact of Kidney Disease on Drug Clearance. Clin. Pharmacol. Ther. 2017, 102, 436–449. [Google Scholar] [CrossRef]
- Wen, S.; Wang, C.; Duan, Y.; Huo, X.; Meng, Q.; Liu, Z.; Yang, S.; Zhu, Y.; Sun, H.; Ma, X.; et al. OAT1 and OAT3 Also Mediate the Drug-Drug Interaction between Piperacillin and Tazobactam. Int. J. Pharm. 2018, 537, 172–182. [Google Scholar] [CrossRef]
- Tjandramaga, T.B.; Mullie, A.; Verbesselt, R.; De Schepper, P.J.; Verbist, L. Piperacillin: Human Pharmacokinetics after Intravenous and Intramuscular Administration. Antimicrob. Agents Chemother. 1978, 14, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kestenbaum, B. Proximal Tubular Secretory Clearance: A Neglected Partner of Kidney Function. Clin. J. Am. Soc. Nephrol. 2018, 13, 1291–1296. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhaese, S.A.M.; Hoste, E.A.; De Waele, J.J. Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the ‘Maximum Tolerable Dose’. Antibiotics 2022, 11, 889. https://doi.org/10.3390/antibiotics11070889
Dhaese SAM, Hoste EA, De Waele JJ. Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the ‘Maximum Tolerable Dose’. Antibiotics. 2022; 11(7):889. https://doi.org/10.3390/antibiotics11070889
Chicago/Turabian StyleDhaese, Sofie A. M., Eric A. Hoste, and Jan J. De Waele. 2022. "Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the ‘Maximum Tolerable Dose’" Antibiotics 11, no. 7: 889. https://doi.org/10.3390/antibiotics11070889
APA StyleDhaese, S. A. M., Hoste, E. A., & De Waele, J. J. (2022). Why We May Need Higher Doses of Beta-Lactam Antibiotics: Introducing the ‘Maximum Tolerable Dose’. Antibiotics, 11(7), 889. https://doi.org/10.3390/antibiotics11070889